

Abstract
GM‐CSF is important in regulating acute, persistent neutrophilic inflammation in certain settings, including lung injury. Ligand binding induces rapid internalization of the GM‐CSF receptor (GM‐CSFRα) complex, a process essential for signaling. Whereas GM‐CSF controls many aspects of neutrophil biology, regulation of GM‐CSFRα expression is poorly understood, particularly the role of GM‐CSFRα in ligand clearance and whether signaling is sustained despite major down‐regulation of GM‐CSFRα surface expression. We established a quantitative assay of GM‐CSFRα surface expression and used this, together with selective anti‐GM‐CSFR antibodies, to define GM‐CSFRα kinetics in human neutrophils, and in murine blood and alveolar neutrophils in a lung injury model. Despite rapid sustained ligand‐induced GM‐CSFRα loss from the neutrophil surface, which persisted even following ligand removal, pro‐survival effects of GM‐CSF required ongoing ligand‐receptor interaction. Neutrophils recruited to the lungs following LPS challenge showed initially high mGM‐CSFRα expression, which along with mGM‐CSFRβ declined over 24 hr; this was associated with a transient increase in bronchoalveolar lavage fluid (BALF) mGM‐CSF concentration. Treating mice in an LPS challenge model with CAM‐3003, an anti‐mGM‐CSFRα mAb, inhibited inflammatory cell influx into the lung and maintained the level of BALF mGM‐CSF. Consistent with neutrophil consumption of GM‐CSF, human neutrophils depleted exogenous GM‐CSF, independent of protease activity. These data show that loss of membrane GM‐CSFRα following GM‐CSF exposure does not preclude sustained GM‐CSF/GM‐CSFRα signaling and that this receptor plays a key role in ligand clearance. Hence neutrophilic activation via GM‐CSFR may play an important role in neutrophilic lung inflammation even in the absence of high GM‐CSF levels or GM‐CSFRα expression.
Keywords: alveolar, apoptosis, inflammation, LPS, signaling
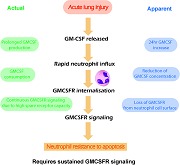
GM‐CSF released during ALI stimulates neutrophil recruitment, induces down‐regulation of GM‐CSFR, and GM‐CSF consumption by neutrophils, yet sustained anti‐apoptotic signals require continued GM‐CSF stimulation.
Abbreviations
- (m)GM‐CSFRα/β
(murine) granulocyte‐macrophage colony stimulating factor receptor‐α/β
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BALF
bronchoalveolar lavage fluid
1. INTRODUCTION
Neutrophils are a key component of the inflammatory response and play a central role in the pathogenesis of the acute respiratory distress syndrome (ARDS).1 Indeed, the extent and duration of alveolar airspace neutrophilia in ARDS is a strong predictor of outcome.2 Whereas the presence of neutrophils within an inflamed tissue does not mandate a pathogenic role for these cells, in ARDS we and others have shown that the neutrophils within the alveolar airspace have a highly primed and pro‐survival phenotype with enhanced superoxide anion and protease release, preserved neutrophil‐extracellular trap (NET) formation, and delayed apoptosis3, 4 and as such are considered to be important drivers of lung injury.
GM‐CSF is a 14.7 kDa heavily glycosylated protein, and one of the four recognized myeloid CSFs. GM‐CSF is produced from a variety of cells including monocyte/macrophages, T cells, fibroblasts, and lung epithelial cells5, 6 and as well as being a key myeloid growth factor, has important functional effects on a range of fully mature cells including neutrophils, monocytes, and eosinophils. This cytokine is also essential for alveolar macrophage function7, 8 and lung surfactant homeostasis.9, 10 Ligand binding to its receptor GM‐CSFRα results in dimerization with, and signaling via, the GM‐CSFRβ chain, also called the common β receptor, which is shared with the IL‐3 and IL‐5 cytokine signaling pathways.11, 12 Activation of this receptor complex results in JAK‐mediated receptor tyrosine phosphorylation and subsequent interaction with a Shc adaptor protein and GRB2/SoS complex to initiate signaling.13
As well as stimulating myeloid cell proliferation and granulocyte release from the bone marrow,14 GM‐CSF has effects on a number of other neutrophil functions including (i) up‐regulation of IgA FcR, FMLPR, CD11b and LTB4 receptor expression; (ii) enhanced chemotaxis, phagocytosis, release of LTB4 and arachidonic acid, and NOX2‐mediated superoxide anion generation; and (iii) a marked pro‐survival effect mediated by PI3K‐dependent inhibition of apoptosis.15, 16 Indeed, GM‐CSF, which is found in abundance during the very early phase of most forms of acute lung injury, has been shown to be the dominant factor inhibiting neutrophil apoptosis in the alveolar airspace of patients with ARDS.3
It is therefore reasonable to propose that GM‐CSF acting at GM‐CSFRα in the lungs of patients with nonviral‐mediated ARDS could be playing a central role in the exuberant immune response evident in the lungs during ALI. However, in cell lines transfected with or selected for high constitutive expression of GM‐CSFRα, it has been shown that ligand binding induces rapid and substantial receptor internalization (t 1/2 = 11 ± 4 min in erythroblast TF‐1 cells; 8 ± 2 min in FD‐hGMR (FDCP‐1 cells overexpressing human GM‐CSFR) cells, a mouse fibroblast cell line expressing human GM‐CSFRs).17 Likewise, agonism of the cytokine‐specific GM‐CSFα chains in TF‐1‐F11 (TF‐1 cells selected for high expression of GM‐CSFR) cells causes marked proteasome‐dependent degradation of the GM‐CSFβ common β chain,18 which terminates signaling via the receptor complex. One further key, and as yet unresolved question, is whether the very transient nature of the increased alveolar GM‐CSF levels seen in ARDS reflects transient GM‐CSF generation and/or persistent production but enhanced ligand clearance.
Addressing this question, and understanding GM‐CSFR dynamics in human neutrophils, especially in those cells recovered from an inflammatory setting, is therefore crucial to further establish a predominant role for GM‐CSF in ALI. For example, internalization of the GM‐CSFRα might lead to cessation of GM‐CSF signaling and predict an early loss of GM‐CSF mediated effects; in contrast, if the GM‐CSFRα complex remains highly active despite a reduction in cell surface abundance and plays a key role in ligand removal, then single time point measures of GM‐CSF abundance and/or GM‐CSFRα expression in clinical samples might severely underestimate the functional importance of this signaling pathway.
To address this, we established a new quantitative assay of GM‐CSFRα expression to study GM‐CSFRα kinetics in human neutrophils and used a murine lung injury model to explore the dynamics of GM‐CSFRα expression in blood and alveolar neutrophils in vivo. Our data show that loss of cell membrane GM‐CSFRα following GM‐CSF does not preclude sustained GM‐CSF/GM‐CSFRα signaling and that this receptor plays a key role in ligand clearance. These findings have important implications for the interpretation of translational data such as GM‐CSF concentrations measured in disease samples, and of studies investigating the pathogenesis of neutrophilic disease using GM‐CSFRα blockade.
2. MATERIALS AND METHODS
2.1. Study participants
Human peripheral blood neutrophils were isolated from adult healthy non‐medicated volunteers. Neutrophils were also isolated from the blood and the bronchoalveolar lavage fluid (BALF) of patients requiring mechanical ventilation for ARDS as previously detailed.4 All studies complied with the Declaration of Helsinki and were approved by the Cambridge Research Ethics Committee (08/H03306/17); written informed consent was obtained from all subjects or their legal surrogate.
2.2. Isolation of human neutrophils
Peripheral neutrophils were isolated from sodium citrate anti‐coagulated venous blood, using dextran sedimentation and discontinuous Percoll gradients as described19 and resuspended in IMDM supplemented with 10% human serum and penicillin/streptomycin. BALF neutrophils were isolated by negative selection (Robosep).4, 15 The purity of the isolated blood neutrophils was > 95%, with less than 1% mononuclear cells and 4% eosinophils.
2.3. TF‐1 cell viability assays
TF‐1 cells (erythroleukemic cell line; R&D Systems, Abingdon, UK) maintained in 4 ng/ml human GM‐CSF (as supplier's instructions, in RPMI‐1640 with 5% FBS (heat inactivated) and penicillin/streptomycin) were washed 3 times to ensure complete removal of GM‐CSF. The cells were then treated with 0.25 ng/ml GM‐CSF (R&D Systems), in the presence or absence of a serial dilution of CAM3001 (blocking antibody specific to human GM‐CSFRα, MedImmune Ltd, Cambridge, UK) or isotype control (NIP228, MedImmune), with both ligand and antibody being added at the same time to the cultures. The cells were incubated for 72 hr. CellTiter‐Glo (Promega UK, Southampton, UK, G7570) was used to measure ATP as an indirect measure of the number of viable cells according to the manufacturer's instructions.
2.4. Quantification of GM‐CSFRα
TF‐1 cells (R&D Systems) that had been maintained in human GM‐CSF (4 ng/ml, R&D Systems) were washed 3 times to remove GM‐CSF. GM‐CSFRα expression was assessed on the cells following culture for a further 18 hr in the presence or absence of GM‐CSF (4 ng/ml).
GM‐CSFRα expression was quantified on human neutrophils cultured in the presence of GM‐CSF (0.001‐10 ng/ml, R&D Systems), LPS (100 ng/ml, Sigma Aldrich UK, Poole, UK), TNFα (20 ng/ml, R&D Systems) or appropriate vehicle control. In certain experiments neutrophils were pretreated for 30 min with CAM‐3001 (blocking antibody specific to human GM‐CSFRα) or for 1 hr with the proteasomal inhibitor MG132 (20 μM, Sigma Aldrich), brefeldin A (10 μg/ml) to block lysosomal degradation, or the transcriptional inhibitor actinomycin D (2 μg/ml); in certain experiments IL‐8 was measured in the supernatants using an in‐house ELISA.20 GM‐CSFRα levels were also quantified on blood neutrophils and BALF neutrophils derived from patients with ARDS.
2.4.1. Flow cytometry
Pelleted TF‐1 cells were resuspended in 100 μl FACS buffer (eBioscience, ThermoFisher Scientific UK, Loughborough, UK supplemented with 0.1 μM EDTA) containing anti‐human GM‐CSFRα (CD116) antibody or isotype control (both BD Pharmingen, BD Bioscience, Wokingham, UK) (0.5 μg/stain) for 30 min in the dark on ice. TF‐1 cells were then washed in FACS buffer and fixed with 200 μl 4% formaldehyde in PBS for 10 min at RT and analyzed by flow cytometry (LSRII Fortessa, BD Biosciences).
Pelleted neutrophils were resuspended in 100 μl FACS buffer (eBioscience, supplemented with 0.1 μM EDTA) containing phycoerythrin (PE)‐mouse anti‐human GM‐CSFRα (CD116) antibody (0.04 μg/ml) (BD Pharmingen) for 30 min in the dark on ice. Neutrophils were then washed in FACS buffer and fixed with 500 μl 4% formaldehyde in PBS for 10 min at RT and analyzed by flow cytometry (LSRII Fortessa, BD Pharmingen). To determine the absolute number of GM‐CSFRα copies expressed per neutrophil, the mean fluorescence intensity (MFI) values for CD116 (GM‐CSFRα) staining were interpolated against a standard curve obtained by staining 5 bead populations concurrently (1 blank and 4 with increasing antibody binding capacity) (Quantum Simply Cellular anti‐mouse IgG, Bangs Laboratories, Fishers, Indiana, USA) (Supplemental Fig. S1). These microspheres acted as external standards to enable the standardization of fluorescence intensity units irrespective of the detecting instrument, settings or software. Unknowns were read against the calibration curve using the manufacturer's QuickCal analysis template, after confirmation of detection threshold and linearity. Auto‐fluorescence of neutrophils accounted for a portion of the observed fluorescence intensity and this was corrected for by subtraction of the interpolated receptor number from a parallel neutrophil control, minus CD116 antibody (unstained).
2.4.2. Confocal microscopy
Cytospins of freshly isolated healthy volunteer blood neutrophils were prepared,21 stained with AlexaFluor647‐CAM‐3001 (1:100 dilution; MedImmune) and mounted with Pro‐Long Gold Anti‐Fade Mountant with DAPI (ThermoFisher Scientific, Loughborough, UK) prior to imaging (Leica TCS SP5).
2.5. GM‐CSFRα mRNA analysis
Total RNA was isolated from neutrophils purified from 10 healthy volunteer donors (treated for 6 hr in the presence or absence of 1 ng/ml recombinant human GM‐CSF) using TRI‐reagent (Sigma, Aldrich, UK) and RNeasy mini‐columns (Qiagen, Manchester, UK); complimentary DNA (cDNA) was prepared, fragmented, labelled and hybridized onto GeneChip Human Genome U133 Plus 2.0 oligonucleotide arrays as detailed.4 The data, as submitted on GSE76293, were processed using R/Bioconductor and normalized using RMA from the “affy” package. The fold change values, and the negative logarithm of the adjusted P values were computed using the “limma” package, where empirical Bayes statistics and the Benjamini‐Hochberg correction method were used.
2.6. Assessment of neutrophil apoptosis in vitro
Neutrophils were cultured for 20 hr with GM‐CSF, CAM‐3001 (1 μM), or buffer (as detailed above) and apoptosis assessed by flow cytometry following double staining with FITC‐Annexin V and PI (FITC‐Annexin V Apoptosis Detection kit I, BD Pharmingen).22 Apoptotic neutrophils were identified as being Annexin V positive and PI negative. Previous studies from our group had confirmed the tight agreement between apoptosis values obtained in neutrophils using this method and several other standard assessments of apoptosis including direct morphologic quantification.16, 21
2.7. LPS‐induced lung injury
Pathogen‐free female C57BL/6 or BALBc/JBomTac mice were obtained from Charles River Laboratories, Margate, UK or Taconic Europe, Laven, Denmark, respectively, and studied at 8–9 weeks of age with a body weight of circa 20 g. Mice were supplied with food and water ad libitum and observed carefully after the LPS challenge for any adverse effects. In vivo procedures performed in the United Kingdom were conducted under the authority of a Home Office issued Project License in accordance with the Animals [Scientific Procedures] Act 1986 with appropriate ethical approval, and procedures in Sweden conducted under AZ permit number 31–11684/08 and ethics approval M104/08; group sizes were determined either using the MFI for GM‐CSFRα in BALF neutrophil numbers or total cell influx to LPS seen in preliminary studies, with α set at 0.05, β to 0.2, and power to 80%. LPS was delivered where indicated via nebulized aerosol to induce lung inflammation. LPS, P. aeruginosa, serotype 10, phenol extracted (Sigma Aldrich), was dissolved in physiologic saline (9 mg/ml NaCl). The mice were placed in a semi‐open inhalation box (max. 10 mice) and exposed once to nebulized aerosol of P. aeruginosa LPS (1 mg/ml) for 10 min. The aerosol was generated by a Pari LC Jet Star nebulizer, using 5 ml LPS suspension and a flow of 5 l/min (pressure = 2 Bar). The control group was exposed to saline according to the same procedure. Animals were dosed intranasally (i.n.) with CAM‐3003 or isotype control 3 hr before LPS challenge. Budesonide control was administered (3 mg/kg p.o.) 3 hr prior to LPS challenge.
In other instances, lung inflammation was induced by instillation of 10 μg LPS (E. coli 026:B6, Sigma Aldrich) per mouse delivered i.n. in 25 μl of PBS (vehicle control) to groups of 6 mice under light isoflurane anesthesia,23 the optimal dose having previously been confirmed in a study comparing 0.1, 1, or 10 μg per mouse (data not shown). In some experiments, mouse groups were treated with CAM‐3003 (mouse equivalent to CAM‐3001, MedImmune, Lot SP08‐013; 400 μg in 40 μl, i.n. as above) or PBS 3 hr prior, or 6 hr following, LPS instillation (10 μg LPS).
Mice were terminally anesthetized via intraperitoneal administration of Euthatal at 3, 6, or 24 hr post LPS and blood drawn from the vena cava. BALF was collected via an endotracheal cannula placed proximal to the larynx and aliquots of recovered PBS (approx. 1 ml total) were pooled and used for cytokine profiling, flow cytometry, and cytospins. Cytospins (Shandon Cytospin 3) from BALF were methanol fixed and stained with REASTAIN Quick‐Diff Kit (Reagena, Toivala, Finland). Femurs were collected, and the bone marrow flushed to collect cells for flow cytometry. A staggered dosing strategy was used to ensure that all cellular samples were collected, stained and analyzed in parallel where required.
2.8. Assessment of LPS‐induced lung injury
Total and differential cell counts were quantified either using cytospins, and/or (following red blood cell lysis) blood, BALF and bone marrow samples were stained with a panel of fluorescent antibodies and the percentage of neutrophils calculated: BUV395‐CD45 (4 μg/ml, clone 30‐F11, BD Horizon, BD Bioscience, Wokingham, UK), BUV421‐Ly6G (12 μg/ml, clone 1A8, BD Horizon), PE‐Ly6C (12 μg/ml, clone AL‐21, BD Pharmingen), and eF780 viability dye (1:1000 dilution, eBioscience). The cells were fixed in 4% formaldehyde before being analyzed (LSRII Fortessa, BD Horizon). Neutrophils were identified as CD45+, Ly6G high, CD11b high and Ly6C low. In addition, a total cell count was performed on BALF by flow cytometry following a 1:9 dilution in PBS (MACSQuant, Bergisch Gladbach, Germany); leukocytes were identified by their characteristic FSC/SSC distribution.
Mouse GM‐CSF in BALF was measured by ELISA (DuoSet ELISA kits, R&D Systems) and IL‐1β in lung homogenate was measured using MSD multiplex analysis (Mouse Pro‐inflammatory 7‐plex, MesoScale Discovery, Rockville, Maryland, USA).
2.9. Measurement of murine GM‐CSFRα and GM‐CSFRβ expression in neutrophils
GM‐CSFRα and β expression was quantified in mouse BALF, blood and bone marrow neutrophils by flow cytometry and expressed as the MFI geometric mean. Cells were stained for GM‐CSFRα with APC‐CAM‐3003 (labelled with Lightning‐Link APC as per the manufacturer's instructions, Innova Biosciences, Cambridge, UK), GM‐CSFRβ with PE‐CD131 (JORO50, BD Pharmingen), and BUV395‐CD45 (4 μg/ml, clone 30‐F11, BD Horizon), BV421‐Ly6G (12 μg/ml, clone 1A8, BD Horizon), AlexaFluor488‐Ly6C (12 μg/ml, clone HK1.4, Biolegend, San Diego, California, USA), AlexaFluor488‐CD11b (12 μg/ml, clone M1/70, BD Pharmingen) and eF780 viability dye (1:1000 dilution, eBioscience). All flow cytometry was performed on one day on a single instrument for each study.
2.10. In vitro ligand depletion
Human neutrophils were cultured in the presence of GM‐CSF (30 pg/ml, R&D Systems) or vehicle alone and supernatant was collected over a 24 hr time course. Where appropriate, cells were pretreated (30 min) with Sivelestat (10 μM, Sigma Aldrich) and EDTA (R&D Systems). The level of GM‐CSF remaining in the supernatant was determined by ELISA (R&D Systems).
2.11. Statistical analysis
Data are expressed as mean ± sem for (n) separate experiments, each conducted in triplicate unless otherwise indicated. Assessment of statistical difference was undertaken by 2‐way ANOVA with Bonferroni's test adjusted for multiple comparisons and a P value of < 0.05 considered significant.
3. RESULTS
3.1. GM‐CSFRα quantification and kinetics following GM‐CSF treatment
We first observed that an antagonistic anti‐GM‐CSFRα antibody (CAM‐3001) dose dependently inhibited the ability of GM‐CSF to protect against TF‐1 cell apoptosis (Fig. 1A), and yet GM‐CSFRα was barely detectable on the cell surface of cells that had been cultured with GM‐CSF (Fig. 1B). Once GM‐CSF had been withdrawn for 18 hr, GM‐CSFRα was then detectable at the cell surface (Fig. 1B). These data were consistent with previously published data suggesting that GM‐CSF drives the internalization of its own receptor. Given that the antibodies and GM‐CSF were added together to the TF‐1 viability assays, this raised questions as to how CAM‐3001 acts as an effective inhibitor when its target is actively down‐regulated by the ligand, and how receptor kinetics might impact its therapeutic use. In view of this, we designed experiments in a therapeutically relevant context to explore this question in more detail.
Figure 1.
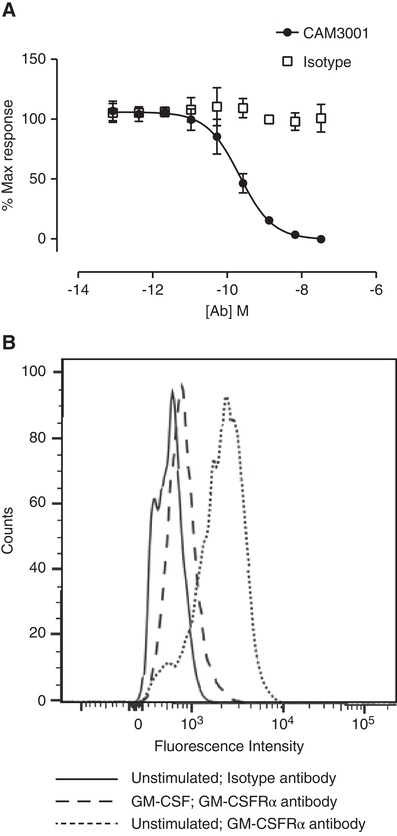
GM‐CSFR blockade and quantification in TF‐1 cell line. (A) TF‐1 cells, washed to remove residual human recombinant GM‐CSF from the routine culture conditions, were treated with 0.25 ng/ml GM‐CSF, in the presence of a serial dilution of CAM3001 or isotype control and cultured for 72 hr. CellTiter‐Glo was used to measure ATP as an indirect measure of number of viable cells. Data represent mean ± sem of n = 4 independent experiments. (B) TF‐1 cells that had been maintained in 4 ng/ml human GM‐CSF were washed three times to ensure complete removal of GM‐CSF. The cells were then returned to culture in the presence or absence of 4 ng/mL human GM‐CSF for 18 hr. The cells were stained with CAM3001 followed by PE‐conjugated secondary antibody to assess surface levels of GM‐CSFRα. In the cells that had been cultured in the absence of GM‐CSF the GM‐CSFRα was detectable above background. In the cells that had been maintained in GM‐CSF the GM‐CSFRα was considerably lower. Image shown is a representative experiment of four independent experiments
Measurement of GM‐CSFRα expression in human neutrophils is readily achievable using confocal imaging (Fig. 2A) or standard flow cytometry using GM‐CSFRα‐selective antibodies. However, for more accurate quantification of cell surface GM‐CSFRα number we utilized microspheres with known binding affinities as external standards, which enables the standardization of fluorescence intensity units irrespective of staining variability between experiments, instrument, and software (Supplemental Fig. S1). This approach was considered of importance for measurements made in patient‐derived neutrophils. After correction for nonspecific staining, a mean surface GM‐CSFRα number of 7141 ± 474 (mean ± sem; n = 28) receptors per cell was calculated (Fig. 2B).
Figure 2.
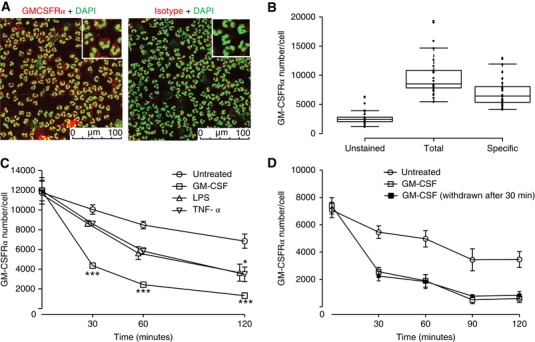
GM‐CSFRα quantification and kinetics in human neutrophils. (A) Neutrophils were incubated with DAPI and AlexaFluor647‐CAM‐3001 (GM‐CSFRα antibody, left panel) or AlexaFluor647‐NIP228 (Isotype control, right panel) and the presence of GMCSF receptor expression determined by immunofluorescence. Images represent one of three independent experiments. (B) Neutrophils were incubated with AlexaFluor647‐CAM‐3001 and GM‐CSFRα receptor density was measured by flow cytometry using Quantum Simply Cellular beads. Center lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. n = 28 donors assessed in 28 independent experiments. (C and D) Human neutrophils were treated with the indicated cytokine (GM‐CSF [1 ng/ml], TNFα [20 ng/ml] and LPS [100 ng/ml]) before GM‐CSFRα number/cell was assessed by flow cytometry (as for B) at the indicated time and compared to baseline. In some experiments (D) samples treated with GM‐CSF (1 ng/ml) were washed (x2) and resuspended in GM‐CSF free media for the remainder of the experiment. Data represent mean ± sem of n = 3 (C) or n = 4 (D) independent experiments. Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at ***P < 0.001, **P < 0.01, *P < 0.05)
Following stimulation of isolated neutrophils with 1 ng/ml GM‐CSF, we observed a marked and time‐dependent reduction in mean cell surface GM‐CSFRα number, decreasing by 64% (P < 0.001) 30 min after stimulation, and by 89% (P < 0.001) at 2 hr (Fig. 2C). A more modest loss of cell surface GM‐CSFRα number was observed when the cells were stimulated with TNFα (20 ng/ml) or LPS (100 ng/ml); in addition, this effect was noticeably slower (P < 0.05 at 2 hr; Fig. 2C). When GM‐CSF was removed after 30 min by washing, GM‐CSFRα number failed to recover, suggesting that cell surface GM‐CSFRα numbers were either not recycled or recycled only very slowly after withdrawal of ligand (Fig. 2D).
Interrogation of our recently generated human neutrophil transcriptomic data set, generated using neutrophils isolated in an identical way and treated with human recombinant GM‐CSF at 1 ng/ml for 6 hr (GEO accession number GSE76293), revealed a 1.9‐mean fold increase in GM‐CSFRα mRNA abundance using 4 independent probes for the GM‐CSFRα (mean adjusted P = 8.7 × 10−5; Table 1). This contrasts to the very major GM‐CSF stimulated increase (25‐fold) in CD69 mRNA (Table 1) (Zhang et al., 2004). Together, these data indicate that GM‐CSFRα is rapidly lost from the neutrophil cell surface following GM‐CSF stimulation, and to a lesser extent, following LPS and TNFα; whereas GM‐CSF stimulation appears to increase GM‐CSFRα transcription, this was not associated with early recovery of cell surface GM‐CSFRα expression after ligand removal.
Table 1.
Fold change in mRNA for CD69 and GM‐CSFRα in neutrophils stimulated with GM‐CSF for 6 hrFreshly isolated human neutrophils were incubated with recombinant human GM‐CSF (1 ng/ml) or vehicle control for 6 hr and cDNA prepared as previously detailed.4 Labelled cDNA was hybridized onto GeneChip Human Genome U133 Plus 2.0 oligonucleotide arrays (Affymetrix, Santa Clara, CA, USA). The data, as submitted on GSE76293, were processed as detailed above and the fold change values, and the negative logarithm of the adjusted P values computed using the “limma” package, where empirical Bayes statistics and the Benjamini‐Hochberg correction method were used
| Gene | Probe ID | Fold Change | –LOG(adjPValue) | adjPValue |
|---|---|---|---|---|
| CD69 | 209795_at | 25.3 | 17.1 | 8.57e‐18 |
| GMCSFRA | 210340_s_at | 1.99 | 7.5 | 3.54e‐8 |
| GMCSFRA | 207085_x_at | 2.16 | 5.7 | 1.95e‐6 |
| GMCSFRA | 211286_x_at | 1.82 | 5.3 | 5.22e‐6 |
| GMCSFRA | 211287_x_at | 1.65 | 3.5 | 3.41e‐4 |
Previous studies have suggested roles for both the lysosome and proteasome in the related CSF receptor G‐CSFRα ligand‐mediated internalization,25 and for similar mechanisms to operate for the shared common β chain.18 However, we were unable to block GM‐CSF‐mediated GM‐CSFRα internalization nor GM‐CSF mediated IL‐8 release with MG132 (cell‐permeable proteasome inhibitor), and likewise actinomycin D and brefeldin A had no effect on receptor internalization (Supplemental Fig. S2). Proteosomal degradation, recycling from the golgi or endosomal compartments, and lysosomal degradation do not therefore appear to affect GM‐CSFRα cell surface kinetics.
3.2. The pro‐survival effect of GM‐CSF in neutrophils requires prolonged GM‐CSFRα stimulation
Given the above data, which show that neutrophils lose approximately 90% of their cell surface GM‐CSFRα when stimulated with GM‐CSF for 2 hr, we wished to examine if this correlated with a loss of receptor signaling at longer time points. The functional read out of GM‐CSF‐induced inhibition of constitutive (time‐dependent spontaneous) apoptosis was chosen, which can be readily assessed in vitro using dual Annexin V and PI staining. Hence neutrophils maintained in the continuous presence of GM‐CSF for 20 hr at 37°C show a marked (> 70%) and concentration‐dependent (EC50 0.03 ng/ml; n = 8) inhibition of apoptosis (Fig. 3A). When these cells were pretreated with 0.01–1000 nM CAM‐3001, a human GM‐CSFR blocking antibody, this effect was completely abolished, again in a concentration‐dependent manner (IC50 CAM‐3001 inhibition of GM‐CSF treatment 0.05 ± 0.03 μM; n = 3) (Fig. 3B). Most instructively, when GM‐CSF was removed by washing 1.5 or 6 hr into these incubations, that is, at a time when there was a profound loss in cell surface GM‐CSFRα expression, the pro‐survival effect of GM‐CSF was almost entirely lost (Fig. 3C); likewise, when CAM‐3001 was added at a maximally effective concentration (1 μM) 1, 2, 4, and even 6 hr after GM‐CSF, full inhibition of the GM‐CSF‐induced pro‐survival effect was still observed (Fig. 3D). These data indicate: (i) the critical need for “sustained” GM‐CSFRα signaling to affect the anti‐apoptotic function of GM‐CSF and (ii) together with the very low EC50 for this response (0.03 ng/ml), a high degree of GM‐CSFRα receptor “spareness.” Hence, even substantial receptor loss does not appear to prevent sustained and effective GM‐CSFRα signaling in the human neutrophil.
Figure 3.
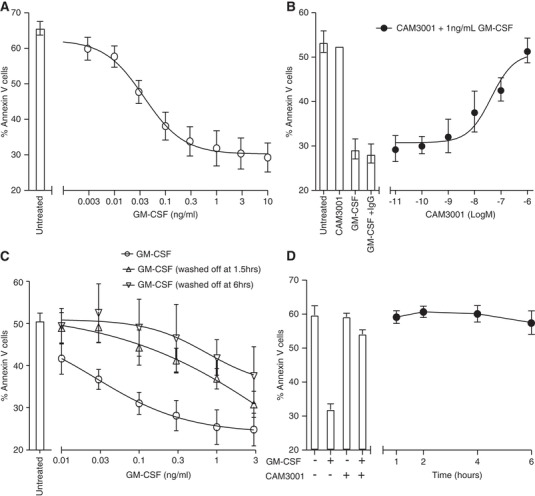
Sustained GM‐CSF signaling required for GM‐CSF‐mediated neutrophil survival. (A–D) Freshly isolated neutrophils were treated with the indicated concentration of GM‐CSF and the percentage of apoptotic cells analyzed after 20 hr culture by flow cytometry. (B) Neutrophils were pretreated with increasing concentrations of CAM‐3001 for 20 min prior to GM‐CSF treatment. (C) Cells were maintained in media containing the indicated concentration of GM‐CSF or the culture media were replaced after 1.5 or 6 hr of treatment with GM‐CSF‐free media for the remainder of the incubation time. Media change alone did not impact apoptosis of control samples without GM‐CSF incubation (data not shown). (D) Human neutrophils were treated with GM‐CSF (1 ng/ml) and this was followed by addition of CAM‐3001 (1 μM) at the indicated time after GM‐CSF treatment began. The bars indicate the response to GM‐CSF and the effect of pretreatment with CAM‐3001 (20 min) prior to treatment with GM‐CSF. Data represents mean ± sem of n = 8 (A), n = 3 (B), n = 5 (C) or n = 3 (D) independent experiments
3.3. Assessment of GM‐CSFRα kinetics in inflammatory neutrophils in vivo
We next explored the dynamics of GM‐CSFRα expression in inflammatory neutrophils to determine if time‐dependent GM‐CSFRα loss in these cells could be observed in vivo. This was undertaken using an LPS‐induced lung injury model in mice. We demonstrated previously that CAM‐3003, the murine equivalent to CAM‐3001, potently inhibited GM‐CSF‐induced proliferation of mouse FDCP cells in a dose‐dependent manner and reduced smoke‐induced lung inflammation.26 Supporting a role for GM‐CSF signaling in acute lung injury, we show that CAM‐3003 significantly reduced the influx of total inflammatory cells to the lung in response to inhaled LPS (35 ± 10% for 100 μg i.n. dose; 48 ± 8% reduction for 400 μg i.n. dose in total BALF cells compared to isotype control) (Fig. 4A), which consisted predominantly of neutrophils (37 ± 8% for 100 μg i.n. dose; 50 ± 8% reduction for 400 μg i.n. dose in BALF neutrophils compared to isotype control) (Fig. 4B). In addition, CAM‐3003 reduced LPS‐ induced lung concentrations of IL‐1β (Fig. 4C), IL‐6, TNFα, and CXCL2 (data not shown). We hypothesized that the use of CAM‐3003 would allow us to determine the role of GM‐CSFRα in ligand removal in the inflamed lung.
Figure 4.
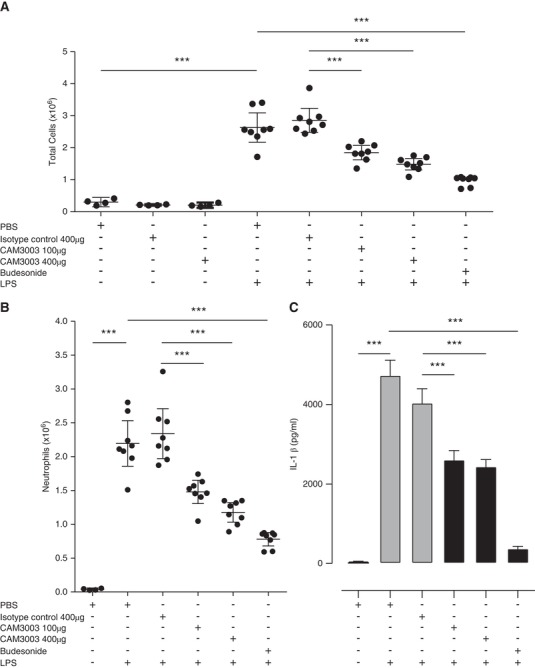
GM‐CSFRα blockade inhibits inflammation in response to inhaled LPS in a mouse model of acute lung injury. Mice were treated with inhaled PBS or LPS (nebulized 1 mg/mL for 10 min) and 24 hr later BALF total cell counts (A), BALF neutrophils (B) and lung homogenate IL‐1β (C) were assessed 24 hr after LPS challenge. Additionally, mice were treated intranasally with PBS, isotype control, or CAM‐3003 (at doses indicated), or budesonide (3 mg/kg, p.o.) 3 hr prior to LPS exposure as indicated. n = 8 mice per group, except control groups without LPS (n = 4 mice per group). Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at ***P < 0.001, **P < 0.01, *P < 0.05 compared to either PBS group, isotype control or LPS group as indicated by bars)
Following i.n. instillation of LPS, the percentage of neutrophils in the BALF increased in a time‐dependent manner from < 1% in the PBS control group to 70 ± 8%, 90 ± 2% and 90 ± 2% at 3, 6, and 24 hr post‐LPS, respectively. This was associated with a blood neutrophilia and small but consistent decline in the overall percentage of neutrophils within the bone marrow (baseline 38 ± 2% neutrophils; 24 hr after LPS 26 ± 1% neutrophils; P < 0.01; Fig. 5A). The concentration of GM‐CSF in the BALF peaked 3 hr after LPS treatment (294 ± 34 pg/ml) but declined thereafter to near baseline values by 24 hr (Fig. 5B). Of note, this reduction in BALF GM‐CSF at 24 hr was significantly attenuated following the administration of CAM‐3003 administered either 3 hr before, or 6 hr after, the LPS challenge (Fig. 5C), implying that the removal of GM‐CSF from the airspace is at least in part GM‐CSFRα‐mediated.
Figure 5.
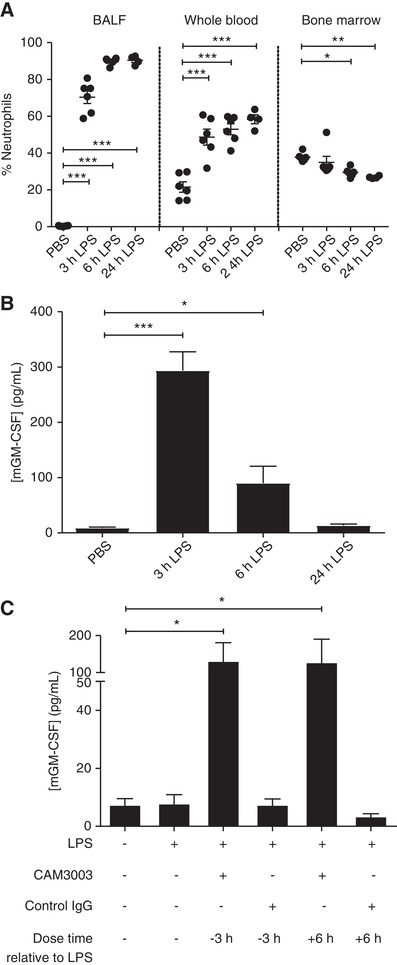
GM‐CSFRα blockade causes a sustained increase in LPS‐induced alveolar GM‐CSF concentration. Mice were treated with PBS or LPS (10 μg, intranasally [i.n.]) for the indicated time before the percentage of neutrophils (as a percentage of CD45+ cells) in the blood, bone‐marrow and BALF was determined by (A) flow cytometry and (B) the concentration of GM‐CSF in the BALF measured by ELISA. (C) CAM‐3003 (anti‐mouse GM‐CSFRα mAb) or isotype control (400 μg, i.n.) was administered either 3 hr prior or 6 hr post‐LPS administration (10 μg, i.n.) and the concentration of GM‐CSF after 24 hr was determined by ELISA. Data show mean ± sem for each mouse group (A, n = 6; B, n = 6; C n = 5). Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at ***P < 0.001, **P < 0.01, *P < 0.05, compared to PBS group (A and B) or LPS group (C)). Data in A are representative of 2 independent experiments
Cell surface expression of GM‐CSFRα and β was measured on the BALF neutrophils (CD45+, Ly6G+, CD11b+, Ly6C−) by flow cytometry. We observed a time‐dependent reduction in the cell surface expression of GM‐CSFRα on BALF neutrophils (Fig. 6A) following LPS challenge. In contrast, bone marrow neutrophils increased GM‐CSFRα expression following LPS challenge (Fig. 6A). GM‐CSFRβ expression was also far lower in BALF neutrophils compared to blood neutrophils even at the earliest time point (Fig. 6B). Although Figure 6 shows the data for GM‐CSFRα and GM‐CSFRβ expression in BALF neutrophils following PBS challenge, the extremely small number of neutrophils recovered in this control group of animals makes accurate quantification of GM‐CSFRα and β in these cells challenging and hence uncertain. Figure 6 also suggests a dissociation in the time‐course for GM‐CSFRα and β loss from the neutrophil cell surface, with faster kinetics observed for the common β chain.
Figure 6.
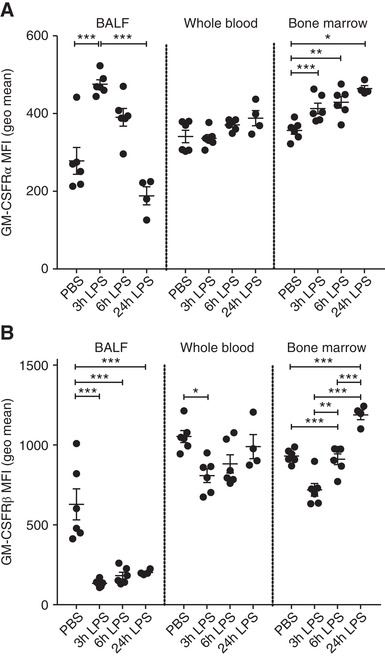
Dynamic changes in BALF neutrophil GM‐CSFRα and common β chain expression during LPS‐induced acute lung injury. Mice were treated with PBS or LPS (10 μg, intranasally [i.n.]) for the indicated time. The expression of GM‐CSFRα (A) or GM‐CSFRβ (B) was then determined, using flow cytometry, on neutrophils isolated from BALF, whole blood and bone marrow. Data are expressed as geometric mean fluorescent intensity. Data show single points as well as mean ± sem for each mouse group (n = 4–6). Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at ***P < 0.001, **P < 0.01, *P < 0.05)
These data indicate that in GM‐CSF rich environments in vivo, GM‐CSFRα and β expression on infiltrating neutrophils is rapidly down‐regulated, the former being consistent with our human in vitro stimulations. This hypothesis is supported by a preliminary assessment of surface neutrophil GM‐CSFRα in a small number of patients with ARDS (n = 7) where a greater variance and lower mean receptor number was observed in GM‐CSFRα expression in BALF neutrophils (5006 ± 1303 [mean ± sem] GM‐CSFRα receptors/neutrophil) compared to GM‐CSFRα expression in patient‐matched blood neutrophils (8026 ± 847 [mean ± sem]; Supplemental Fig. S3). Blood neutrophils from patients with ARDS when treated with GM‐CSF ex vivo showed increased survival similarly to healthy donor neutrophils (Supplemental Fig. S3) showing there was no disease‐dependent deficiency in their GM‐CSF responsiveness. The limited number of samples (purified BALF neutrophils) available in the ARDS group precluded any further analysis of GM‐CSFRα expression.
3.4. Neutrophils deplete GM‐CSF in vitro
To further support the hypothesis that neutrophils are a key contributor to GM‐CSF depletion, we demonstrated the ability of human neutrophils to deplete exogenously added recombinant human GM‐CSF (Fig. 7). The time‐course of ligand depletion was consistent with previously observed decreases in GM‐CSFR and could not be explained by ligand degradation by proteases (e.g., released by activation of neutrophils) because protease inhibitors had no impact on ligand depletion in vitro.
Figure 7.
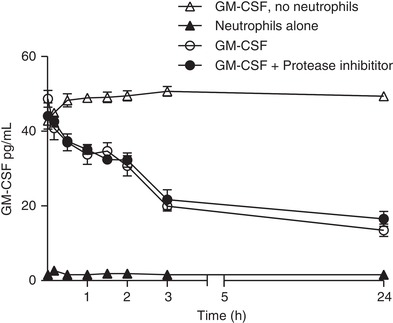
Human neutrophils deplete exogenously added GM‐CSF from media independent of ligand degradation. Human neutrophils were treated with 30 pg/mL GM‐CSF for the indicated time before the concentration of GM‐CSF was determined from cell supernatant by ELISA. Where indicated, neutrophils were pretreated with protease inhibitors (10 μM Sivelestat and 2 mM EDTA) prior to treatment with GM‐CSF. Data are expressed as mean ± sem (Data shown are for n = 3 donors in a single experiment; another donor showed the same effect in a further independent experiment)
We conclude that intra‐alveolar concentrations of GM‐CSF are depleted by GM‐CSFRα‐mediated consumption of ligand, as well as, we assume, by its rate of production by resident and influxing cells while continuing to activate GM‐CSFRα signaling at low surface receptor numbers; thus, measurements of GM‐CSF at a single time point are unlikely to reflect the true biologic relevance of this pro‐inflammatory growth factor.
4. DISCUSSION
Whereas it is well established that GM‐CSF induces a strong priming and pro‐survival effect in human neutrophils and plays an important role in the pathogenesis of ARDS, the local interplay between GM‐CSF concentration, GM‐CSFR dynamics, and the temporal mapping of functional effects of this ligand on neutrophils has not been fully explored, particularly in a disease‐relevant context.
Here we show that, following ligand‐mediated receptor internalization, neutrophil GM‐CSFRα numbers at the cell surface fall to very low levels (circa 10% of the receptor numbers seen in unstimulated cells) within a short time frame and are not replenished following ligand removal. A similar sustained reduction of eosinophil GM‐CSFRα expression in response to GM‐CSF (but not to IL‐3 or IL‐5) was also observed by Gregory et al.27; however, these authors did not explore the mechanisms of this response or examine this in an in vivo setting. In our study, rather surprisingly inhibition of the proteasome did not impact on α chain dynamics nor GM‐CSF‐dependent IL‐8 production and the null effect of actinomycin D and brefeldin A also suggests that there is little contribution from newly synthesized GM‐CSFRα or through receptor recycling.
Importantly, even very low levels of GM‐CSFRα appear able to maintain active GM‐CSF signaling in neutrophils because the continued presence of the ligand was required to maintain the pro‐survival effects of GM‐CSF on neutrophils despite the concomitant and major reductions in receptor number. Despite previous published data from the Chilvers’ group demonstrating detectable pAKT activity at 60 mins after GM‐CSF stimulation,16 we have been unable to demonstrate continued signaling at longer time frames consistent with the requirement for sustained GM‐CSFR activation for neutrophil survival, using conventional assays measuring pAKT, pErk, pStat5, or phosphorylation of GM‐CSFRβ (data not shown). However, our observations are compatible with a previous report in murine bone marrow cells that only 10% of available GM‐CSFRs need to be bound by GM‐CSF to elicit a maximal response28; therefore the signaling events that link GM‐CSFR activity at later time points may be important but also below our detection threshold, or the signaling pathway responsible may be undetermined. Our lines of evidence suggest a large spare receptor capacity for GM‐CSFR in neutrophils. In in vivo models, complete GM‐CSFR blockade using an antagonist approach, or very major lowering in the free GM‐CSF concentration would be required to induce therapeutic blockade of this axis in a neutrophil‐dominated disease process. This may also explain why the administration of recombinant GM‐CSF to patients with ALI‐ARDS (to restore neutrophil phagocytic activity) does not worsen outcomes,29, 30 that is, the concentration of GM‐CSF required to maintain neutrophil survival and GM‐CSF‐dependent cytokine release may already be sufficient to saturate low levels of membrane GM‐CSFRα and maintain signaling.
We show that infiltrating neutrophils deplete free ligand via a receptor‐mediated event, most likely internalization. A previous report31 of receptor‐mediated internalization of CXCL8 by neutrophils in LPS‐induced local skin inflammation suggests that neutrophils may be programmed to limit pro‐inflammatory signals in the setting of infection and inflammation by local ligand depletion. Mice lacking the GM‐CSF receptor, but not wild‐type controls, developed high circulating levels of GM‐CSF following endotoxin challenge,32 supporting the role for receptor‐ligand internalization as a method of limiting inflammatory responses. Furthermore, loss‐of‐function mutations in the human CSFRA gene lead to pulmonary alveolar proteinosis (due to failure of alveolar macrophages to clear surfactant) and are associated with markedly increased circulating GM‐CSF concentrations,33 suggesting that even in the absence of an inflammatory stimulus, ligand internalization is required for GM‐CSF homeostasis. In the context of inflammation, reported time‐courses for pulmonary (BALF) GM‐CSF accumulation in ALI patients have indicated that GM‐CSF concentrations are increased early in disease but subsequently decline3; in our mouse model of LPS, the GM‐CSF levels peak at 3 hr, and thereafter diminish sharply. However, when a receptor blocking antibody was added, either before or even 6 hr post‐LPS challenge, measured concentrations of GM‐CSF in BALF were maintained, and the antibody significantly inhibited cell influx to the lung in a dose‐dependent manner. Given that the decline in detectable GM‐CSF levels reduces in concert with the time‐course of neutrophil infiltration into the lung, it is likely that infiltrating neutrophils are a key consumer of free GM‐CSF. In support of this hypothesis, we have been able to demonstrate rapid and significant GM‐CSF depletion (exogenously added recombinant ligand) by human neutrophils in vitro, which is protease independent.
Our data might also suggest that receptor‐mediated ligand depletion is a significant factor in determining detectable concentrations of free GM‐CSF in clinical samples from the lung in other disease states and may alter interpretation of studies that have described no or only modest increases in the concentrations of GM‐CSF in inflammatory situations. Furthermore, ligand internalization may contribute to temporal regulation of inflammatory responses and local tissue injury; for example, GM‐CSF confers acute protection in a mouse model of influenza infection, but animals that continuously secrete high levels of GM‐CSF develop desquamative interstitial pneumonia that impairs long‐term recovery.34 For the same reasons, the role of GM‐CSF production and signaling may have been underestimated in several other disease settings such as cryptogenic organizing pneumonia, which is also characterized by intense inflammation.35 Clinical studies using recombinant GM‐CSF in ALI and other diseases are ongoing and will help us elucidate this matter in more detail; however, our data suggest that the degree of neutrophilic infiltrate and the precise timing of therapeutic administration may determine the response to such treatments. Our data could help to design more effective in vivo studies to understand the interplay between appropriate responses to infection, and chronic inappropriate neutrophilic responses that may be driven by prolonged GM‐CSF secretion.
In summary, these data show that GM‐CSF exposure results in a rapid and sustained loss of cell membrane GM‐CSFRα yet this does not preclude sustained G‐CSF/GM‐CSFRα signaling. Moreover, the GM‐CSFRα receptor appears to play a key role in ligand clearance. Hence neutrophilic activation via GM‐CSFR may play an important role in neutrophilic lung inflammation even in the absence of high GM‐CSF levels or GM‐CSFRα expression.
DISCLOSURES
All authors with MedImmune affiliation are (or were) employees of MedImmune and may have received AstraZeneca shares as part of their remuneration. There are no other conflicts of interest to declare.
Supporting information
GM‐CSFRα expression on human neutrophils was quantified by the interpolation of unknown values (MFI measured in neutrophils stained with PE‐anti‐CD116 (anti‐GM‐CSFRα) with a calibration curve obtained using MFI values of IgG binding bead populations with increasing binding capacities. These beads are stained concomitantly to neutrophils for each experiment and measured by flow cytometry using the same flow cytometer and settings as the cells. (A) Flow cytometry histogram showing different bead populations MFI (normalized to Mode). Blank bead population has no IgG binding capacity (Dark green; blank beads, light green; bead set #1, orange; bead set #2, blue; bead set #3, red; bead set #4). (B)Representative example of a standard curve generated using MFI measured for each bead population. Unknown values from neutrophil MFIs are automatically interpolated from the calibration curve to obtain absolute numbers of receptor stained per cell.
Freshly isolated neutrophils were pre‐incubated for 1 hour with MG132 (20 μM) (A), actinomycin D (2 μg/ml) (C), or brefeldin A (10 μg/ml) (D) and then treated with GM‐CSF (1 ng/ml) or buffer alone. At the indicated time the neutrophils were incubated with AlexaFluor647‐CAM‐3001 and GM‐CSFRα receptor density was measured by flow cytometry using Quantum Simply Cellular beads. Data shown are mean ± SEM of n = 3 independent experiments. Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test; no significant difference observed between inhibitor treated samples and controls. (B) Supernatants from MG132 treated samples were collected and IL‐8 content measured by ELISA. Data represent mean ± SEM of n = 3 independent experiments. Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at *** p < 0.001, ** p < 0.01).
(A) Neutrophils from healthy volunteer peripheral blood, ARDS peripheral blood or ARDS BALF were incubated with AlexaFluor647‐CAM‐3001. GM‐CSFRα receptor density was measured by flow cytometry using Quantum Simply Cellular beads. Data are expressed as GM‐CSFRα copies per cell for each donor and as mean ± SEM for each group. For each ARDS patient an age‐ and sex‐matched healthy donor was enrolled in the study and GM‐CSFRα measured in whole blood neutrophils at the same time. (B) Freshly isolated neutrophils from the blood of patients with ARDS or matched healthy controls were left untreated or pre‐incubated with CAM‐3001 (1 μM) for 30 minutes before being incubated with GM‐CSF (1 ng/ml) or buffer for 20 hours. Following incubation apoptosis was measured using Annexin V/PI staining by flow cytometry and expressed as percentage of total neutrophil population. Data represent single data points for each patient and the mean ± SEM, n = 8 independent experiments. Data analyzed using paired t test for paired sampled (A) and 2‐way ANOVA with Bonferroni's post‐test (B) (Significant at *p < 0.05).
AUTHORSHIP
D.K.F., E.R.C., A.S.C., A.M.C., and M.A.S. were responsible for study conceptualization, supervision, data interpretation, manuscript preparation, and critical review; S.A. and G.J.F. also contributed to manuscript preparation. S.A., G.J.F., J.K.J., S.P., O.W., H.K., and R.S. were responsible for experimental delivery, data analysis, and interpretation of human neutrophil and/or cell line in vitro studies. S.A., A.J.D., D.J.C., E.S.C., and D.K.F. were responsible for experimental delivery and/or experimental design and supervision, and data analysis and interpretation of LPS studies in mice. A.D., A.P., and T.R.D.J.R. were responsible for mRNA analysis in GM‐CSF stimulated neutrophils.
S.A., G.J.F., D.K.F., and E.R.C. are joint first/senior authors.
ACKNOWLEDGMENTS
We would like to thank healthy volunteers, ARDS patients, and their legal surrogates for their consent and participation in this study. We also acknowledge the expert technical assistance of the In Vivo Sciences team, MedImmune Ltd., and Biosciences, AstraZeneca, for mouse studies. This work was funded by a studentship from MedImmune Ltd., to S.A., Papworth Hospital, Wellcome Trust, and the NIHR Cambridge Biomedical Research Centre.
De Alessandris S, Ferguson GJ, Dodd AJ, et al. Neutrophil GM‐CSF receptor dynamics in acute lung injury. 2019;105:1183–1194. 10.1002/JLB.3MA0918-347R
REFERENCES
- 1. Thille AW, Esteban A, Fernandez‐Segoviano P. Chronology of histological lesions in acute respiratory distress syndrome with diffuse alveolar damage: a prospective cohort study of clinical autopsies. Lancet Respir Med. 2013;1:395‐401. [DOI] [PubMed] [Google Scholar]
- 2. Weiland JE, Davis WB, Holter JF, Mohammed JR, Dorinsky PM, Gadek KE. Lung neutrophils in the adult respiratory distress syndrome. Clinical and pathophysiologic significance. Am Rev Respir Dis. 1986;133:218‐225. [DOI] [PubMed] [Google Scholar]
- 3. Matute‐Bello G, Liles WC, Radella F. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997;156:1969‐1977. [DOI] [PubMed] [Google Scholar]
- 4. Juss JK, House D, Amour A. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3‐kinase inhibition. Am J Respir Crit Care Med. 2016;194:961‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Root RK, Dale DC. Granulocyte colony‐stimulating factor and granulocyte‐macrophage colony‐stimulating factor: comparisons and potential for use in the treatment of infections in nonneutropenic patients. J Infect Dis. 1999;179:S342‐S352. [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto K, Ahyi AN, Pepper‐Cunningham ZA. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am J Respir Cell Mol Biol. 2014;50:253‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ballinger MN, Paine R III, CH Sereza. Role of granulocyte macrophage colony‐stimulating factor during gram‐negative lung infection with Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:766‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. LeVine AM, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T. Surfactant protein‐A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103:1015‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki T, Sakagami T, Rubin BK. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703‐2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kitamura T, Tanaka N, Watanabe J. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony‐stimulating factor. J Exp Med. 1999;190:875‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitamura T, Takaku F, Miyajima A. IL‐1 up‐regulates the expression of cytokine receptors on a factor‐dependent human hemopoietic cell line, TF‐1. Int Immunol. 1991;3:571‐577. [DOI] [PubMed] [Google Scholar]
- 12. Woodcock JM, Zacharakis B, Plaetinck G. Three residues in the common beta chain of the human GM‐CSF, IL‐3 and IL‐5 receptors are essential for GM‐CSF and IL‐5 but not IL‐3 high affinity binding and interact with Glu21 of GM‐CSF. EMBO J. 1994;13:5176‐5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lieschke GJ, Burgess AW. Granulocyte colony‐stimulating factor and granulocyte‐macrophage colony‐stimulating factor. N Engl J Med. 1992;327:28‐35. [DOI] [PubMed] [Google Scholar]
- 14. Kamp VM, Leentjens J, Pillay J. Modulation of granulocyte kinetics by GM‐CSF/IFN‐γ in a human LPS re‐challenge model. J Leukoc Biol. 2013;94:513‐520. [DOI] [PubMed] [Google Scholar]
- 15. Cowburn AS, Summers C, Dunmore BJ. Granulocyte/macrophage colony‐stimulating factor causes a paradoxical increase in the BH3‐only pro‐apoptotic protein Bim in human neutrophils. Am J Respir Cell Mol Biol. 2011;44:879‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Juss JK, Hayhoe RP, Owen CE. Functional redundancy of class I phosphoinositide 3‐kinase (PI3K) isoforms in signaling growth factor‐mediated human neutrophil survival. PLoSOne. 2012;7:e45933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vainshtein I, Roskos LK, Cheng J, Sleeman MA, Wang B, Liang M. Quantitative measurement of the target‐mediated internalization kinetics of biopharmaceuticals. Pharm Res. 2015;32:286‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinez‐Moczygemba M, Huston DP. Proteasomal regulation of betac signaling reveals a novel mechanism for cytokine receptor heterotypic desensitization. J Clin Invest. 2001;108:1797‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haslett C, Guthrie LA, Kopaniak MM, Johnston Jr RB, Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol. 1985;119:101‐110. [PMC free article] [PubMed] [Google Scholar]
- 20. Cowburn AS, Deighton J, Walmsley SR, Chilvers ER. The survival effect of TNF‐alpha in human neutrophils is mediated via NF‐kappa B‐dependent IL‐8 release. Eur J Immunol. 2004;34:1733‐1743. [DOI] [PubMed] [Google Scholar]
- 21. Murray J, Barbara JA, Dunkley SA. Regulation of neutrophil apoptosis by tumor necrosis factor‐alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. 1997;90:2772‐2783. [PubMed] [Google Scholar]
- 22. Cowburn AS, Cadwallader KA, Reed BJ, Farahi N, Chilvers ER. Role of PI3‐kinase‐dependent Bad phosphorylation and altered transcription in cytokine‐mediated neutrophil survival. Blood. 2002;100:2607‐2616. [DOI] [PubMed] [Google Scholar]
- 23. Bozinovski S, Anthony D, Vlahos R. Targeting pro‐resolution pathways to combat chronic inflammation in COPD. J Thorac Dis. 2014;6:1548‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang X, Kluger Y, Nakayama Y. Gene expression in mature neutrophils: early responses to inflammatory stimuli. J Leukoc Biol. 2004;75:358‐372. [DOI] [PubMed] [Google Scholar]
- 25. Kindwall‐Keller TL, Druhan LJ, Ai J. Role of the proteasome in modulating native G‐CSFR expression. Cytokine. 2008;43:114‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Botelho FM, Nikota JK, Bauer C. A mouse GM‐CSF receptor antibody attenuates neutrophilia in mice exposed to cigarette smoke. Eur Respir J. 2011;38:285‐294. [DOI] [PubMed] [Google Scholar]
- 27. Gregory B, Kirchem A, Phipps S. Differential regulation of human eosinophil IL‐3, IL‐5 and GM‐CSF receptor alpha‐chain expression by cytokines: iL‐3, IL‐5 and GM‐CSF down‐regulate IL‐5 receptor alpha expression with loss of IL‐5 responsiveness, but up‐regulate IL‐3 receptor alpha expression. J Immunol. 2003;170:5359‐5366. [DOI] [PubMed] [Google Scholar]
- 28. Nicola NA, Peterson L, Hilton DJ, Metcalf D. Cellular processing of murine colony‐stimulating factor (Multi‐CSF, GM‐CSF, G‐CSF) receptors by normal hemopoietic cells and cell lines. Growth Factors. 1988;1:41‐49. [DOI] [PubMed] [Google Scholar]
- 29. Paine R III, TJ Standifo, RE Deche. A randomized trial of recombinant human granulocyte‐macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med. 2012;40:90‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinder EM, Rostron AJ, Hellyer TP. Randomised controlled trial of GM‐CSF in critically ill patients with impaired neutrophil phagocytosis. Thorax. 2018;73:918‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basran A, Jabeen M, Bingle L. Roles of neutrophils in the regulation of the extent of human inflammation through delivery of IL‐1 and clearance of chemokines. J Leukoc Biol. 2013;93:7‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Metcalf D, Nicola NA, Mifsud S, Di Rago L. Receptor clearance obscures the magnitude of granulocyte‐macrophage colony‐stimulating factor responses in mice to endotoxin or local infections. Blood. 1999;93:1579‐1585. [PubMed] [Google Scholar]
- 33. Suzuki T, Sakagami T, Young LR. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. J Respir Crit Care Med. 2010;182:1292‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sever‐Chroneos Z, Murthy A, Davis J. GM‐CSF modulates pulmonary resistance to influenza A infection. Antiviral Res. 2011;92:319‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saha S, Doe C, Mistry V. Granulocyte‐macrophage colony‐stimulating factor expression in induced sputum and bronchial mucosa in asthma and COPD. Thorax. 2009;64:671‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GM‐CSFRα expression on human neutrophils was quantified by the interpolation of unknown values (MFI measured in neutrophils stained with PE‐anti‐CD116 (anti‐GM‐CSFRα) with a calibration curve obtained using MFI values of IgG binding bead populations with increasing binding capacities. These beads are stained concomitantly to neutrophils for each experiment and measured by flow cytometry using the same flow cytometer and settings as the cells. (A) Flow cytometry histogram showing different bead populations MFI (normalized to Mode). Blank bead population has no IgG binding capacity (Dark green; blank beads, light green; bead set #1, orange; bead set #2, blue; bead set #3, red; bead set #4). (B)Representative example of a standard curve generated using MFI measured for each bead population. Unknown values from neutrophil MFIs are automatically interpolated from the calibration curve to obtain absolute numbers of receptor stained per cell.
Freshly isolated neutrophils were pre‐incubated for 1 hour with MG132 (20 μM) (A), actinomycin D (2 μg/ml) (C), or brefeldin A (10 μg/ml) (D) and then treated with GM‐CSF (1 ng/ml) or buffer alone. At the indicated time the neutrophils were incubated with AlexaFluor647‐CAM‐3001 and GM‐CSFRα receptor density was measured by flow cytometry using Quantum Simply Cellular beads. Data shown are mean ± SEM of n = 3 independent experiments. Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test; no significant difference observed between inhibitor treated samples and controls. (B) Supernatants from MG132 treated samples were collected and IL‐8 content measured by ELISA. Data represent mean ± SEM of n = 3 independent experiments. Statistical analysis was performed by 2‐way ANOVA with Bonferroni's post‐test (Significant at *** p < 0.001, ** p < 0.01).
(A) Neutrophils from healthy volunteer peripheral blood, ARDS peripheral blood or ARDS BALF were incubated with AlexaFluor647‐CAM‐3001. GM‐CSFRα receptor density was measured by flow cytometry using Quantum Simply Cellular beads. Data are expressed as GM‐CSFRα copies per cell for each donor and as mean ± SEM for each group. For each ARDS patient an age‐ and sex‐matched healthy donor was enrolled in the study and GM‐CSFRα measured in whole blood neutrophils at the same time. (B) Freshly isolated neutrophils from the blood of patients with ARDS or matched healthy controls were left untreated or pre‐incubated with CAM‐3001 (1 μM) for 30 minutes before being incubated with GM‐CSF (1 ng/ml) or buffer for 20 hours. Following incubation apoptosis was measured using Annexin V/PI staining by flow cytometry and expressed as percentage of total neutrophil population. Data represent single data points for each patient and the mean ± SEM, n = 8 independent experiments. Data analyzed using paired t test for paired sampled (A) and 2‐way ANOVA with Bonferroni's post‐test (B) (Significant at *p < 0.05).