

Abstract
The ydj gene cluster is found in 80% of sequenced Escherichia coli genomes and other closely related species in the human microbiome. On the basis of the annotations of the enzymes located in this cluster, it is expected that together they catalyze the catabolism of an unknown carbohydrate. The focus of this investigation is on YdjI, which is in the ydj gene cluster of E. coli K-12. It is predicted to be a class II aldolase of unknown function. Here we describe a structural and functional characterization of this enzyme. YdjI catalyzes the hydrogen/deuterium exchange of the pro-S hydrogen at C3 of dihydroxyacetone phosphate (DHAP). In the presence of DHAP, YdjI catalyzes an aldol condensation with a variety of aldo sugars. YdjI shows a strong preference for higher-order (seven-, eight-, and nine-carbon) monosaccharides with specific hydroxyl stereochemistries and a negatively charged terminus (carboxylate or phosphate). The best substrate is L-arabinuronic acid with an apparent kcat of 3.0 s−1. The product, L-glycero-L-galacto-octuluronate-1-phosphate, has a kcat/Km value of 2.1 × 103 M−1 s−1 in the retro-aldol reaction with YdjI. This is the first recorded synthesis of L-glycero-L-galacto-octuluronate-1-phosphate and six similar carbohydrates. The crystal structure of YdjI, determined to a nominal resolution of 1.75 Å (Protein Data Bank entry 6OFU), reveals unusual positions for two arginine residues located near the active site. Computational docking was utilized to distinguish preferable binding orientations for L-glycero-L-galacto-octuluronate-1-phosphate. These results indicate a possible alternative binding orientation for L-glycero-L-galacto-octuluronate-1-phosphate compared to that observed in other class II aldolases, which utilize shorter carbohydrate molecules.
Graphical Abstract
There are an estimated 1014 microbial cells living within every human whose combined number of genes is predicted to outnumber human genes by two orders of magnitude.1 A growing body of evidence suggests that individual human health is dependent upon a symbiotic relationship among the variety of bacterial species comprising the human microbiome.2,3 Indeed, a number of disease states, such as colorectal cancer, diabetes, obesity, and cardiovascular problems, have been linked to a dysbiosis, or an imbalance, in the human gut microbiota.4 One example is the pathogenesis of inflammatory bowel disorders, specifically Crohn’s disease and ulcerative colitis, with colonization of adherent-invasive Escherichia coli (AIEC).5 Strikingly, prevention of Crohn’s disease regression is positively influenced by colonization of Faecalibacterium prausnitzii.6 However, the exact mechanisms behind the prevention and progression of chronic colitis and the source of the dysbiosis between these species in an individual’s gut are unknown.
One possible explanation is that metabolic differences among gut microbiota allow for certain species to gain a selective advantage over others. Humans do not metabolize or absorb most of the carbohydrates consumed.7 However, bacterial species in the human gut have evolved the ability to metabolize many of these carbohydrates.8 Indeed, this is one mechanism by which both commensal and pathogenic species enhance their colonization.9–11 For commensal species, this is a mechanism for protection from harmful pathogenic species known as colonization resistance.10,12 To date, however, the metabolic utilization of alternative carbohydrate sources of many of these bacterial species remains poorly characterized.
Many carbohydrate catabolic pathways utilize an aldolase in the final step.13,14 Specifically, aldolases, which are essential and ubiquitous, catalyze the stereospecific formation or breaking of carbon−carbon bonds.13,14 The most well-known example is D-fructose-1,6-bisphosphate aldolase (FbaA), which catalyzes the reversible cleavage of D-fructose-1,6-bisphosphate to dihydroxyacetone phosphate and D-glyceraldehyde-3-phosphate (Scheme 1).15 Other well-studied aldolases include D-tagatose-1,6-bisphosphate aldolase (KbaY and GatY), L-rhamnulose-1-phosphate aldolase (RhaD), L-fuculose-1-phosphate aldolase (FucA), and 2-deoxy-5-keto-D-gluconate-6-phosphate (IolJ), which is essential for myo-inositol degradation in Bacillus subtilis.16,17
Scheme 1.
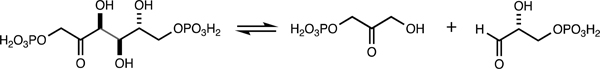
Reaction Catalyzed by D-Fructose-1,6-bisphosphate Aldolase
Common aldolases are divided into two classes on the basis of the mechanism by which the reactions are catalyzed.15 Class I aldolases utilize an active site lysine residue to form a Schiffbase intermediate. Class II enzymes employ an active site transition metal, usually zinc, to stabilize an enolate intermediate. These two classes of enzymes evolved separately and are mechanistically unrelated. Class II aldolases are subdivided into two clusters of orthologous groups (COGs).18 COG0191 contains, for example, FbaA, KbaY, GatY, and IolJ, whereas RhuD and FucA are found in COG0235. E. coli K12 has four enzymes currently annotated as members of COG0191, and three of these enzymes have been well-characterized. One is fructose-1,6-bisphosphate aldolase used in glycolysis (FbaY, Uniprot entry P0AB71); the other two are tagatose-1,6-bisphosphate aldolases (GatY, Uniprot entry P0C8J6; KbaY, Uniprot entry P0AB74). The fourth example is YdjI (Uniprot entry P77704), an enzyme of unknown function.
YdjI is localized in a cluster containing genes encoding five enzymes, two transporters, and a regulator (Figure 1). On the basis of the putative annotations of these enzymes, this cluster is likely to be responsible for the catabolism of a carbohydrate of unknown structure. In this gene cluster, YdjL and YdjJ are zinc-dependent alcohol dehydrogenases. YdjH is annotated to be in the ribokinase family.19 YdjG is an aldo-keto reductase family member, which was previously shown to catalyze the reduction of methyl glyoxal to hydroxyacetone.20 YdjK and YdjE are members of the major facilitator superfamily of transporters.19 YdjF is a helix−turn−helix, Deo-R type transcriptional regulator.19,21 This gene cluster is highly conserved among E. coli strains, appearing in approximately 80% of sequenced E. coli genomes, including E. coli K12, the probiotic strain E. coli Nissle 1917, the adherent-invasive E. coli LF82, and the enterohemorrhagic E. coli O157:H7.21,22 It is also found in species that are closely related to E. coli, including the Gram-negative Shigella dysenteriae and the Gram-positive Clostridium bolteae.
Figure 1.

Organization of the ydj gene cluster in E. coli. The cluster includes genes encoding five enzymes [YdjL, YdjJ, YdjI, YdjH, and YdjG (blue)], two transporters [YdjK and YdkE (green)], and a transcriptional regulator [YdjF (red)].
The ydj gene cluster was discovered as part of our larger initiative to identify carbohydrate catabolic pathways with unknown functions. Enzymes of potential interest were identified by first searching species of the human gut microbiome for proteins commonly found in carbohydrate catabolic pathways as predicted by their COG assignments. A computer script was written to retrieve the genes of interest from selected species, and then the sequences were compared to a self-compiled database of known carbohydrate catabolic enzymes based on annotation scores from the Uniprot database.23 This computational approach of identifying uncharacterized carbohydrate catabolic pathways is fast and less dependent on high-quality genome annotation and offers the advantage of considering the genomic context of an enzyme without adding additional complexity. As an initial validation for this methodology, the E. coli K12 genome was interrogated and four potential unknown carbohydrate catabolic pathways were identified with the ydj gene cluster being one. The initial goal was to quickly characterize the five enzymes of the gene cluster, but complete characterization of this metabolic pathway has remained elusive. Here, we describe the functional characterization of the substrates and three-dimensional structure of YdjI, the remaining uncharacterized class II aldolase from E. coli K12.
EXPERIMENTAL METHODS
Materials and Equipment.
All materials used in this study were obtained from Sigma-Aldrich, Carbosynth, or GEHealthcare Bio-Sciences, unless stated otherwise. E. coli strains XL1 Blue and BL21-Gold (DE3) were obtained from New England Biolabs. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 400 MHz system equipped with a broadband probe and sample changer or a Bruker Avance 500 MHz system equipped with a tripleresonance cyroprobe. Mass spectrometry samples were collected using an MDS-Sciex 4000 Qtrap system or a Thermo Scientific Q Exactive Focus system run in negative ion mode. Ultraviolet (UV) spectra were recorded with a SpectraMax340 UV−visible plate reader and 96-well NucC plates.
Sequence Similarity Networks.
Sequence similarity networks (SSNs) were generated by submitting the InterPro number for the protein family of interest or a list of the Uniprot IDs assigned to an individual cluster of orthologous genes (COG) to the EFI-EST webtool.24 The initial SSN was generated with an alignment score cutoff set such that each connection (edge) represents approximately 30% sequence identity. Sequences that share a high level of sequence identity (>90%) were grouped into a single node. All network layouts were created and visualized using Cytoscape 3.4.25 Within Cytoscape, more stringent SSNs were created by increasing the alignment score cutoff in small increments (usually by 5−10). This process was continued until the clusters were estimated to be isofunctional, where each cluster represents enzymes that catalyze the same reaction. Isofunctionality was determined by mapping the known functions onto the SSNs and following their movements as the alignment cutoff is increased until each cluster contains a single known catalytic activity.
Cloning of YdjI from E. coli K12.
The gene for YdjI (Uniprot entry P77704) was amplified from the genomic DNA of E. coli K12 using the following primer pair: 5′-ACGCATATGCTCGCAGATATCAGATATTGGGAAAACGATG-3′ and 5′-ATTAAGCTTTTCCGCTTTGCCATCAGAACCGAACA-3′. Restriction sites for NdeI and HindIII (underlined) were introduced into the forward and reserve primers, respectively. These sites allow for the addition of a C-terminal six-His tag in the pET-30a+ expression vector.
Polymerase chain reaction (PCR) amplification was carried out with Phusion DNA polymerase following the manufacturer’s specifications. Amplified PCR products were purified using the QIAquick PCR purification kit from Qiagen. Subsequently, restriction enzymes (New England Biolabs) and CutSmart buffer were added and allowed to incubate for 2 h at 37 °C. Digested genes were purified using agarose (1%) gel electrophoresis and the QIAquick gel extraction kit from Qiagen. The pET-30a vector (Novagen) was subjected to the same restriction enzyme digestion and gel purification. Additionally, the 5′-end phosphate was removed by incubating with recombinant shrimp alkaline phosphatase (rSAP) for 2 h after restriction digest. The genes were cloned into the digested vector using DNA ligase (New England BioLabs) following the supplied protocol. After ligation, high-salt buffer was removed by the PCR purification kit and eluted with 20 μL of deionized H2O. The resulting plasmid (2 μL) was used to transform E. coli XL1-blue cells (40 μL) via electroporation, and the cells were allowed to recover in 2 mL of lysogeny broth (LB) medium at 37 °C. After 2 h, the cells were harvested by centrifugation, resuspended in 100 μL of LB medium, and plated on kanamycin selective LB medium. Individual colonies were selected and grown in 5 mL of liquid LB medium overnight at 37 °C, and recombinant plasmids were harvested using the GenElute Plasmid Miniprep Kit from Sigma-Aldrich. Plasmids were sequenced to identify the correct cloned genes in the appropriate vector.
Expression and Purification of YdjI.
Recombinant plasmids containing the ydjI gene were used to transform E. coli BL21 (DE3) cells via a heat-shock method, and the cells were plated on LB medium containing 30 mg/L kanamycin.26 A single colony was selected to inoculate a 20 mL starter culture and grown overnight at 37 °C. This culture was used to inoculate 2 L of LB medium (1.0 L of medium in two 2.8 L Fernbach flasks) and grown at 37 °C until an OD600 of 0.6 was obtained. Zinc acetate (1.0 mM) was added to the growth medium when the OD600 was ∼0.1−0.2. Protein expression was induced by the addition of 1.0 mM isopropyl β-thiogalactopyranoside (IPTG) at 37 °C for 4−5 h. Cells were harvested by centrifugation at 8000g for 10 min. Typicallys, cell pellets (∼3 g) were frozen and stored at −80 °C until they were purified.
Frozen cells were resuspended in 50 mL of 50 mM HEPES/K+ (pH 8.5) with 250 mM KCl and 10 mM imidazole and then lysed with at least seven rounds of sonication at 4 °C. DNA and RNA were degraded by the addition of 1.0 mg of bovine DNase and RNase followed by stirring on ice for 10 min. Cell debris was removed by centrifugation at 12000g for 40 min followed by syringe filtering with a 0.2 μm filter. The resulting supernatant solution was loaded onto a 5 mL HisTrap HP (GE Healthcare) nickel affinity column that had been pre-equilibrated with resuspension buffer using a Bio-Rad FPLC system. Protein was eluted from the column using 50 mM HEPES/K+ (pH 8.5) with 250 mM KCL and 500 mM imidazole over a gradient of 30 column volumes. Fractions of 4.0 mL were collected and tested for purity by sodium dodecyl sulfate−polyacrylamide gel electrophoresis. Typically, protein eluted between 8 and 15 column volumes. Fractions containing the purest samples were pooled and buffer exchanged by dialysis into 50 mM HEPES/K+ (pH 8.5) with 250 mM KCl overnight at 4 °C. Homogeneous protein was concentrated using Vivaspin 20 centrifugal concentrators (GE Healthcare) with the final concentration of enzyme determined by the absorbance at 280 nm. The extinction coefficient (E280 = 30440 M−1 cm−1) was estimated on the basis of the protein sequence, including the C-terminal His tag and linker.27 Typically, the protein was divided into 100 μL aliquots, flash-frozen, and stored at −80 °C. Yields were typically 75−100 mg of YdjI from 2.0 L of cell culture. The metal content was determined using ICP-MS. For this, the purified protein was digested with concentrated nitric acid by refluxing for 2 h and then diluted with metal-free water to give a final nitric acid concentration of 1% (v/v).
Synthesis of Noncommercial Carbohydrates.
The methods for the synthesis of compounds not commercially available are found in the Supporting Information. These compounds include penturonic acids (D-arabinuronate, L-arabinuronate, D-xyluluronate, L-lyxuronate, and D-riburonate), teturonic acids (L-threouronate, D-threouronate, and D-erythruronate), DHAP, hydroxyacetone phosphate, tartronate semialdehyde, glycoaldehyde, L-threose-4-P, D- and L-arabinose-5-P, and D-xylose-5-P.
YdjI-Catalyzed Solvent−Deuterium Exchange.
YdjI was placed in 25 mM imidazole/D2O buffer using Vivaspin 500 concentrators with 10 kDa cutoff filters. Potential substrates for YdjI (10 mM) were mixed with the enzyme (10 μM) in 20 mM imidazole/D2O buffer (pH 7.4). The compounds assayed for catalytic activity included pyruvate, dihydroxyacetone, dihydroxyacetone phosphate, hydroxypyruvate, hydroxyacetone, hydroxyacetone phosphate, methylglyoxal, and acetone. The exchange of the α-protons with deuterium from the solvent was monitored by 1H NMR spectroscopy for ≤48 h. For measurement of the rate of solvent exchange with DHAP catalyzed by YdjI, 10 mM DHAP was mixed with 2 μM YdjI and the 1H NMR spectrum was collected every 90 s for 60 min. The extent of substrate exchange with the solvent was calculated from the peak area with respect to an internal standard of acetate. The change in the concentration of DHAP over time was plotted and fit to eq 1, where λ is the observed first-order rate constant for solvent exchange with the substrate. The turnover rate constant (kcat) was determined using eq 2, assuming a saturating substrate concentration.
| (1) |
| (2) |
Stereochemistry of H/D Exchange in DHAP Catalyzed by YdjI.
DHAP (10 mM) was added to YdjI (2 μM) in 25 mM imidazole/D2O buffer (pH 7.4). Sodium acetate (5.0 mM) was added as a calibration standard. The reaction was allowed to proceed for 2 h, until the DHAP was completely exchanged with deuterium from the solvent. Commercial rabbit muscle aldolase (Sigma-Aldrich) was diluted into 25 mM imidazole buffer to a concentration of 1 unit/μL; 1.0 unit was added to the reaction mixture described above, and the reaction monitored by 1H NMR spectroscopy for an additional 4 h.
pH Dependence of H/D Exchange of DHAP Catalyzed by YdjI.
The exchange reactions were conducted starting from 500 mM imidazole/D2O buffer stocks at pH 6.0, 6.5, 7.0, 7.5, and 8.0 by adding DCl until the desired pH was obtained. These concentrated buffers were diluted to 25 mM imidazole/ D2O working stocks. DHAP (5.0 mM) was added to YdjI (2.0 μM) in 25 mM imidazole/D2O buffer (pH 6.0−8.0). The reactions were followed by 1H NMR spectroscopy by recording a spectrum every 120 s for 30 min. The final pH of reaction was measured after the reaction had reached completion. The change in the concentration of DHAP over time was plotted and fit to eq 1 to yield an apparent rate of turnover as described above.
YdjI Assays for Catalytic Activity.
YdjI was screened for catalytic activity using two methods. The aldol condensation reaction was monitored by mixing 10 mM DHAP, 10 μM YdjI, and 15 mM potential aldehyde substrates in 25 mM imidazole/D2O buffer (pH 7.4) or 50 mM HEPES/K+ (pH 7.4) and monitoring the reaction as a function of time by 31P or 1H NMR spectroscopy at 32 °C. For experiments with DHAP and phosphorylated sugars, each substrate (10 mM) was used to minimize the complexity of the final NMR spectra. Reactions were followed by 31P NMR spectroscopy for 24−72 h. Kinetic experiments were conducted under the same reaction conditions as the original assays with a variable concentration of YdjI (2−10 μM) depending on the observed rate of turnover. Spectra were recorded every 3 min for 1.0 h with the product resonances quantified by integration with respect to an internal standard (phosphate or methyl phosphonate). The concentration of the product was plotted as a function of time and fit to a linear equation or a single exponential.
The retro-aldol reaction was monitored by UV spectroscopy using a coupled assay containing 5.0 μM YdjI, 1−10 mM substrate, glycerol-3-phosphate dehydrogenase (G3PD, 0.1 unit), and 300 μM NADH at 30 °C. If DHAP was not a product of this transformation (e.g., fructose-6-phosphate), triosephosphate isomerase (1 unit) was included to convert D-glyceraldehyde-3-P to DHAP. The values of kcat and kcat/Km were determined by fitting the initial velocity data to eq 3 using GraFit 5, where v is the initial velocity of the reaction, E is the enzyme concentration, kcat is the turnover number, and Km is the Michaelis constant.
| (3) |
Stereochemistry of the YdjI-Catalyzed Reaction.
YdjI was used to enzymatically synthesize fructose-1,6-bisphosphate(FBP). DHAP was generated in situ using 50 mM dihydroxyacetone, 1.0 mM ATP, 2.0 mM MgCl2, and 1 unit of glycerol kinase. The ATP was recycled using 60 mM PEP and 5 units of pyruvate kinase. DHAP was converted to G3PD using triosephosphate isomerase (100 units). To generate FBP, 20 μM YdjI was added to a final reaction volume of 1.0 mL in 50 mM HEPES/K+ buffer (pH 8.0). The reaction was monitored for 24 h using 31P NMR. For purification of FBP, enzymes were removed using a PALL-Omega 10K centrifugal filtration column. The eluent flow-through was loaded onto a DEAE Sephadex A25 column (1.5 cm × 10 cm) that had been pre-equilibrated with 500 mM NH4HCO3 (pH 8.5) and washed with water until the pH was neutral. FBP was eluted from the column using a 100 mL gradient of 0 to 500 mM NH4HCO3. Fractions were monitored using the phenolsulfuric acid test for carbohydrates with the most concentrated fractions pooled and concentrated under reduced pressure.28,29 The NMR spectrum of the purified FBP was compared to that for commercially obtained FBP and comparison of catalytic activities with commercially obtained FBP aldolase using the coupled assay described above for the retro-aldol reaction.
Purification of YdjI-Catalyzed Reaction Products.
Selected products of the YdjI-catalyzed reactions were purified so they could be further characterized or assayed. After the reactions were complete, YdjI was removed using a PALL-Omega 10K centrifugal filtration column. The flow-through was purified using DEAE anion exchange chromatography following the procedure outlined above. Depending on the number of negative charges, most compounds eluted between 100 and 250 mM NH4HCO3. Fractions were tested for the presence of carbohydrates using the phenol−sulfuric acid test. Those that tested positive were pooled and acidified using Dowex H+ resin to a pH of 3−4 to remove the ammonium bicarbonate buffer. The Dowex resin was removed by filtration, and the flow-through was neutralized to a pH of 6.0 using NaHCO3 and concentrated to dryness under reduced pressure.
Crystallization and Structural Analysis.
For crystallization purposes, the gene encoding YdjI was recloned and ligated into pET-31(b+) to generate a construct with a C-terminal polyhistidine tag (LEHHHHHH). The pET-31-ydjI plasmid was utilized to transform Rosetta2(DE3) E. coli cells (Novagen). The cultures were grown in lysogeny broth supplemented with ampicillin (100 mg/L) and chloramphenicol (50 mg/L) at 37 °C while being shaken until an optical density of 0.5 was reached at 600 nm. At this time, zinc acetate was added (0.1 mM), and the growth allowed to continue until the optical density reached approximately 0.8. The flasks were cooled in an ice bath, and protein expression was initiated with the addition of 1.0 mM IPTG. The cells were then allowed to grow at 22 °C for 18 h. The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and YdjI was purified with Prometheus Ni-NTA agarose (Prometheus Protein Biology Products) according to the manufacturer’s instructions. The protein was dialyzed against 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl and concentrated to 25 mg/mL on the basis of an extinction coefficient of 0.99 (mg/mL)−1 cm−1 at 280 nm.
Crystallization conditions for YdjI were surveyed at both room temperature and 4 °C by the hanging drop method of vapor diffusion using a laboratory-based sparse matrix screen. X-ray diffraction quality crystals of the enzyme were subsequently grown from precipitant solutions containing 10−12% poly(ethylene glycol) 5000, methyl ether, and 100 mM MES (pH 6.0) at 21 °C in the presence of 5.0 mM DHAP. The crystals belonged to space group P21 with the following unit cell dimensions: a = 80.8 Å, b = 85.7 Å, c = 84.3 Å, and β = 109.9°. The asymmetric unit contained four subunits.
For X-ray data collection, the crystals were transferred to a cryoprotectant solution containing 22% poly(ethylene glycol) 5000 methyl ether, 300 mM NaCl, 5.0 mM DHAP, 16% ethylene glycol, and 100 mM MES (pH 6.0). X-ray data were collected at the Advanced Photon Source, Structural Biology Center Beamline 19-BM. The X-ray data were processed with HKL3000.30 The structure was determined via molecular replacement with PHASER using as a starting model the structure of tagatose-1,6-bisphosphate aldolase [Protein Data Bank (PDB) entry 1GVF].31,32 Iterative cycles of model building with COOT and refinement with REFMAC reduced Rwork and Rfree to 14.1% and 18.2%, respectively, from 50 to 1.75 Å resolution.33–35 Relevant X-ray data collection and model refinement statistics are listed in Table 1.
Table 1.
X-ray Data Collection and Refinement Statistics
| resolution limits | 50−1.75 (1.78−1.75)b |
| space group unit cell dimensions | P21 |
| a (Å) | 80.8 |
| b (Å) | 85.7 |
| c (Å) | 84.3 |
| β (deg) | 109.9 |
| no. of independent reflections | 104686 (7205) |
| completeness (%) | 96.5 (90.6) |
| redundancy | 3.6 (2.6) |
| average I/average σ(I) | 33.2 (4.4) |
| Rsyma (%) | 7.1 (21.3) |
| R-factorc (overall) (%)/no. of reflections | 14.3/104686 |
| R-factor (working) (%)/no. of reflections | 14.1/99486 |
| R-factor (free) (%)/no. of reflections | 18.2/5200 |
| no. of protein atoms | 8142 |
| no. of heteroatoms average B value (Å2) | 1006 |
| protein atoms | 24.1 |
| metals | 43.9 |
| solvent | 34.5 |
| weighted root-mean-square deviations from ideality | |
| bond lengths (Å) | 0.011 |
| bond angles (deg) | 1.4 |
| general planes (deg) | 0.009 |
| Ramachandran regionsd (%) | |
| most favored | 99.0 |
| additionally allowed | 1.0 |
| generously allowed | 0.0 |
| PDB entry | 6OFU |
.
Statistics for the highest-resolution bin.
R-factor = where FO is the observed structure factor amplitude and FC is the calculated structure factor amplitude.
Distribution of the Ramachandran angles according to PROCHECK.36
Computational Docking of Ligands to YdjI.
Computational docking was conducted using the AutoDock Vina plugin (version 2.1.1) for Pymol (version 1.8 or higher) or using the command-line scripts from the AutoDock Tools Package provided by The Scripps Research Institute.37–40 The receptor was chosen to be chains A and B in the X-ray coordinate file. Chains A and B served as the active site and the secondary subunit, respectively. The search space was confined to a 24 Å × 20 Å × 27 Å box, centered near residue Phe52 of chain A, and sized such that two arginine residues, Arg84 and Arg254′, from the adjacent chain B subunit, could be included as flexible residues. The docking output was set to provide 10 poses. A modified version of the flexible side chain method for the covalent docking technique was used after the initial AutoDock Vina plugin failed to produce realistic results with DHAP and L-glycero-L-galacto-octuluronate-1-P.41 For the docking optimization of L-glycero-L-galacto-octuluronate-1-P, DHAP was manually positioned in the YdjI structure using an overlay of bound DHAP from a homologous structure (PDB entry 5VJD).13 The DHAP moiety was then included as part of the receptor and held fixed, and the hydrate of L-arabinuronate was docked using AutoDock Vina. The top results were manually positioned near the DHAP moiety, and a bond between C3 of DHAP and C1 of L-arabinuronate was created in silico, to yield L-glycero-L-galacto-octuluronate-1-P. The resulting eight-carbon monosaccharide was designated to be part of a new receptor file and allowed to act as a flexible side chain with all bonds among C1−C4 held fixed and inactive during the docking routine. The remaining bonds of L-glycero-L-galacto-octuluronate-1-P were allowed to be optimized during the docking calculations. For this experiment, a water molecule served as the ligand to be bound.
RESULTS
Bioinformatic Analysis of YdjI.
YdjI is assigned to COG0191, which is comprised of 2280 unique protein sequences as of January 3, 2019. The SSN for COG0191 with the functionally characterized enzymes and proteins of interest, mapped at two different alignment score cutoffs, 55 and 100, is presented in Figure S1. At an arbitrary cutoff of 55 (∼34% sequence identity), YdjI is found in the major cluster containing tagatose-1,6-bisphosphate aldolase from E. coli (GatY and KbaY) and 2-deoxy-5-keto-D-gluconate-6-phosphate aldolase (IolJ) from B. subtilis. At a cutoff of 100 (∼60% sequence identity), the SSN is essentially isofunctional, indicating that each cluster likely represents a single substrate profile. Here, YdjI is found in a small cluster containing six nodes representing a total of 19 enzymes. In this small cluster of enzymes, all nodes share at least 70% sequence identity and are found in the same genomic context and gene cluster as YdjI from different bacterial species, including E. coli, S. dysenteriae, Salmonella enterica, and C. bolteae.
Stereospecific Hydrogen Exchange.
The gene encoding YdjI from E. coli K12 was cloned and expressed, and the enzyme purified to homogeneity. ICP-MS analysis showed that YdjI contained 0.90 ± 0.03 equiv of Zn2+ per monomer. Eight ketones were screened as possible substrates for the aldol condensation reaction by monitoring hydrogen exchange with deuterium from the solvent by 1H NMR spectroscopy. Pyruvate, hydroxypyruvate, methyl glyoxal, acetone, hydroxyacetone, dihydroxyacetone, hydroxyacetone phosphate, and dihydroxyacetone phosphate (DHAP) were all tested as potential substrates for YdjI (Figure S2). The hydrogen exchange reactions for each compound were monitored for ≤72 h, but measurable catalytic activity was observed only with DHAP.
In solution, DHAP exists in two interexchangeable forms, where 37% of the keto group is hydrated (Figure 2a).42 Hydrogen/deuterium exchange at C3 of DHAP was observed by following the disappearance of the hydrogen resonance for one of the prochiral hydrogens at 3.55 ppm and the appearance of a broader resonance at 3.53 ppm. Alternatively, exchange of a hydrogen at C3 of the keto form of DHAP was monitored at 4.54 ppm. The apparent rate constant for the hydrogen exchange reaction was 8.4 ± 0.3 s−1 (Figure 2b,c). To determine which of the two prochiral hydrogens at C3 of DHAP is exchanged by YdjI, rabbit muscle aldolase was added to the reaction mixture. After an incubation period of 4 h, no additional hydrogen exchange with the solvent was observed (Figure 2d). For comparison, 1.0 unit of rabbit muscle aldolase was shown to catalyze the exchange of one of the C3 hydrogens of DHAP (10 mM) in 60 min (data not shown). Therefore, YdjI catalyzes the removal of the same prochiral hydrogen from DHAP as rabbit FBP aldolase, and thus, the pro-S hydrogen from C3 is abstracted (Scheme 2).14,43 The effect of changes in pH on the rate of deuterium exchange was also examined. YdjI was shown to be most active at pH 7.1 with a 1.6-fold loss of activity at pH 6.1 and a 3.6-fold loss of activity at pH 8.0 (Figure S9).
Figure 2.

Solvent−deuterium exchange within DHAP. (a) 1H NMR spectrum of DHAP in 25 mM imidazole/D2O buffer (pH 7.4). (b) 1H NMR spectrum of DHAP (10 mM) after a 21 min incubation with YdjI (2.0 μM). (c) 1H NMR spectrum of DHAP (10 mM) after a 90 min incubation with YdjI (2 μM). (d) 1H NMR spectrum of DHAP (10 mM) after a 4 h incubation with YdjI (2.0 μM) and FBP aldolase (1.0 unit). The reaction is illustrated in Scheme 2.
Scheme 2.

Solvent−Deuterium Exchange Reaction within DHAP Catalyzed by YdjI
Product Stereochemistry at C4.
The stereochemistry at C4 after condensation with an aldose sugar remained to be defined. The reaction can occur in a manner similar to that of the reaction catalyzed by FBP aldolase, resulting in a product with R stereochemistry at C4, or alternatively, with the opposite stereochemistry in a reaction similar to that catalyzed by tagatose-1,6-bisphosphate (TBP) aldolase.43 YdjI was mixed with DHAP and D-glyceraldehyde-3-P (D-G3P) to generate either FBP or TBP. DHAP was generated in situ using glycerol kinase, ATP, and dihydroxyacetone. D-G3P was generated from DHAP by the addition of triosephosphate isomerase (TIM). Upon addition of YdjI, FBP is the primary product, although small amounts of uncharacterized side products are visible in the 31P NMR spectrum (Figure 3a). The same reaction with FBP aldolase produces the α- and β-anomeric forms of FBP (Figure 3b). To demonstrate that the reaction product is FBP, it was purified by anion exchange chromatography (Figure 3c) and compared with FBP obtained commercially (Figure 3d). The YdjI-generated FBP was also demonstrated to be a substrate for FBP aldolase when the reaction was monitored by a coupled spectroscopic assay. Therefore, the YdiI-catalyzed condensation of DHAP and D-G3P produces FBP with formation of an S stereocenter at C3 and an R stereocenter at C4.
Figure 3.

31P NMR spectra of YdjI and FPB aldolase reaction products. (a) Reaction of YdjI with DHAP and TIM to generate D-fructose-1,6-bisphosphate (FBP). (b) Reaction of FBP aldolase with DHAP and TIM to generate FBP. (c) FPB purified from the reaction mixture depicted in panel a. (d) 31P NMR spectrum of FBP obtained commercially. Peak assignments for FBP obtained from previously published reports.44,45
Screening for Aldose Sugar Substrates.
After determining that DHAP is the ketose component for the YdjI-catalyzed reaction, we screened potential aldose fragments for the condensation reaction by 1H and 31P NMR spectroscopy. In total, 49 compounds were tested as potential substrates for YdjI in the presence of DHAP (Figure S3). It was initially determined that YdjI will accept methyl glyoxal (13) as a substrate with formation of 6-deoxy-5-keto-fructose-1-P [DKFP (40)]. Methyl glyoxal is a degradation product of DHAP and a common contaminate in commercially available DHAP.46,47 Unlike the high specificity for the keto substrate, significant promiscuity was observed within the aldehydecontaining substrates. YdjI catalyzed the condensation of 33 different aldose substrates with DHAP (Figure 4). Activity was determined by observation of NMR resonances that could be readily distinguished from those of either DHAP or DKFP (40). The compounds that showed the most robust activity had their turnover numbers quantified by following the reaction over time using 31P NMR spectroscopy (Table 2). The isolated products were subjected to mass spectrometry and were confirmed to have the expected molecular weight (Figure 5).
Figure 4.

Structures of aldoses found to be substrates for YdjI in the aldose condensation reaction with DHAP.
Table 2.
Turnover Numbers for Substrates with YdjI and DHAPa
| substrate | expected product |
apparent kcat (s−1)b |
MW (M) |
found MW (M − H)− |
|---|---|---|---|---|
| 3 | 34 | 0.40 ± 0.05 | 243.99 | 242.99 |
| 5 | 35 | 2.2 ± 0.2 | 244.03 | 243.03 |
| 6 | 36 | 4.8 ± 0.6 | 244.03 | 243.03 |
| 9/10 | 37/38 | 0.50 ± 0.02 | 274.00 | 273.00 |
| 13 | 40 | 0.64 ± 0.03 | 242.01 | 241.01 |
| 14 | 41 | 0.71 ± 0.06 | 290.03 | 289.03 |
| 15 | 42 | 0.48 ± 0.04 | 290.03 | 289.03 |
| 17 | 43 | 0.68 ± 0.04 | 304.01 | 303.01 |
| 20 | 44 | 0.66 ± 0.04 | 370.00 | 369.00 |
| 25 | 45 | 3.0 ± 0.3 | 334.02 | 333.02 |
| 26 | 46 | 2.3 ± 0.2 | 334.02 | 333.02 |
| 28 | 47 | >0.7c | 334.02 | 333.02 |
| 29 | 48 | 0.50 ± 0.04 | 400.01 | 399.01 |
| 30 | 49 | 1.60 ± 0.09 | 400.01 | 399.01 |
| 32 | 50 | 0.050 ± 0.004 | 364.03 | 363.03 |
| 33 | 51 | 1.8 ± 0.1 | 364.03 | 363.03 |
Reactions were monitored by 31P NMR spectroscopy and quantified by integration with an internal phosphate standard.
Errors were calculated from the standard deviation of the fitting results as described.
Overlap between substrate and product peaks prevented accurate integrations. The value reported is a lower limit based on the amount of product formed in 30 min and corrected for enzyme concentration.
Figure 5.

Structures of products formed from the condensation of DHAP with selected aldehyde substrates by YdjI.
To measure the specificity constant (kcat/Km) for each substrate, selected compounds were synthesized on a larger scale (∼50 mg) by mixing YdjI and DHAP with an excess of the aldose substrate, and the products purified by anion exchange chromatography. These compounds were used as substrates for YdjI in the retro-aldol reaction. The formation of DHAP was monitored in a coupled enzyme assay with glycerol-3-dehydrogenase (Table 3).
Table 3.
Steady-State Kinetic Parameters of Selected Substrates with YdjIa
| substrate | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|
| D-fructose-1-Pb | 0.042 ± 0.003 | 7.8 ± 1.0 |
| 39b | 0.060 ± 0.001 | 30.5 ± 0.7 |
| 40c | <0.010 | – |
| 41c | <0.001 | – |
| 42b,c | <0.001 | – |
| 44 | 0.011 ± 0.001 | 26.6 ± 3.5 |
| 45 | 0.860 ± 0.002 | (2.1 ± 0.1) × 103 |
| 46 | 0.021 ± 0.012 | 14.7 ± 1.9 |
| 47 | 0.260 ± 0.084 | (4.2 ± 1.6) × 102 |
| 50 | 0.038 ± 0.001 | (2.0 ± 0.2) × 102 |
| 51 | 0.089 ± 0.002 | (2.0 ± 0.2) × 102 |
Errors were calculated from the standard deviation of the fitting results.
Commerically purchased compounds.
Estimated on the basis of the lower limit of detection.
Structural Characterization of Selected Substrates.
To further demonstrate that the proposed stereochemistry at C4 is consistent with the broad substrate profile of YdjI, two additional experiments were conducted. In the first experiment, the retro-aldol activities of the well-characterized FBP aldolase with some of the products generated enzymatically by YdjI were quantified. The high degree of promiscuity for aldolase enzymes in the aldol condensation reaction is well-documented.15,16,43 However, the retro-aldol reaction has been shown to be quite selective for a specific stereochemical arrangement at C3 and C4.16 For example, FBP aldolase has a 1400-fold higher specificity for cleavage of FBP39 than for cleavage of TBP.43 FBP was shown to be a relatively poor substrate for YdjI with a kcat/Km of 30 M−1 s−1 (Table 3).
Six compounds synthesized by YdjI [D-xyluluronate-1-P (34), D-glycero-L-galacto-octuluronate-1-P (46), L-glycero-L-galacto-octuluronate-1-P (45), L-glycero-D-altro-octuluronate-1P (47), D-erythro-L-galacto-nonuluronate-1-P (50), and L-threo-L-galacto-nonuluronate-1-P (51)] were shown to be substrates for FBP aldolase. These compounds showed a range of activity with D-xyluluronate-1-P (34) being 115-fold slower than FBP (39), while L-glycero-L-galacto-octuluronate-1-P (45) is only 23-fold slower than FBP (Table S1). Compared to FBP, these are poor substrates, but these experiments further support the conclusion that the stereochemical arrangements at C3 and C4 are the same for FBP aldolase and YdjI.
NMR spectroscopy was used to further characterize the reaction product D-xyluluronate (34) with previously published chemical shift values. D-Xyluluronate-1-P (34) was synthesized by mixing YdjI with DHAP and glyoxylate (3). The resulting product was dephosphorylated using rSAP and purified by anion exchange chromatography. The 13C NMR chemical shifts of the dephosphorylated product are very similar to the published 13C chemical shifts for authentic D-xyluluronate synthesized enzymatically by a different route.48 The chemical shifts for D-riburonate (the C3 epimer) are reproduced in Table 4 for comparison.
Table 4.
13C NMR Chemical Shifts (parts per million)
| substrate | C1 | C2 | C3 | C4 | C5 |
|---|---|---|---|---|---|
| D-xyluluronatea | 66.02 | 212.37 | 77.05 | 73.15 | 176.90 |
| D-xyluluronateb | 65.99 | 212.38 | 76.88 | 73.05 | 177.13 |
| D-ribuluronateb | 66.21 | 211.13 | 77.88 | 74.62 | 176.43 |
Produced from glyoxylate and DHAP with YdjI after dephosphorylation.
As reported previously.48
Three-Dimensional Structure of YdjI.
The crystals utilized in this investigation contained four subunits in the asymmetric unit, which packed together to form a tetrameric quaternary structure similar to that observed for tagatose-1,6-bisphosphate aldolase from E. coli.32 Subsequent gel filtration chromatography confirmed that YdjI migrates as a tetramer. The final model of YdjI was refined to an overall R-factor of 14.3% at 1.75 Å resolution. Shown in Figure 6a is a ribbon representation of the tetramer, which can be envisioned as a dimer of dimers. The total buried surface area between monomers A and B is 3500 Å2, whereas that between monomers A and C is 1500 Å2.
Figure 6.

(a) Tetrameric ribbon structure of YdjI. The zinc ion is shown as a gray sphere. (b) Active site of YdjI. Key residues are labeled and shown as sticks.
Each subunit is characterized by eight β-strands and 11 α-helices that form a classical TIM barrel motif.49 The electron density for Cys234 in each subunit clearly revealed that it has been oxidized to cysteic acid. It is located on α-helix 10 and positioned near the subunit−subunit interfaces formed by monomers A and B and monomers C and D. In three of the subunits, there were breaks in the electron density corresponding to the polypeptide chain between α-helices 6 and 7 and between β-strand 6 and α-helix 8. These regions of flexibility are located at the C-terminal region of the TIM barrel. In monomer B, there was only a single break in the electron density between α-helices 6 and 7.
As expected for a class II aldolase, in each subunit of the tetramer there is a zinc ion located at the C-terminal region of the β-barrel. Ligation about the zinc is variable in each subunit as described for other structures.13 In subunit A, the zinc is coordinated by His82, His178, His206, and two water molecules. This surface-exposed zinc site is typically found when the ligand is bound at the active site, as previously described.13 With respect to subunit B, however, the zinc is ligated in an octahedral coordination sphere by His82, His178, His206, Glu133 (bidentate), and a water molecule. This buried site is found when the ligand is absent, as previously described.13 Upon binding of the ligand, the zinc is proposed to undergo an ∼3.7 Å movement from the buried site to the surface-exposed site facilitated by rotation of the coordinating histidine residues and movement of adjacent loop regions.13 There are two zinc ions in subunit C. One is held in place by His82, His206, Glu133 (bidentate), and two water molecules, whereas the second zinc interacts with His82 and three waters. Finally, in subunit D, the zinc coordination geometry is octahedral with His82, His206, Glu133 (bidentate), and two waters functioning as ligands.
Most class II aldolases contain a highly conserved aspartate residue positioned near the active site zinc ion. In YdjI, this residue is Asp81 (Figure 6b). It has previously been proposed that this conserved aspartate residue is responsible for abstracting or donating the proton to the C4 hydroxyl group of the substrate.32,50 However, a recent publication suggests that a histidine residue (equivalent to His178 in YdjI) may be responsible for the proton abstraction and a second histidine residue (equivalent to His82 in YdjI) participates in stabilizing the negatively charged intermediate.13 A conserved glutamate residue has also been proposed to function as an active site base by abstracting a proton from DHAP. In the model of YdjI, this glutamate residue (mostly likely Glu144, based on the sequence alignment, but Glu145 cannot be ruled out) is located in a disordered region of the electron density map for monomers A, B, and D. In monomer C, however, electron density is observed for Glu144 and shows that it is located >20 Å from the active site zinc. This positioning is most likely due to crystalline packing interactions. The phosphate binding pocket is expected to be found near Ser209 and Thr231 on the basis of structural alignments with other family members with substrates bound at their active sites. Unfortunately, no electron density was observed for DHAP at any of the active sites despite being present during crystal growth. However, YdjI was shown to be catalytically active in 100 mM MES buffer (pH 6.0) and 300 mM NaCl, with a 3-fold decrease in activity in the retro-aldol reaction with L-glycero-L-galacto-octuluronate-1-P, relative to that seen with 50 mM HEPES (pH 7.4) and 250 mM KCl.
Docking of Ligands into the Active Site of YdjI.
Computational docking was conducted to better understand the differences in substrate binding between FBP aldolase and YdjI, specifically for the best substrate, L-glycero-L-galactooctuluronate-1-P (45). Chain A of the YdjI tetramer was chosen as the primary active site due to the position of the zinc and orientation of active site residues, previously described as the active conformation.13 The adjacent chain B provided the secondary active site features. Initial docking studies placed DHAP and L-glycero-L-galacto-octuluronate-1-P in the active site but failed to position either substrate with the anticipated coordination of the carbonyl and hydroxyl groups at C2 and C3, respectively, or place the phosphate group in an appropriate location (Figure S4A,B). Attempts to dock DHAP using AutoDockZn, which allows for Zn coordination, failed to improve these results.51 A structural overlay between YdjI and five other FBP aldolase enzymes with either DHAP or FBP bound in the active site (PDB entries 1B57, 5VJD, 3Q94, 3GB6, and 3ELF) provides a reasonable consensus for how DHAP coordinates to the zinc in the active sites of these enzymes (Figure S5). The distance between the carbonyl group at C2 and the zinc should be between 2.2 and 3.2 Å, and the distance between the hydroxyl group at C3 and the zinc should be between 2.1 and 2.3 Å. Using these interactions as a guide, DHAP was manually positioned in the active site of YdjI (Figure S6) such that the C2 carbonyl group and the C3 hydroxyl group are 2.7 and 2.2 Å from the zinc, respectively. The phosphate residue should be placed near Ser209 and Thr231, which are conserved in PDB entries 1B57, 5VJD, and 3ELF and shown to interact with the phosphate moiety of bound substrates. Subsequently, DHAP was included to be a fixed part of the YdjI active site, to which the hydrate of L-arabinuronate (25) was docked. The hydrate of L-arabinuronate (25) was used to enable flexibility of the hydroxyl groups and for easier in silico C−C bond formation to yield L-glycero-L-galacto-octuluronate-1-P (45).
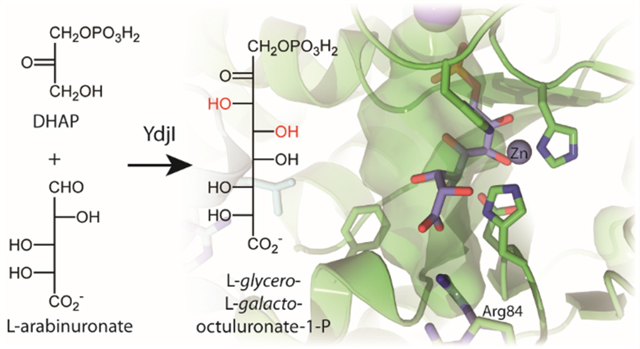
L-Arabinuronate (25) or its hydrate does not dock particularly well to this modified active site (Figure S7), but the results were used to position the molecule such that a C−C bond between C3 of DHAP and C1 of L-arabinuronate (25) could be formed in silico, yielding a more realistic position of L-glycero-L-galacto-octuluronate-1-P (45) bound in the active site. With this version of L-glycero-L-galacto-octuluronate-1-P bound in the active site, a modified version of covalent docking using flexible side chains was used for the docking predictions.41 During the docking routine, all bonds among the first four carbons (C1−C4) were held fixed in the same orientation that was found in structures containing FBP bound at the active site.13 All remaining bonds (C5−C8) were considered as a flexible “side chain” of the receptor and allowed to be computationally optimized by the docking function. To minimize effects due to the addition of a ligand, a water molecule was chosen as the ligand. The best pose is shown with blue sticks in Figure 7a. A two-dimensional illustration with measured distances of interactions is shown in Figure 7b.
Figure 7.

(a) Modified covalent docking of L-glycero-L-galacto-octuluronate-1-P into YdjI. Shown in blue sticks is the top result from the docking routine. Shown in black dotted lines are predicted interactions between the substrate and YdjI receptor. Shown in green space filling is the predicted active site cavity in which the substrate binds. (b) Two-dimensional illustration of the active site and interactions with docked L-glycero-L-galacto-octuluronate-1-P. Interactions are shown with the dotted lines and measured in angstroms.
This orientation of L-glycero-L-galacto-octuluronate-1-P (45) appears to be more reasonable than the pose produced by the regular docking routine (Figure S4B). The phosphate group is hydrogen bonded to Thr231 and Ser209, which are located 2.5 and 2.6 Å, respectively, from the nearest oxygen of the phosphate moiety. The C2 and C3 hydroxyls are within coordinating distances of the zinc as defined above. The C2 hydroxyl is predicted to interact with His178, and the C3 hydroxyl interacts with His82 and Asp81. The conserved Asp81 is 2.7 Å from the C3 hydroxyl. The hydroxyl group on C4 is 3.3 Å from Asp81 and 3.6 Å from His178 (measured from the attached proton). This position supports the chemical mechanism for the retro-aldol reaction by which Asp81 and/or His178 would remove the proton attached to the hydroxyl group at C4.13,14,52 The C4 hydroxyl is also found to be 3.4 Å from His82, allowing it to participate in the stabilization of the negatively charged intermediate. The hydroxyl at C5 is predicted to interact with His230. Asp81 may also interact with the hydroxyl at C6. The distance between Arg84 and the C8 carboxylate moiety is 3.3 Å. The multiple predicted contacts of Asp81 to the substrate suggest its importance in substrate binding as previously described.13 This distance could not be reduced further by allowing Arg84 to be flexible during docking. No pose that predicts an interaction between the carboxylate group and Arg254 could be achieved, even with the nine-carbon carbohydrates. The preference of the carboxylate interaction with Arg84 versus Arg254 is further supported by the active site cavity extending toward Arg84. Access to Arg254 is apparently blocked by Phe52 and Leu250′ and partially obstructed by His230. In the Mycobacterium tuberculosis FBP aldolase (PDB entry 3ELF), Arg314 is used to bind the P6 phosphate of FBP (Figure S8). Arg314 is found closer to the position of Leu250′ in YdjI. In the M. tuberculosis FBP aldolase, Arg314 is 4.5 Å closer to the active site than Arg254 in YdjI.
DISCUSSION
In this investigation, YdjI, an aldolase of unknown function from E. coli K12, has been characterized and its three-dimensional structure determined to 1.75 Å resolution. YdjI was shown to catalyze the condensation of aldose sugars with DHAP to form an array of novel carbohydrates. The initial analysis of the SSN and experimental data yielded two conclusions about enzymes from COG0191. First, all members of COG0191 appear to utilize DHAP as one component of the substrates for the condensation reaction. YdjI was assayed with eight keto-containing compounds for activity and exhibited exclusive activity with DHAP. Second, all previously characterized members of this COG remove the pro-S hydrogen from DHAP, and YdjI was shown to catalyze the exchange of this hydrogen with solvent. Aldolases that remove the pro-R hydrogen from DHAP, such as rhamnulose-1-P aldolase (Uniprot entry P32169) and fuculose-1-P aldolase (Uniprot entry P0AB87), are located in COG0235. Members of the COG191 family exhibit relatively high sequence identity (>35%) but can have different substrate profiles. For example, YdjI is 38% identical to TBP aldolase, but YdjI is functionally more similar to FBP aldolase (22% sequence identity). The two characteristics described above may explain the apparent high degree of sequence conservation among the family members with different substrate preferences. The distinguishing features of substrate preferences will be dictated by two factors: (a) the stereochemical preference at C4 and (b) the chemical modifications within the rest of the substrate (e.g., deoxy at C5, phosphate at C6, etc.).
For the aldol condensation reaction, there are two possible stereochemical outcomes at C4, depending on the face of attack on the aldehyde. FBP aldolase undergoes a si-face attack resulting in an R stereocenter, while TBP aldolase undergoes re-face attack resulting in an S stereocenter at C4.43 YdjI yields products with the same stereochemistry at C4 as FBP aldolase. The stereochemistry at C4 was determined by showing that YdjI synthesizes FBP when given DHAP and D-G3P, and D-xyluluronate-1-P (34) when given DHAP and glyoxylate (3). Moreover, FBP aldolase was shown to catalyze the retro-aldol reaction when given six different compounds synthesized by YdjI (Table S1). On the basis of these data, YdjI catalyzes the cleavage of a keto sugar with a phosphate group at C1 where the stereochemical arrangements of the hydroxyl groups at C3 and C4 are the S and R configurations, respectively.
Aldolases are known to display high variability with the aldehyde component for the aldol condensation reaction.15,16,43 Similarly, YdjI was shown to condense an array of aldose sugars with DHAP. Potential substrates were tested, from short two carbon compounds, such as acetaldehyde, to longer carbohydrates such as L-iduronic acid (Table S2). Not surprisingly, YdjI combines DHAP with many of the same aldehyde compounds as FBP aldolase. These include D-glyceraldehyde-3-P (11), methylglyoxal (13), D-erythrose (16), D-ribose (24), D-ribose-5-P (31), D-lactaldehyde (6), and L-lactaldehyde (5). Direct comparisons of YdjI and FBP aldolase show that FBP aldolase catalyzes the condensation of D-ribose-5-P approximately twice as fast as YdjI, approximately equal with the D/L-glyceraldehyde, but YdjI is ∼2 orders of magnitude faster than FBP aldolase with methylglyoxal. One significant difference was found with D-arabinose and its derivatives. Previously, FBP aldolase was shown to utilize D-arabinose-5-P to give D-glycero-D-ido-octulo-1,8-bisphosphate.53 This finding was reproduced during this study, and the product was confirmed by comparison with the published NMR chemical shifts (data not shown). However, YdjI shows no measurable activity with D-arabinose-5-P, D-arabinose, or D-arabinuronic acid. This is somewhat surprising given the high degree of chemical similarity between FBP aldolase and YdjI.
There are two other key differences between FBP aldolase and YdjI. First, YdjI prefers longer chain monosaccharides. Among the molecules assayed with YdjI, the carboxylic acid series provides the best insight into this trend toward longer molecules (Figure 8a). Glyoxylate (3) is the slowest substrate (0.4 s−1), followed by D/L-tartronate semialdehyde (9/10, 0.5 s−1) and L-threouronic (17, 0.7 s−1). The penturonic acids, L-arabinuronate (25) and D-xyluronate (26), are the fastest at 3.0 and 2.3 s−1, respectively. There is a slight loss of activity with L-iduronate (33) at 1.8 s−1. The rate data for the retro-aldol reaction agree with this trend, as well. YdjI has a strong preference for a terminal negative charge versus a hydroxyl group (Figure 8b). This is quite apparent when comparing the substrate activity of L-arabinose (21), L-arabinuronate (25), and L-arabinose-5-P (29). L-Arabinose (21) is a very slow substrate, and only an upper limit for kcat could be estimated (<0.003 s−1). However, with L-arabinose-5-P (29) or L-arabinuronate (25), the rate constants are 2−3 orders of magnitude faster. The same trend is observed for D-xylose (22), D-xyluluronate (26), and D-xylose-5-P (30). For longer carbohydrates, YdjI exhibits a preference for a carboxylate terminus relative to phosphate. YdjI is faster with L-arabinuronate (25) and D-xyluronate (26) than with L-arabinose-5-P (29) and D-xylose-5-P (30). No activity was detected with D-glucose-6-P, but D-glucuronate (32) is active. With shorter substrates (fewer than four carbons), there are no detectable differences. For example, L-threouronate (17) and L-threose-4-P (20) exhibit the same rate of turnover.
Figure 8.

Summary of the substrate profile for YdjI. (a) Relative rates of turnover for the carboxylate series of compounds. (b) Relative rates of turnover comparing terminal moieties of compounds. N.D., rate determined; N.A., no activity observed.
The outliers in these trends are D- and L-lactaldehyde (6 and 5, respectively). These compounds contain terminal methyl groups; however, D-lactaldehyde (6) has the highest turnover number (4.8 s−1), nearing the rate of hydrogen/deuterium exchange of DHAP (8.4 s−1). It is unclear how a terminal methyl group would be handled in longer substrates. However, given the very low activity with terminal hydroxyl groups, it seems unlikely that a terminal methyl group with longer substrates would show increases in activity.
An initial investigation of the three-dimensional crystal structure suggests minimal differences between FBP aldolases and YdjI. One noticeable difference is the location and environment of Arg254 (analogous to Arg331 in E. coli FBP aldolase). In E. coli FBP aldolase, Arg331 can be nearly superimposed with Arg314 from M. tuberculosis and is ∼4.5 Å closer to the active site than Arg254 in YdjI. Moreover, the extra space is occupied by Phe52 and Leu250′. These two hydrophobic residues appear to restrict access to Arg254 and prevent binding of the substrate in YdjI as observed in the structure of FBP aldolase (Figure S8). A second arginine residue, Arg84, found near the active site in YdjI, suggests a possible alternative binding mode for these longer substrates. Arg84 is strictly conserved in the cluster of enzymes containing YdjI in the SSN from COG0191. Computational docking was conducted to investigate the possible substrate binding modes in YdjI. Unfortunately, many of the standard docking routines failed to provide a reasonable positioning of the substrate in the active site. Using a combination of manual placement and a modified covalent docking approach, a more realistic docking pose was achieved with L-glycero-L-galacto-octuluronate-1-P (Figure 7). These results lend additional support to the idea of Arg84 being responsible for binding the negative terminus of the preferred substrates for YdjI.
Given these data, the preferred substrate for YdjI is more apparent. The top portion resembles DHAP as a 2-ketose with a phosphate group attached to C1. The stereocenters of C3−C6 are preferred in the L-galacto configuration, and the terminal carbon is either carboxylated or phosphorylated. For substrates with eight carbons, the stereochemistry at C7 can be S or R. Currently, the best substrate for YdjI is L-glycero-L-galacto-octuluronate-1-P (45) with a kcat/Km value of 2 × 103 M−1s−1.
The genomic context of YdjI suggests that it participates in a catabolic pathway for the degradation of carbohydrates. L-glycero-L-galacto-Octuluronate-1-P is, to the best of our knowledge, not a known carbohydrate. There are, however, four known molecules that resemble the best substrate for YdjI (Figure 9). 3-Deoxy-D-arabino-heptulosonate-7-P (52, DAHP) is an intermediate in the shikimate pathway for the synthesis of aromatic amino acids.54 3-Deoxy-D-manno-octulosonate-8-P (53, KDO) is a carbohydrate used in the synthesis of lipopolysaccharides (LPS) in bacteria.55 D-glycero-D-talo-Octulosonate-8-P (54, KO) is an uncommon substitute for KDO in LPS.56 3-Deoxy-D-glycero-D-galacto-nonulosonate-9-P (55, KDN) is a desamino sialic acid derivative found within glycoproteins of vertebrates and bacteria.57,58 While KDN has a known degradation pathway, DAHP and KDO/KO do not.58 However, E. coli was shown to utilize KDO as a sole carbon source after a spontaneous mutation to the cell. It was proposed that the mutation enabled KDO to be decarboxylated in the medium and subsequently transported into the cell and degraded via an unknown catabolic reaction pathway.59
Figure 9.

Structures of known keto-carboxylate carbohydrates.
As shown, none of these molecules can be substrates for YdjI. The carbon adjacent to the phosphate moiety must be oxidized to a ketone, and it is unclear how a ketone moiety adjacent to the carboxylate terminus would be handled by YdjI. It is possible that some of the dehydrogenases found within the ydi gene cluster may be responsible for the oxidation or reduction of the penultimate carbons at either end of these carbohydrates. Nevertheless, YdjI is the last remaining uncharacterized class II aldolase found in E. coli K12. This enzyme shows a preference for higher-order monosaccharides containing an L-galacto stereochemistry and a negatively charged terminus. In the subsequent paper (DOI: 10.1021/acs.biochem.9b00327), the substrate profile for YdjH, the kinase that apparently phosphorylates the ultimate substrate for YdjI, is addressed.
Supplementary Material
Funding
This work was supported in part by grants from the Robert A. Welch Foundation (A-840) and the National Institutes of Health (GM122825).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00326.
Methods for the synthesis of penturonic acids, DHAP, hydroxyacetone phosphate, teturonic acids, tartronate semialdehyde and glycolaldehyde, L-threose-4-phosphate, L-arabinose-5-phosphate, D-xylose-5-phosphate, and L-arabinose-5-phosphate; chemical structures for the eight ketone molecules tested with YdjI; chemical structures for all of the aldo sugars tested with YdjI and DHAP; activity of FBP aldolase with selected substrates; summary of observed activities with YdjI; SSN of COG0191; docking results for DHAP and L-glycero-L-galacto-octuluronate-1-phosphate using standard AutoDock Vina methods; overlay of crystal structures containing DHAP or FBP in the active site of YdjI; structure of YdjI showing the manual placement of DHAP; docking result for the hydrate of L-arabinuronate in the YdjI active site occupied by DHAP; and structure of FBP aldolase with FBP bound (PDF)
Accession Codes
YdjI, Uniprot entry P77704.
REFERENCES
- (1).Clemente JC, Ursell LK, Parfrey LW, and Knight R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, and Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. [DOI] [PubMed] [Google Scholar]
- (3).Round JL, and Mazmanian SK (2009) The gut microbiome shapes intestinal immune responses during health and disease. Nat. Rev. Immunol 9, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sekirov I, Russell SL, Antunes LCM, and Finlay BB (2010) Gut microbiota in health and disease. Physiol. Rev 90, 859–904. [DOI] [PubMed] [Google Scholar]
- (5).Chassaing B, Rolhion N, Vallee A. d., Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Soderholm JD, Hugot J-P, Colombel J-F, and Darfeuille-Michaud A. (2011) Crohn disease-associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J. Clin. Invest 121, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Flint HJ, Scott KP, Louis P, and Duncan SH (2012) The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol 9, 577–589. [DOI] [PubMed] [Google Scholar]
- (7).Chassard C, and Lacroix C. (2013) Carbohydrates and the human gut microbiota. Curr. Opin. Clin. Nutr. Metab. Care 16, 453–460. [DOI] [PubMed] [Google Scholar]
- (8).Hooper LV, Midtvedt T, and Gordon JI (2002) How host-microbial interactions shape the nutrient environment of the mammaliam instestine. Annu. Rev. Nutr 22, 283–307. [DOI] [PubMed] [Google Scholar]
- (9).Kamada N, Kim Y-G, Sham HP, Vallance BA, Puente JL, Martens EC, and NUnez G. (2012) Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336, 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Blumberg R, and Powrie F. (2012) Microbiota, disease, and back to health: a metastable journey. Sci. Transl. Med 4, 137rv7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kamada N, Chen GY, Inohara N, and Núñez G. (2013) Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol 14, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rolhion N, and Chassaing B. (2016) When pathogenic bacteria meet the intestinal microbiota. Philos. Trans. R. Soc., B 371, 20150504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jacques B, Coincon M, and Sygusch J. (2018) Active site remodeling during the catalytic cycle in metal-dependent fructose-1,6-bisphosphate aldolases. J. Biol. Chem 293, 7737–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hall DR, Leonard GA, Reed CD, Watt CI, Berry A, and Hunter WN (1999) The crystal structure of Escherichia coli class II fructose-1,6-bisphosphate aldolase in complex with phosphoglycolohydroxamate reveals details of mechanism and specificity. J. Mol. Biol 287, 383–394. [DOI] [PubMed] [Google Scholar]
- (15).Heron PW, and Sygusch J. (2017) Isomer activation controls stereospecificity of class I fructose-1,6-bisphosphate aldolases. J. Biol. Chem 292, 19849–19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schoevaart R, van Rantwijk F, and Sheldon RA (2000) Stereochemistry of nonnatural aldol reactions catalyzed by DHAP aldolases. Biotechnol Bioeng 70, 349–352. [DOI] [PubMed] [Google Scholar]
- (17).Yoshida K.-i., Yamaguchi M, Morinaga T, Kinehara M, Ikeuchi M, Ashida H, and Fujita Y. (2008) myo-Inositol catabolism in Bacillus subtilis. J. Biol. Chem 283, 10415–10424. [DOI] [PubMed] [Google Scholar]
- (18).Galperin MY, Makarova KS, Wolf YI, and Koonin EV (2015) Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43, D261–D269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mitchell A, Chang H-Y, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong S-Y, Bateman A, Punta M, Attwood TK, Sigrist CJA, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, and Finn RD (2015) The Inter Pro protein families database: the classification resource after 15 years. Nucleic Acids Res 43, D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Di Luccio E, Elling RA, and Wilson DK (2006) Identification of a novel NADH-specific aldo-keto reductase using sequence and structural homologies. Biochem. J 400, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).NCBI Resource Coordinators (2013) Database resources of the National Center for Biotechnology Information (NCBI). Nucleic Acids Res 41, D8–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Scaldaferri F, Gerardi V, Mangiola F, Lopetuso LR, Pizzoferrato M, Petito V, Papa A, Stojanovic J, Poscia A, Cammarota G, and Gasbarrini A. (2016) Role and mechanisms of action of Escherichia coli Nissle 1917 in the maintenance of remission in ulcerative colitis patients: An update. World J. Gastroenterol 22, 5505–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).The UniProt Consortium (2015) UniProt: A hub for protein information. Nucleic Acids Res 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta Proteins Proteomics 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang P-L, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, and Bader GD (2007) Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc 2, 2366–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Inoue H, Nojima H, and Okayama H. (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96, 23–28. [DOI] [PubMed] [Google Scholar]
- (27).Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server In The Proteomics Protocols Handbook (Walker JM, Ed.) pp 571–607, Humana Press, Totowa, NJ. [Google Scholar]
- (28).Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S-I, and Lee YC (2005) Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal. Biochem 339, 69–72. [DOI] [PubMed] [Google Scholar]
- (29).Nielsen SS (2010) Phenol-sulfuric acid method for total carbohydrates In Food Analysis Laboratory Manual (Nielsen SS, Ed.) pp 47–53, Springer US, Boston. [Google Scholar]
- (30).Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- (31).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hall DR, Bond CS, Leonard GA, Watt CI, Berry A, and Hunter WN (2002) Structure of tagatose-1,6-bisphosphate aldolase: insight into chiral discrimination, mechanism, and specificty of class II aldolases. J. Biol. Chem 277, 22018–22024. [DOI] [PubMed] [Google Scholar]
- (33).Emsley P, and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (34).Emsley P, Lohkamp B, Scott WG, and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (36).Laskowski RA, Moss DS, and Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol 231, 1049–1067. [DOI] [PubMed] [Google Scholar]
- (37).Trott O, and Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Seeliger D, and de Groot BL (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput.- Aided Mol. Des 24, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).The PyMOL Molecular Graphics System, version 1.8 (2015) Schrodinger, LLC. [Google Scholar]
- (40).Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bianco G, Forli S, Goodsell DS, and Olson AJ (2016) Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci 25, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ślepokura K, and Lis T. (2010) Dihydroxyacetone phosphate, DHAP, in the crystalline state: monomeric and dimeric forms. Carbohydr. Res 345, 512–529. [DOI] [PubMed] [Google Scholar]
- (43).Williams GJ, Domann S, Nelson A, and Berry A. (2003) Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc. Natl. Acad. Sci. U. S. A 100, 3143–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gray GR (1971) Examination of D-fructose 1,6-diphosphate and related sugar phosphates by Fourier transform phosphorus-31 nuclear magnetic resonance spectroscopy. Biochemistry 10, 4705–4711. [DOI] [PubMed] [Google Scholar]
- (45).Tyagi RK, Azrad A, Degani H, and Salomon Y. (1998) Stimulation of fructose 1,6-bisphosphate production in melanoma cells by α-melanocyte-stimulating hormone. Eur. J. Biochem 258, 68–77. [DOI] [PubMed] [Google Scholar]
- (46).White RH (2008) Biochemical origins of lactaldehyde and hydroxyacetone in Methanocaldococcus jannaschii. Biochemistry 47, 5037–5046. [DOI] [PubMed] [Google Scholar]
- (47).White RH, and Xu H. (2006) Methylglyoxal is an intermediate in the biosynthesis of 6-Deoxy-5-ketofructose-1-phosphate: A precursor for aromatic amino acid biosynthesis in Methanocaldococcus jannaschii. Biochemistry 45, 12366–12379. [DOI] [PubMed] [Google Scholar]
- (48).Adachi O, Hours RA, Shinagawa E, Akakabe Y, Yakushi T, and Matsushita K. (2011) Enzymatic synthesis of 4-pentulosonate (4-keto-D-pentonate) from D-aldopentose and D-pentonate by two different pathways using membrane enzymes of acetic acid bacteria. Biosci., Biotechnol., Biochem 75, 2418–2420. [DOI] [PubMed] [Google Scholar]
- (49).Wierenga RK (2001) The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett 492, 193–198. [DOI] [PubMed] [Google Scholar]
- (50).Plater AR, Zgiby SM, Thomson GJ, Qamar S, Wharton CW, and Berry A. (1999) Conserved residues in the mechanism of the E. coli class II FBP-aldolase. J. Mol. Biol 285, 843–855. [DOI] [PubMed] [Google Scholar]
- (51).Santos-Martins D, Forli S, Ramos MJ, and Olson AJ (2014) AutoDock4Zn: An improved autodock force field for small-molecule docking to zinc metalloproteins. J. Chem. inf. Model 54, 2371–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pegan SD, Rukseree K, Franzblau SG, and Mesecar AD (2009) Structural basis for catalysis of a tetrameric class IIa fructose 1,6-bisphosphate aldolase from Mycobacterium tuberculosis. J. Mol. Biol 386, 1038–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Franke FP, Kapuscinski M, Macleod JK, and Williams JF (1984) A 13C-N.M.R study of intermediates in the l-type pentose phosphate cycle. Carbohydr. Res 125, 177–184. [DOI] [PubMed] [Google Scholar]
- (54).Görlach J, Schmid J, and Amrhein N. (1994) Abundance of transcripts specific for genes encoding enzymes of the prechorismate pathway in different organs of tomato (Lycopersicon esculentum L.) plants. Planta 193, 216–223. [DOI] [PubMed] [Google Scholar]
- (55).Radaev S, Dastidar P, Patel M, Woodard RW, and Gatti DL (2000) Structure and mechanism of 3-deoxy-D-mannooctulosonate 8-phosphate synthase. J. Biol. Chem 275, 9476–9484. [DOI] [PubMed] [Google Scholar]
- (56).Hashimoto M, Ozono M, Furuyashiki M, Baba R, Hashiguchi S, Suda Y, Fukase K, and Fujimoto Y. (2016) Characterization of a novel D-glycero-D-talo-oct-2-ulosonic acid-substituted lipid A moiety in the lipopolysaccharide produced by the acetic acid bacterium Acetobacter pasteurianus NBRC 3283. J. Biol. Chem 291, 21184–21194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Varki A. (2008) Sialic acids in human health and disease. Trends Mol. Med 14, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hopkins AP, Hawkhead JA, and Thomas GH (2013) Transport and catabolism of the sialic acids N-glycolylneuraminic acid and 3-keto-3-deoxy-D-glycero-D-galactonononic acid by Escherichia coli K-12. FEMS Microbiol. Lett 347, 14–22. [DOI] [PubMed] [Google Scholar]
- (59).Ögren S, and Nyberg K. (1984) A mutant of Escherichia coli capable of utilizing 3-deoxy-D-manno-2-octulosonic acid as sole carbon source. FEMS Microbiol. Lett 23, 77–81. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.