

Abstract
Connexin (Cx) mimetic peptides derived from extracellular loop II sequences (e.g., Gap27: SRPTEKTIFII; Peptide5: VDCFLSRPTEKT) have been used as reversible, Cx-specific blockers of hemichannel (HCh) and gap junction channel (GJCh) function. These blockers typically require high concentrations (~5 µM, <1 h for HCh; ~100 µM, >1 h for GJCh) to achieve inhibition. We have shown that addition of a hexadecyl (Hdc) lipid tail to the conserved SRPTEKT peptide sequence (SRPTEKT-Hdc) results in a novel, highly efficacious, and potent inhibitor of mechanically induced Ca2+-wave propagation (IC50 64.8 pM) and HCh-mediated dye uptake (IC50 45.0 pM) in Madin-Darby canine kidney cells expressing rat Cx43 (MDCK43). The lack of similar effect on dye coupling (NBD-MTMA) suggested channel conformation-specific inhibition. Here we report that SRPTEKT-Hdc inhibition of Ca2+-wave propagation, dye coupling, and dye uptake depended on the functional configuration of Cx43 as determined by phosphorylation at serine 368 (S368). Ca2+-wave propagation was enhanced in MDCK cells expressing single-site mutants of Cx43 that mimicked (MDCK43-S368D) or favored (MDCK43-S365A) phosphorylation at S368. Furthermore, SRPTEKT-Hdc potently inhibited GJCh-mediated Ca2+-wave propagation (IC50 230.4 pM), dye coupling, and HCh-mediated dye uptake in MDCK43-S368D and -S365A cells. In contrast, Ca2+-wave propagation, dye coupling, and dye uptake were largely unaffected (IC50 12.3 μM) by SRPTEKT-Hdc in MDCK43-S368A and -S365D cells, mutations that mimic or favor dephosphorylation at S368. Together, these data indicate that SRPTEKT-Hdc is a potent inhibitor of physiological Ca2+-wave signaling mediated specifically by the pS368 phosphorylated form of Cx43.
Keywords: Ca2+-wave propagation, conformation-specific gap junction channel inhibitor, connexin 43, hemichannel inhibitor
INTRODUCTION
Connexins (Cxs), the proteins comprising hemichannels (HChs) and gap junction channels (GJChs), facilitate coordinated cellular and tissue functions through channel-dependent mechanisms involving transmembrane and intercellular signaling, as well as channel-independent mechanisms involving protein-protein interactions (13). The 21 members of the connexin gene family share a similar topology consisting of four transmembrane domains, intracellular amino terminus (NT) and carboxyl terminus (CT), two extracellular loops (ECLs), and a cytoplasmic loop (CL). HChs are formed from a hexamer of Cx subunits, and when two HChs from neighboring cells dock via their ECLs, they form a GJCh, creating a cytosolic link between the cells. HChs mediate transmembrane diffusion of electrical and chemical signals, while GJChs support intercellular communication of such signals. Depending on the Cx isotype, phosphorylation of the CT can regulate these channel functions as well as influence intracellular signaling cascades by modulating protein-protein interactions (68). Here we focus on Cx43, which is expressed by a diverse array of cell types and forms channels that are size-selective, but not charge-selective. Thus, Cx43 channels are permeated by ions (e.g., K+ and Cl−) and, in a size-dependent manner, second messenger molecules [e.g., inositol trisphosphate (IP3) and cAMP], nucleotides (e.g., ATP), and other small molecules (1, 6, 16).
The controlled intercellular exchange of these small molecules supports tissue homeostasis (e.g., metabolic coupling) and coordinated communication of stimulated responses (including electrical and second messenger-mediated responses). In settings of cell and tissue injury (e.g., wounding or ischemia), cell death signals can spread through GJChs (and via HChs). This so-called bystander effect (i.e., an injured cell influencing a noninjured cell) can lead to apoptosis and necrotic tissue damage (16, 44, 46). While the regulatory mechanisms involved in governing the types of signals (e.g., death or survival signals) communicated through Cx channels remain unclear, it is becoming increasingly apparent that phosphorylation can regulate channel conformation, thereby influencing channel gating and permeability for various molecules (26). Of the 16 known phosphorylation sites in Cx43, the serine 368 (S368) site is of particular interest, because PKC-mediated phosphorylation at this site has been observed to increase during ischemia and other wound settings, which, as suggested by ischemic preconditioning studies, may be protective against the spread of injury. Importantly, when phosphorylated at S368, the GJChs favor a ~50-pS subconductance state that is well permeated by larger dye molecules (26, 47). These changes in channel behavior and intercellular communication may serve to modulate the wound healing response.
Several studies have shown that inhibiting GJCh and/or HCh function can be protective and promote cell and tissue recovery, particularly in wound repair settings (27, 29, 53, 54, 59, 74, 76). However, many commonly used inhibitors (e.g., carbenoxolone, halothane, octanol, acidic pH) have nonspecific, off-target effects that impact other cellular processes and pathways (18, 28, 43). Additionally, connexin mimetic peptides, which mimic sequences in the Cx protein and inhibit Cx channels without off-target consequences, require high concentrations (>150 μM) and lack the ability to inhibit or promote the activity of specific connexin isotypes or the specific functional states or phosphorylated forms of a given connexin type. Furthermore, it has been presumed that connexin mimetic peptides (e.g., Gap26, Gap27, Peptide5) inhibit all Cx43 channels with equal efficacy, despite the fact that channels can exist in multiple conformation states. Evidence suggests that, within a population of membrane channels, there can exist multiple phosphorylation- and voltage-dependent conductance states, and each of these conductance states differs not only in their permeation by current-carrying ions but, also, in their permeation by larger substances (25, 26). As these channel configurations may play different roles in health and disease, one could imagine that the development of peptides or agents with the capacity to potently target specific functional states, such as that conferred by S368 phosphorylation, could be very important therapeutically.
Previously, we generated a novel Cx inhibitor, SRPTEKT-Hdc, through the addition of a hexadecyl (Hdc) lipid moiety to the conserved SRPTEKT sequence of ECL2-directed peptides and found that lipidation increased the peptide’s potency and inhibitory efficacy in Cx43 GJCh-mediated Ca2+-wave propagation and HCh-mediated dye uptake by approximately five orders of magnitude (9). However, despite the extensive (80%) reduction in Ca2+-wave propagation, SRPTEKT-Hdc had no significant effect on dye coupling, raising the question of whether the peptide was targeting a subset of Cx channels. Since the phosphorylation state of S368 alters the selective profile of Cx43 gap junctions, we hypothesized that SRPTEKT-Hdc might selectively interfere with this phosphorylated isoform of Cx43. To explore this possibility, we used Madin-Darby canine kidney (MDCK) cells expressing wild-type (WT) Cx43 (MDCK43) or single-site mutants of Cx43 in which S365 or S368 was replaced by either alanine, which is structurally similar to serine but cannot be phosphorylated, or aspartate, which has a charge and structure similar to phosphorylated serine (MDCK43-S365A, -S365D, -S368A, and -S368D, respectively). We evaluated the inhibitory efficacy of SRPTEKT-Hdc to selectively block mechanically induced Ca2+-wave propagation, GJCh-mediated dye coupling, and HCh-mediated dye uptake in MDCK43 and in MDCK43-S368D and -S365A cells (which mimic or favor phosphorylation at S368) and MDCK43-S368A and -S365D cells (which mimic or favor dephosphorylation at S368) (69). We found that SRPTEKT-Hdc preferentially inhibited Cx43 GJChs (Ca2+-wave propagation and dye coupling) and HChs (dye uptake) in the conformation conferred by phosphorylation at S368. In fact, the potency of inhibition by SRPTEKT-Hdc was approximately five orders of magnitude greater in the S368 phosphorylation-mimicking mutants than in those mimicking dephosphorylation. These results suggest that SRPTEKT-Hdc is a potent inhibitor of HCh function and physiological Ca2+-wave propagation mediated specifically by the pS368 phosphorylated form of Cx43, and inhibition is likely due to the interaction of SRPTEKT-Hdc with channels in the structural conformation governed by phosphorylation at S368.
MATERIALS AND METHODS
Construction of connexin mimetic peptides.
The lipidated analog of Gap27 (SRPTEKT-Hdc) was prepared in our laboratory by semimanual solid-phase peptide synthesis performed in fritted syringes using a Domino manual synthesizer (Torviq, Niles, MI). Lipidated compounds were prepared on 4-(4-formyl-3-methoxyphenoxy)butyrylaminomethyl resin (aldehyde resin; 0.9 mmol/g) using the fluorenylmethoxycarbonyl-t-butyl (Fmoc/tBu) synthetic strategy, as previously published (9). Briefly, the hexadecyl lipid chain was attached to the aldehyde resin by reductive alkylation. Secondary amine-bound resin formed in reductive alkylation was coupled with Fmoc-protected CT amino acid using N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide in the presence of diisopropylethylamine coupling reagents, and elongation of the peptide chain continued with the standard Fmoc/tBu strategy. All compounds were fully deprotected and cleaved off the resin by treatment with 91% trifluoroacetic acid (3% water, 3% triisopropylsilane, and 3% thioanisole). After ether-hexane extraction of scavengers and protecting group residues, compounds were purified on a Waters 600 HPLC using a reverse-phase column (Vydac C18, 15–20 mm, 22 × 250 mm). Peptides were eluted with a linear gradient of CH3CN-0.1% CF3CO2H at a flow rate of 5.0 mL/min. Separation was monitored at 230 and 280 nm. The purity of products was checked by analytical reverse-phase HPLC using a Waters Alliance 2695 Separations Module with a Waters 2487 dual-wavelength (220 and 280 nm) detector on a reverse-phase column (Waters Symmetry C18, 3.5 mm). Structures were characterized by electrospray ionization using a ThermoQuest Finnigan liquid chromatography quadrupole ion trap or by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) using a Bruker Reflex III MALDI-TOF mass spectrometer with cyanocinnamic acid as a matrix. For internal calibration, an appropriate mixture of standard peptides with an average resolution of 8,000–9,000 was used. High-resolution mass measurements were carried out on a Bruker Ultraflex MALDI-TOF-TOF mass spectrometer and an Apex Qh Fourier transform ion cyclotron resonance (9.4 T) high-resolution instrument. Lyophilized peptides were reconstituted in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) at 10 or 3 mM stock concentration and diluted in the appropriate buffer solution to final concentrations of 1 pM–30 μM.
Cell culture.
MDCK parental cells were cultured in 100-mm tissue culture dishes in Dulbecco’s modified Eagle’s medium (D-1152, Sigma) supplemented with 10% FBS (Gemini Bio-Products, Sacramento, CA) and maintained at 37°C in a humidified 5% CO2 incubator. MDCK cells expressing wild-type Cx43 (MDCK43 WT or, simply, MDCK43) or serine-substitution mutants of Cx43 (MDCK43-S365A, -S365D, -S368A, and -S368D) (69) were cultured under the same conditions but with selection maintained in the presence of 200 μg/mL hygromycin B (Invitrogen, Carlsbad, CA). Adherent cells were passed using trypsin-EDTA (Gibco BRL, Grand Island, NY) and replated at 3.0 × 104 cells/cm2. Prior to immunocytochemistry or mechanical stimulation experiments, wild-type or mutant MDCK43 cells were plated and cultured on 15-mm glass coverslips until groups of 2–15 cells or confluent monolayers were formed, respectively. Cells treated with 12-O-tetradeconylphorbol-13-acetate (TPA; Sigma), a PKC agonist (5), alone were incubated with 50 ng/mL TPA for 30 min at 37°C before experimentation, while cells treated with TPA in conjunction with PD98059 (InvivoGen, San Diego, CA), a MEK1/2 inhibitor (20), or MG132 (MilliporeSigma, Burlington, MA), a proteosome inhibitor (72), were first treated with PD98059 for 30 min or MG132 for 1 h before addition of TPA. Cells treated with bisindolylmaleimide I (BIM; Sigma), a PKC inhibitor (71), were treated with 0.2 μM BIM for 1 h at 37°C before experimentation.
Immunocytochemistry.
Glass coverslips with adherent wild-type or mutant Cx43-expressing MDCK cells were fixed in a 4% paraformaldehyde (Ted Pella, Redding, CA) solution containing PBS, 5 mM MgCl2, and 4% sucrose for 10 min at room temperature, washed in PBS, permeabilized with PBS containing 0.2% Triton X-100 for 5 min, and then blocked in a PBS solution containing 4% fish skin gelatin, 1% normal donkey serum, and 0.2% Triton X-100 for 1 h at 4°C. Coverslips were incubated for ≥30 min to 1 h at room temperature or overnight at 4°C with the appropriate primary antibodies diluted in blocking reagent. Antibodies included mouse anti-Cx43 monoclonal antibody (mAb, 1:250 dilution; MAB3067, MilliporeSigma) and rabbit anti-Cx43-pS368 polyclonal antibody (pAb, 1:250 dilution; ab30559, Abcam, Cambridge, MA). Coverslips were washed three times in PBS for 5 min and incubated with the appropriate fluorophore-labeled secondary antibodies for ≥1 h or overnight. Donkey anti-mouse-Alexa 647 (1:200 dilution; 715-605-150) and donkey anti-rabbit-Alexa 488 (1:200 dilution; 711-545-152) were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Secondary labeling was washed three times in PBS, and nuclei were labeled with Hoechst dye (diluted 1:10,000 in PBS; Invitrogen Molecular Probes). After final washes, coverslips were mounted on slides in VectaShield mounting medium (Vector Laboratories, Burlingame, CA). Cells were imaged using an Olympus BX51 fluorescence microscope fitted with a CoolSnap ES2 camera (Photometrics, Tucson, AZ). Images are presented as merged channels.
Immunoblotting.
Whole cell protein was isolated as described elsewhere (7). Protein concentration was determined using the Pierce bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA). Samples containing 25 μg (chemiluminescence detection) or 10 μg (fluorescence detection) of protein or 50 ng of GST-Cx43 fusion protein [positive control for Cx43 (62)] were prepared in sample buffer with β-mercaptoethanol and boiled at 100°C for 5 min. Proteins were separated via SDS-PAGE using a 12% precast gel (Bio-Rad, Hercules, CA) and transferred to a nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-Rad). Blots were blocked with 5% nonfat milk in 1× Tris-buffered saline containing 0.1% Tween 20 (TBST; Sigma) for 1 h (chemiluminescence) or AquaBlock (EastCoast Bio, North Berwick, ME) overnight (fluorescence) at 4°C and then incubated for ≥2 h or overnight with the appropriate primary antibodies diluted in blocking reagent. Antibodies included mouse anti-GAPDH mAb (1:1,000 dilution; ZG003, Invitrogen) as a protein loading control, rabbit anti-Cx43 pAb (1:4,000 dilution; C6219, Sigma), and rabbit anti-Cx43-pS368 pAb (1:250 dilution; ab30559, Abcam). Blots were washed twice in TBST for 5 min and twice in PBS for 5 min and then incubated in the appropriate horseradish peroxidase (HRP)-conjugated or infrared (IR) dye-conjugated secondary antibodies for 1.5–2 h. Goat anti-mouse-HRP (1:3,750 dilution; W4021) was obtained from Promega (Madison, WI) and donkey anti-rabbit-HRP (1:5,000 dilution; NA9340) from GE Healthcare UK (Little Chalfont, Buckinghamshire, UK). Goat anti-mouse-IRDye 800 (1:20,000 dilution; 926-32210) and goat anti-rabbit-IRDye 680 (1:20,000 dilution; 926-68071) were obtained from LI-COR (Lincoln, NE). HRP conjugate enzymes were stimulated with SuperSignal West Dura chemiluminescence substrate (Thermo Fisher Scientific), and blots were imaged using the G:BOX Chemi XR5 imaging system and GeneSys image acquisition software (Syngene, Cambridge, UK). Western blot bands labeled with dye-conjugated antibodies were detected and recorded using an Odyssey infrared scanner (LI-COR). The use of secondary antibodies detectable at different infrared wavelengths (680 and 800 nm) permitted simultaneous detection of either total Cx43 or the pS368 phosphorylated form of Cx43 with the GAPDH loading control. The intensities of protein bands were quantified by densitometry (ImageJ software, National Institutes of Health, Bethesda, MD) and included blots obtained using chemiluminescent and fluorescent approaches. Total Cx43 levels were determined as the ratio of Cx43 to GAPDH, and pS368-Cx43 levels were determined as the ratio of pS368 to total Cx43.
Mechanical stimulation and intracellular Ca2+ concentration measurements.
Glass coverslips with adherent cells were washed in Hanks’ balanced saline solution (HBSS: 1.3 mM CaCl2, 5.0 mM KCl, 0.3 mM KH2PO4, 0.5 mM MgCl2, 0.4 mM MgSO4, 137.9 mM NaCl, 0.3 mM Na2HPO4, and 1% glucose additionally buffered with 25 mM HEPES, pH 7.4; Gibco BRL) and loaded with the Ca2+ indicator fura 2 by incubation in fura 2-acetoxymethyl ester (fura 2-AM, 5 μM in HBSS; Calbiochem, La Jolla, CA). For optimal loading, wild-type or mutant MDCK43 cells were incubated in fura 2-AM for 70 min at room temperature. Cells were washed with HBSS and allowed to sit for ≥20 min at room temperature before digital imaging. Coverslips were then mounted in a custom-made chamber and placed on the stage of an inverted microscope (model IX70, Olympus America, Center Valley, PA) equipped for differential interference contrast and fluorescence observation. Fura 2 fluorescence was observed with a ×40 oil-immersion objective after alternating excitation at 340 and 380 nm by a 75-W xenon lamp linked to a Delta Ram V illuminator (Photon Technologies, Monmouth Junction, NJ) via a gel optic line. Images of emitted fluorescence >510 nm were recorded by an Evolve 512 charge-coupled device (CCD) camera (Photometrics) and simultaneously displayed on a 21.5-inch LG Flatron color monitor. The imaging system was under software control (EasyRatioPro, Photon Technologies).
Intercellular Ca2+-wave propagation was induced by mechanical stimulation of a single cell with a 1.5-mm borosilicate glass micropipette (~1-μm tip diameter) positioned near the apical membrane by a hydraulically driven micromanipulator (model MX750R, Siskiyou Design Instruments, Grants Pass, OR). The pipette was briefly deflected downward to deform the cell membrane, and the elicited response was recorded for 1 min beginning 3 s before mechanical stimulation. To assess the effects of peptidomimetic incubation time, responses were recorded in different areas of the coverslip stimulated at 3- to 5-min intervals over the course of a 90-min incubation.
Intracellular Ca2+ concentration ([Ca2+]i) for each individual cell in the field of view was calculated by ratiometric analysis of fura 2 fluorescence using equations published elsewhere (37). A change in [Ca2+]i was considered positive if [Ca2+]i increased by 200 nM or more, a two- to fourfold change over resting values. The stimulated cell was not counted in data analysis. Thus a Ca2+-wave of zero cells represents a response only by the stimulated cell and no intercellular communication. If the cell membrane was broken by mechanical stimulation (recognized by rapid loss of fura 2 dye), the experiment was not included in analysis. This prevented analysis of Ca2+-wave propagation triggered by lost intracellular contents such as ATP (6). The number of cells in the field of view was variable: ~50–80 cells. On occasion, wave propagation would exit the field of view. In such cases, the Ca2+-wave propagation was given a total score of 50 cells. Because maximum numbers were imposed on wave counts, the number of cells participating in Ca2+-wave propagation in unblocked conditions may be slightly underrepresented.
Dye coupling.
Glass coverslips with adherent MDCK43 cells were washed with external solution (142.5 mM NaCl, 4 mM KCl, 1 mM MgCl2, 5 mM glucose, 2 mM sodium pyruvate, 10 mM HEPES, 15 mM CsCl, 10 mM tetraethylammonium chloride, and 1 mM CaCl2, adjusted to 315 mosM with H2O and pH 7.2), mounted in a custom-made chamber, and placed on the stage of an Olympus IX71 inverted microscope equipped with a ×40 objective for phase-contrast and fluorescence observation. Injection micropipettes were pulled from 1-mm borosilicate, filamented, glass capillary tubes on a puller (Sutter Instruments, Novato, CA). Micropipette tips were filled with gap junction-permeant (final concentration ~4.6 mM in internal solution) and impermeant (final concentration 3 mM) dyes via capillary action and backfilled with dye-free internal solution (124 mM KCl, 3 mM MgCl2, 5 mM glucose, 9 mM HEPES, 9 mM EGTA, 14 mM CsCl, 9 mM tetraethylammonium chloride, 5 mM Na2ATP, and 0.5 mM CaCl2, adjusted to 315 mosM with H2O and pH 7.2). The GJCh-permeant dye was NBD-MTMA (N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium; molecular mass = 280 Da, net charge +1, λex = 343,458 nm, λem = 530 nm) (4); the GJCh-impermeant dye was tetramethylrhodamine dextran (mol wt = 3,000, λex = 555 nm, λem = 580 nm; Invitrogen Molecular Probes). Dye was microinjected with a micromanipulator (model MGB-3.0, Siskiyou Design Instruments) into the cytoplasm of individual cells. Injections were visualized with phase-contrast optics, and intercellular dye diffusion was monitored using epifluorescence optics. Fluorescence was visualized using the Olympus BX50WI microscope outfitted with a U-MNB filter set (Olympus) for NBD-MTMA detection and a U-MWIG2 filter set (Olympus) for rhodamine dextran detection. At 5 min postinjection, a CCD camera (model DFC7000T, Leica Microsystems, Wetzlar, Germany) captured digital images onto a personal computer. The number of coupled cells was determined from these images using LAS X software (Leica Microsystems). Any cell with greater than background levels of fluorescence was considered coupled to the injected cell.
Dye uptake.
Dye uptake experiments were completed as described elsewhere (35). Briefly, wild-type or mutant MDCK43 cells were plated at a low density of 2.0 × 104 cells/well in six-well plates. All cells were rinsed with culture medium followed by external solution (1 mM CaCl2) and then incubated for 15 min in “low-Ca2+ solution” (external solution with 5 mM EGTA) with or without peptide. Rings made from the tops of 14-mL Falcon tubes (Corning Life Sciences, Tewksbury, MA) were adhered to the center of each well with petroleum jelly, creating a smaller well area. Dye solution with or without peptide was added to each tube-top well for 15 min while plates were kept on ice and protected from light. The dye solution contained 1.25 mg/mL NBD-MTMA and 0.125 mg/mL tetramethylrhodamine dextran dissolved in external solution with no added CaCl2. After 15 min of exposure to dye in the presence or absence of peptide, dye and the rings were removed and cells were rinsed with culture medium and external solution and then immediately imaged with the Olympus IX71 fluorescence microscope. Phase-contrast, NBD-MTMA (41001HQ filter), and rhodamine dextran (WIGA3 filter) images were acquired using the SenSys CCD camera and V++ software. Each field imaged was scored for number of NBD-positive/rhodamine dextran-negative cells and total number of cells (typically 9–10) within the visualized fields. Cell clusters of two or more were counted as one cell to account for likely dye diffusion through gap junctions. At least three fields for each well were imaged with at least three wells per experimental condition. The percentage of NBD-MTMA-positive cells per field is reported.
Statistical analysis.
All statistical analyses were performed with GraphPad Prism software (San Diego, CA) with α set at 0.05. Immunoblot bar graphs, obtained by densitometric analysis, are presented as means ± SE (SD reported in text) and represent the ratio of total Cx43 to GAPDH or pS368 to total Cx43, both normalized to this same ratio in wild-type MDCK43 cells. Ca2+-wave propagation data are expressed as means ± SE (SD reported in text). The concentration-response curve was generated with a nonlinear regression using the log (inhibitor) vs. response (3 parameters) model, and the IC50 value and 95% confidence interval (CI) were determined from the fit curve. Dye coupling bar graphs display number (means ±SE) of cells receiving dye within 5 min of injection (SD reported in text). Dye uptake bar graphs display percentage (means ± SE) of NBD-MTMA-positive cells (SD reported in text). Differences between control and experimental groups were analyzed using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc multiple-comparison test or a two-tailed Student’s t-test, as appropriate. P < 0.05 was used to establish a significant difference between samples; precise P values are reported.
RESULTS
Total Cx43 and pS368-Cx43 in wild-type and mutant MDCK43 cells.
To assess differences in the susceptibility of specific phosphorylated forms of Cx43 to inhibition by SRPTEKT-Hdc, we used Cx-deficient MDCK cells transfected to express wild-type Cx43 (MDCK43) or Cx43 containing site-directed mutations in which the serine residues at position 365 or 368 were converted either to alanine to mimic “dephosphorylation” or to aspartate to mimic “phosphorylation” (MDCK43-S368A, -S368D, -S365A, or -S365D). Immunoblots containing whole cell lysates from each of these cell lines were probed to assess total Cx43 expression and Cx43 phosphorylated at S368 (using the phosphorylation-specific pS368 antibody) (Fig. 1, A and B). As shown in Fig. 1A, total Cx43, as represented by a 43-kDa band(s), was present in all Cx43-expressing cell lines, although at apparently reduced levels in S365A- and S365D-expressing cells (69). Densitometric quantification of multiple such blots (n = 4; Fig. 1A) revealed no significant differences in total Cx43 between Cx43-expressing samples. A Cx43-specific band at 20 kDa was detected in all the Cx43-expressing cell lines, and 26- and 29-kDa bands were detected in the MDCK43 WT and MDCK43-S368A and -S368D cells. These bands likely arise from internal ribosomal entry sites, as previously reported (63); the significance of their differential expression between these cell lines, in particular the abundance of the 20-kDa isoform in the S368D mutant, has not been investigated. Data in Fig. 1B (n = 3) are also consistent with previously published data (69) showing that phosphorylation at S368 was detected primarily in MDCK43 WT and MDCK43-S365A cells with two- to threefold higher levels than MDCK43-S365D cells. Additionally, wild-type cells treated with the PKC agonist TPA demonstrated enhanced S368 phosphorylation, evidenced by the significant 1.4-fold increase compared with untreated wild-type cells. No band was detected in either the S368A or S368D mutant, as the pS368 antibody does not cross-react with these substitution mutants.
Fig. 1.

Expression of Cx43 and pS368 in WT and mutant MDCK43 cells. A: immunoblot and quantification (means ± SE, n = 4) of total Cx43 expression [relative to MDCK43 WT cells (abbreviated as 43WT)] in MDCK parental (mean ratio 0.02, SD 0.01), MDCK43 WT, MDCK43-S368A (0.9, SD 0.2), -S368D (1.0, SD 0.3), -S365A (0.5, SD 0.7), and -S365D (0.7, SD 0.5) cells (indicated P values from ANOVA analysis). B: immunoblot and quantification (means ± SE, n = 3) of pS368-Cx43 levels (relative to 43WT) in MDCK43 WT, MDCK43-S368A (0.4, SD 0.2), -S368D (0.3, SD 0.2), -S365A (0.7, SD 0.3), and -S365D (0.4, SD 0.3) cells (P values from ANOVA) and MDCK43 WT cells treated with TPA (1.4, SD 0.1) (P value from t-test). M, molecular mass (in kDa) markers; GST43, positive control for Cx43. ^Significant difference in expression compared with MDCK43 WT. C: immunofluorescence images showing the overlay of total Cx43 (tCx43), pS368-Cx43, and nuclear staining (Hoechst) in MDCK43 WT, MDCK43-S368A, -S368D, -S365A, and -S365D cells. White arrowheads indicate coincident staining of tCx43 and pS368-Cx43 at the gap junction plaque. Scale bar = 20 µm; all images at same magnification.
Figure 1C shows colabeled immunofluorescence images of total Cx43 and pS368 in wild-type and mutant Cx43-expressing MDCK cells. The pS368 antibody revealed distinct, prominent labeling only in wild-type and S365A-expressing cells, again consistent with previously published results (69). Together, these data indicate that MDCK cells expressing wild-type or S365A forms of Cx43 are amenable to phosphorylation at S368, while the S365D mutant is poorly or not phosphorylated at this site.
Phosphorylation at S368 enhances mechanically induced Ca2+-wave propagation.
Mechanical stimulation was used to examine GJCh-mediated Ca2+-wave propagation in wild-type and mutant MDCK43 cells. As we reported previously (9), mechanical stimulation of a single MDCK43 cell resulted in an immediate increase in [Ca2+]i in the stimulated cell that was propagated to 12.6 (SD 6.9, n = 16) neighboring cells (Fig. 2A) within the 1-min recording period. Similarly, for each of the mutants, mechanical stimulation of a single cell resulted in Ca2+-wave propagation to multiple adjacent cells (Fig. 2, B–E). In MDCK43-S368A and -S365D cells, mutations that mimic or favor dephosphorylation at S368, Ca2+-waves were propagated to 16.3 (SD 13.3, n = 96) and 17.7 (SD 10.0, n = 81) neighboring cells, respectively (Fig. 2, B and C). On the other hand, in the MDCK43-S368D and -S365A cells, mutations that mimic or favor phosphorylation at S368, Ca2+-wave propagation was significantly enhanced compared with wild-type cells, with waves propagating to 34.7 (SD 14.3, n = 34) and 26.8 (SD 8.8, n = 26) cells, respectively (Fig. 2, D and E). It should be noted that the enhanced Ca2+-wave propagation in the MDCK43-S368D cells did not involve ATP release through Cx43-HChs and purinergic signaling, as inclusion of apyrase in the extracellular fluid did not reduce the magnitude of the Ca2+-wave.
Fig. 2.

Mechanically induced Ca2+-wave propagation in WT and mutant MDCK43 cells. Left to right: color maps overlaid onto differential interference contrast images of the imaged cells show intracellular Ca2+ concentration ([Ca2+]i) as a function of time following mechanical stimulation of a single cell (arrow). A–E: extent of Ca2+-wave propagation in untreated MDCK43 WT cells (A); MDCK43-S368A cells (B); MDCK43-S365D cells (C); MDCK43-S368D cells (D); and MDCK43-S365A cells (E). Time after stimulation is indicated. Color bar displays approximate [Ca2+]i. Scale bar = 30 µm; all images at same magnification.
SRPTEKT-Hdc selectively inhibits Ca2+-wave propagation mediated by the pS368 phosphorylated form of Cx43.
We showed previously that 60 min of exposure to SRPTEKT-Hdc, a lipidated peptide that mimics a portion of the Cx43 ECL2 sequence, very potently, but reversibly, inhibits mechanically induced Ca2+-wave propagation in rabbit tracheal epithelial cells and MDCK43 cells (9). Similar results are shown in Fig. 3A, where the Ca2+-wave in 100 nM SRPTEKT-Hdc-treated MDCK43 cells was limited to only 1.8 (SD 1.7, n = 11) neighboring cells. In contrast, Ca2+-wave propagation was unaffected by incubation with SRPTEKT-Hdc in MDCK cells expressing Cx43-S368A [15.6 (SD 10.9) cells, n = 27] and Cx43-S365D [12.8 (SD 6.2) cells, n = 17] mutations that mimic or favor dephosphorylation at S368 (Fig. 3, B and C). Similar to its effect in MDCK43 cells, SRPTEKT-Hdc inhibited Ca2+-wave propagation in MDCK cells expressing Cx43-S368D [6.0 (SD 7.0) cells, n = 21] and Cx43-S365A [5.6 (SD 3.7 cells), n = 22] mutations that mimic or favor phosphorylation at S368 (Fig. 3, D and E). These data are summarized in Fig. 4.
Fig. 3.

Mechanically induced Ca2+-wave propagation is blocked by SRPTEKT-Hdc in a phosphorylation-dependent manner. Images (left to right) are as described in Fig. 2. A–E: extent of Ca2+-wave propagation following incubation in 100 nM SRPTEKT-Hdc for the indicated times in MDCK43 WT cells (71 min) (A); MDCK43-S368A cells (63 min) (B); MDCK43-S365D cells (73 min) (C); MDCK43-S368D cells (73 min) (D); and MDCK43-S365A cells (73 min) (E). SRPTEKT-Hdc significantly inhibited propagation of the Ca2+-wave in WT cells (A) and in cells that favored or mimicked phosphorylation at S368 (D and E). Scale bar = 30 µm; all images at same magnification.
Fig. 4.

Inhibition of Ca2+-wave propagation in WT and mutant MDCK43 cells. The mean (± SE) number of cells responding with an increase in intracellular Ca2+ concentration of at least 200 nM after stimulation of a single cell is shown (sample sizes are indicated in each bar) for MDCK43 WT, MDCK43-S368A, -S365D, -S368D, and -S365A cells under (untreated) control conditions or following incubation in 100 nM SRPTEKT-Hdc for 61–75 min. P values comparing controls across cell types are from ANOVA and are indicated above the bars: †significant increase in Ca2+-wave propagation compared with the Ca2+-wave propagated in untreated MDCK43 WT cells. P values comparing untreated with peptide-treated are from t-test: *significant reduction in wave propagation compared with the Ca2+-wave propagated in the absence of agent; ns indicates no significant difference in wave propagation.
To address this apparent phosphorylation-specific effect further, MDCK43 cells were treated with the PKC inhibitor BIM to prevent phosphorylation at S368. Similar to wild-type and mutant-expressing cells in the absence of peptide, mechanical stimulation of BIM-treated MDCK43 cells resulted in Ca2+-waves that propagated to 11.1 (SD 5.2, n = 21) adjacent cells (Fig. 5A). However, unlike untreated cells, incubation in 100 nM SRPTEKT-Hdc failed to inhibit Ca2+-wave propagation in BIM-treated MDCK43 cells [9.1 (SD 4.3) cells, n = 13; Fig. 5A], a result similar to that in MDCK43-S368A and -S365D cells. We also examined Ca2+-wave propagation in MDCK43 cells treated with TPA to increase phosphorylation at S368 (Fig. 1). TPA treatment significantly reduced Ca2+-wave propagation in MDCK43 cells, independent of SRPTEKT-Hdc exposure [TPA: 1.9 (SD 1.8) cells, n = 23; TPA + SRPTEKT-Hdc: 1.2 (SD 1.6) cells, n = 6; Fig. 5A], and this effect was time-dependent, with complete abolition of Ca2+-waves after 10 min (Fig. 5A, inset). TPA treatment has been reported to cause internalization of Cx43 GJChs (21, 22), which could explain this loss of coupling independent of SRPTEKT-Hdc. Indeed, immunofluorescence labeling of Cx43 in untreated MDCK43 WT cells or MDCK43 WT cells that were treated with TPA (30 min) or TPA (30 min) followed by an additional 1.5 h in TPA-free media (to examine longer-term effects) revealed a near-complete loss or redistribution of Cx43 from plasma membrane gap junction plaques into intracellular compartments following TPA treatment compared with untreated cells (Fig. 5B), consistent with previous findings (21, 22). To determine whether this loss of communication was specific to PKC-induced phosphorylation at S368, Ca2+-wave propagation was evaluated in TPA-treated MDCK43-S368A and -S368D cells, which are not amenable to PKC phosphorylation at S368. TPA treatment completely abolished Ca2+-wave propagation in both mutants independent of addition of SRPTEKT-Hdc [in MDCK43-S368A, TPA: 0.3 (SD 0.4) cells, n = 16; TPA + SRPTEKT-Hdc: 0.1 (SD 0.3) cells, n = 12; in MDCK43-S368D cells, TPA: 0.8 (SD 1.4), n = 20; TPA + SRPTEKT-Hdc: 0.4 (SD 0.5) cells, n = 11; Fig. 5A]. These results suggest that the TPA-induced reduction in Cx43 GJCh-mediated Ca2+-wave propagation likely reflects phosphorylation at sites other than S368, possibly by MAPK at S262, S279, or S282 (65, 66), AKT at S373 (21), or Src at tyrosine 247 (65), all of which are involved in regulating Cx43 gap junction size, channel gating, and/or turnover (reviewed in Refs. 67 and 68).
Fig. 5.

Ca2+-wave propagation in BIM- and TPA-treated WT and mutant MDCK43 cells. A: the mean (± SE) number of cells responding with an increase in intracellular Ca2+ concentration of at least 200 nM after stimulation of a single cell is shown (sample sizes are indicated in each bar) for BIM-treated MDCK43 WT cells, TPA-treated MDCK43 WT cells, TPA-treated MDCK43-S368A cells, and TPA-treated MDCK43-S368D cells under (untreated) control conditions or following incubation in 100 nM SRPTEKT-Hdc for 61–75 min. P values (from t-test) are indicated above the bars; ns indicates no significant difference in wave propagation. B: immunofluorescence images showing the overlay of total Cx43 (tCx43) and nuclear staining (Hoechst) in MDCK43 WT cells that were either untreated (control), TPA-treated for 30 min, or TPA-treated for 30 min followed by an additional 1.5 h in TPA-depleted medium. Scale bar = 30 µm; all images at same magnification. C: PD98059 + TPA-treated MDCK43 WT cells and MG132 + TPA-treated MDCK43 WT cells under (untreated) control conditions or following incubation in 100 nM SRPTEKT-Hdc for 61–75 min. P values (from t-test) are indicated above the bars: *significant reduction in wave propagation compared with the Ca2+-wave propagated in the absence of agent.
To better assess the effects of SRPTEKT-Hdc on S368 phosphorylation in MDCK43 cells, an effort was made to alleviate the consequences of TPA on junctional communication and turnover by treating cells with TPA in conjunction with either the MAPK kinase (MEK1/2) inhibitor PD98059 to prevent phosphorylation of Cx43 at S262/279/282 [linked to decreased channel open probability (10) and increased internalization (48, 50)] or with the proteasome inhibitor MG132 to induce AKT-dependent phosphorylation of Cx43 at S373 [associated with accretion and stability of Cx43 in gap junction plaques (21, 22)]. Compared with TPA alone (Fig. 5A), TPA in the presence of PD98059 resulted in slightly greater, although not significantly different (P = 0.0882), Ca2+-wave propagation [3.2 (SD 3.3) cells, n = 29] that was significantly inhibited by SRPTEKT-Hdc [0.8 (SD 1.2) cells, n = 13; Fig. 5C]. Moreover, MDCK43 cells treated with TPA in the presence of MG132 exhibited Ca2+-waves that propagated to 24.2 (SD 12.3) neighboring cells (n = 33; Fig. 5C), a magnitude significantly greater (P = 0.0014) than in cells treated with MG132 alone [14.4 (SD 7.2) cells, n = 22] and similar to that in MDCK43-S368D and -S365A cells (Fig. 2, D and E). Similar to its effect on these mutants, SRPTEKT-Hdc significantly inhibited Ca2+-wave propagation in MDCK43 WT cells treated with MG132 + TPA [2.5 (SD 2.3) cells, n = 28; Fig. 5C]. Overall, the results presented in Figs. 3–5 indicate that SRPTEKT-Hdc preferentially and specifically inhibits Ca2+-wave propagation mediated by Cx43 GJChs in the conformation governed by phosphorylation at S368.
Concentration dependence of SRPTEKT-Hdc inhibition of Ca2+-wave propagation is influenced by phosphorylation at S368.
To quantify and compare the potency of SRPTEKT-Hdc in blocking Ca2+-wave propagation in MDCK43-S368A and -S368D cells, we determined the number of cells in the wave as a function of concentration (pM to µM) following 61–75 min of incubation. As shown in Fig. 6, Ca2+-wave propagation was inhibited by SRPTEKT-Hdc in a concentration-dependent manner for each of the mutants, although a full evaluation of inhibitory concentrations could not be performed for MDCK43-S368A cells due to cytotoxic effects of the peptide at concentrations >10 μM.
Fig. 6.
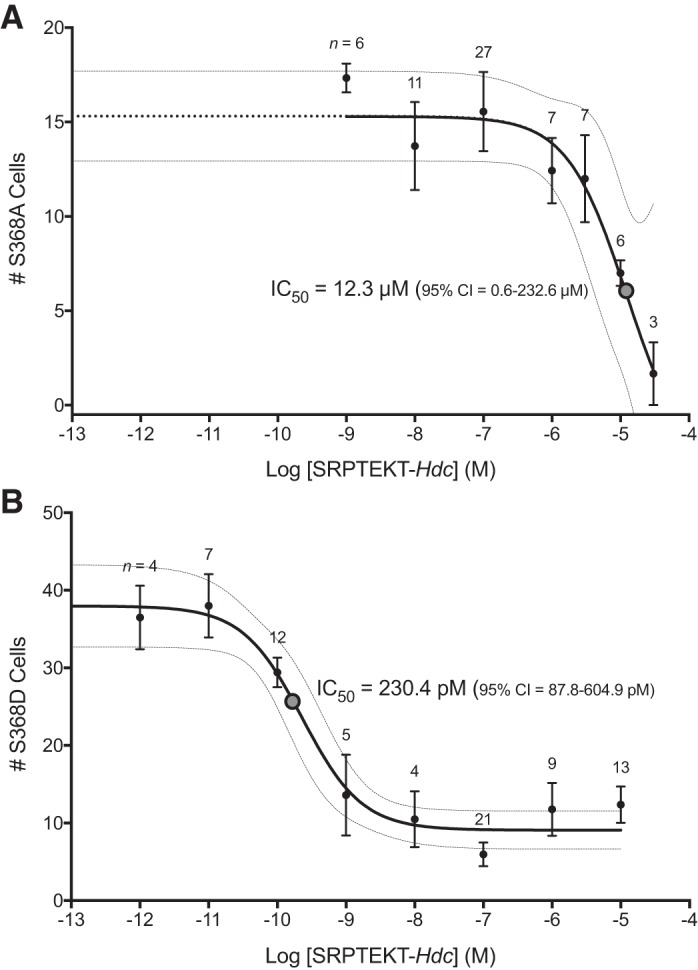
Concentration-response curves for SRPTEKT-Hdc inhibition of Ca2+-wave propagation in MDCK43-S368A and MDCK43-S368D cells. A and B: number of MDCK43-S368A (A) and MDCK43-S368D (B) cells responding with an increase in intracellular Ca2+ concentration of at least 200 nM after 61–75 min of incubation with increasing concentrations of SRPTEKT-Hdc. Values are means ± SE. Sample sizes are indicated. The IC50 value (dotted circle) and the 95% confidence interval (CI; banded lines) were calculated from the nonlinear curve fit for each cell type and are indicated. The S368A curve was extended (dotted line) to align with S368D.
The IC50 for SRPTEKT-Hdc inhibition of wave propagation in MDCK43-S368A cells (Fig. 6A) was 12.3 μM (95% CI 0.6–232.6 μM), nearly five orders of magnitude higher than the IC50 of 230.4 pM (95% CI 87.8–604.9 pM) observed for MDCK43-S368D cells (Fig. 6B). The 95% CI for the S368D phosphomimetic mutant overlaps that previously published for MDCK43 cells (9), in which SRPTEKT-Hdc was found to be five orders of magnitude more potent than Gap27 in inhibiting Ca2+-wave propagation. In contrast, the concentration-response curve for MDCK43-S368A cells is considerably right-shifted, indicating a significant and measurable loss of potency in inhibiting the dephosphorylated form of Cx43. By comparing these two mutants, it is clear that SRPTEKT-Hdc selectively and potently inhibits Ca2+-wave propagation through GJChs containing Cx43 phosphorylated at S368.
Phosphorylation at S368 supports enhanced dye coupling compared with dephosphorylation at S368.
Dye coupling in MDCK43 cells was unaffected by SRPTEKT-Hdc (9). Since Cx43 isoforms mimicking or favoring pS368 preferentially supported GJCh-mediated Ca2+-wave propagation and were selectively inhibited by SRPTEKT-Hdc, we determined whether dye coupling mediated by these isoforms would also be blocked by SRPTEKT-Hdc. Injection of a single MDCK43 cell with the positively charged NBD-MTMA dye and rhodamine dextran, which marks the injected cell (GJCh-impermeant), resulted in diffusion of NBD-MTMA dye to 62.4 (SD 38.8) neighboring cells within 5 min (n = 31; Fig. 7, A and G), while only the injected cell contained rhodamine dextran (data not shown). Dye coupling in dephosphorylation-mimicking MDCK43-S368A cells was significantly limited compared with WT, as dye spread to only 16.5 (SD 27.7) cells (n = 21; Fig. 7, B and G). In contrast, phosphorylation-mimicking MDCK43-S368D cells supported dye coupling [49.6 (SD 23.0) cells, n = 33] that was not significantly different from WT but was considerably increased compared with MDCK43-S368A cells (Fig. 7, C and G).
Fig. 7.

Dye coupling is blocked by SRPTEKT-Hdc in a S368 phosphorylation-dependent manner. A–F: representative images of NBD-MTMA dye coupling in MDCK43 WT cells (A and D); MDCK43-S368A cells (B and E); and MDCK43-S368D cells (C and F). Cells in A, B, and C were untreated, and those in D, E, and F were treated for 61–105 min with 100 nM SRPTEKT-Hdc before dye injection. Images were taken following 5 min of continuous injection; pipettes are evident in each image. Asterisks in fluorescent images denote microinjected cell. Note that coinjected rhodamine dextran did not diffuse to neighboring cells (not shown). Scale bar = 30 µm. G: number of cells receiving dye for each treatment condition. White bar, absence of SRPTEKT-Hdc; black bars, presence of SRPTEKT-Hdc. Values are means ± SE (sample sizes are indicated in each bar). P values comparing controls across cell types are from ANOVA and are indicated above the bars: †significant decrease in dye coupling compared with dye coupling in untreated MDCK43 WT cells; ‡significant increase in dye coupling in MDCK43-S368D cells compared with dye coupling in MDCK43-S368A cells. P values comparing untreated to peptide-treated are from t-test: *significant reduction in dye coupling compared with dye coupling in the absence of agent. ns indicates no significant difference in wave propagation.
SRPTEKT-Hdc selectively inhibits dye coupling mediated by the pS368 phosphorylated form of Cx43.
We showed that SRPTEKT-Hdc failed to inhibit GJCh-mediated NBD-MTMA dye coupling in MDCK43 cells (9), and similar results are shown in Fig. 7, D and G, where dye coupling in MDCK43 cells treated with 100 nM SRPTEKT-Hdc for 61–105 min [63.2 (SD 38.5), n = 20] was not different from untreated cells. Dye coupling in MDCK43-S368A cells [16.4 (SD 21.1) cells, n = 32] was also unaffected by SRPTEKT-Hdc treatment (Fig. 7, E and G). In contrast, SRPTEKT-Hdc significantly inhibited dye coupling in MDCK43-S368D cells [14.4 (SD 10.8) cells, n = 23; Fig. 7, F and G]. Together, these results provide further evidence that SRPTEKT-Hdc preferentially and specifically inhibits Cx43 GJChs in the conformation conferred by phosphorylation at S368.
SRPTEKT-Hdc selectively inhibits dye uptake mediated by HChs composed of the pS368 phosphorylated form of Cx43.
To determine whether SRPTEKT-Hdc is also specific for HChs with phosphorylation at S368, the MDCK43-S368A and -S368D cells were used in dye uptake assays. Our published data showed inhibition of HCh-mediated dye uptake by SRPTEKT-Hdc in MDCK43 cells (9) and are presented again here for comparison with the mutants (Fig. 8B). In the presence of 1 mM extracellular Ca2+, when Cx43 HChs are presumed closed, the percentage of cells taking up NBD-MTMA dye was 7.2% (SD 8.0, n = 17) for MDCK43 cells, 9.0% (SD 6.4, n = 15) for the dephosphorylation-mimicking MDCK43-S368A cells, and 10.2% (SD 6.9, n = 15) for the phosphorylation-mimicking MDCK43-S368D cells (Fig. 8B). In contrast, following exposure to low extracellular [Ca2+] for 15 min followed by nominally Ca2+-free dye solution for 15 min, dye uptake increased to 32.6% (SD 22.9, n = 78) of MDCK43 cells, 28.4% (SD 14.2, n = 16) of MDCK43-S368A cells, and 32.4% (SD 13.1, n = 31) of MDCK43-S368D cells (Fig. 8). Pretreatment of MDCK43-S368A cells with 100 nM SRPTEKT-Hdc had no significant effect on HCh-mediated dye uptake [22.6% (SD 18.7), n = 16; Fig. 8]. However, SRPTEKT-Hdc significantly inhibited dye uptake by MDCK43-S368D cells [12.6% (SD 7.2), n = 36], similar to that observed in MDCK43 cells [7.5% (SD 8.0), n = 60], albeit to a lesser, but not significantly different, extent (Fig. 8). Overall, the data presented in Fig. 8 show that the specificity of SRPTEKT-Hdc for inhibiting Cx43 GJChs phosphorylated at S368 is recapitulated for HChs.
Fig. 8.

Dye uptake is blocked by SRPTEKT-Hdc in a phosphorylation-dependent manner. NBD-MTMA dye uptake under Ca2+-free conditions is shown. A: representative phase-contrast and NBD-MTMA fluorescence images of MDCK43-S368A cells (a) or MDCK43-S368D cells (b) under untreated, control conditions or following 15-min pretreatment in 100 nM SRPTEKT-Hdc followed by 15-min dye uptake in the presence of 100 nM SRPTEKT-Hdc. Rhodamine images are not shown as no cells contained this dye. Scale bar = 100 µm. B: percentage of NBD-MTMA-positive MDCK43 WT [as previously published (9)], MDCK43-S368A and MDCK43-S368D cells under baseline (normal Ca2+) conditions, control (untreated, Ca2+-free) conditions, and following exposure to 100 nM SRPTEKT-Hdc. The mean (± SE) percentage of NBD-MTMA-positive cells is shown (sample sizes are indicated in each bar). P values (from t-test) are indicated above the bars: ^significant increase in dye uptake compared with baseline; *significant reduction in percent dye uptake compared with the absence of agent.
DISCUSSION
In this study we show that the lipidated peptide SRPTEKT-Hdc potently inhibits HCh-mediated dye uptake as well as GJCh-mediated Ca2+-wave propagation and dye coupling and has specificity for signaling associated with channels containing the pS368 phosphorylated form of Cx43 (Fig. 9). Interestingly, Ca2+-wave propagation was enhanced in cells expressing the form of Cx43 favoring or mimicking phosphorylation at S368, suggesting that phosphorylation at this site is involved in mediating the communication of Ca2+-waves. Furthermore, the potency for inhibition of GJCh-mediated Ca2+-wave propagation by SRPTEKT-Hdc was five orders of magnitude greater for Cx43 channels in this phosphorylation state than for channels favoring or mimicking dephosphorylation at S368. Dye coupling was also greater and selectively inhibited in cells mimicking the pS368 form of Cx43 compared with the dephosphorylated form, providing further evidence for the role of phosphorylation in dictating signal-specific communication through Cx43 channels and for the preference of SRPTEKT-Hdc to specifically inhibit pS368-Cx43 GJChs. In addition, this selective phosphorylation-dependent susceptibility for inhibition also held true for the inhibition of HCh-mediated dye uptake. These data suggest that SRPTEKT-Hdc interacts with HChs and GJChs in a phosphorylation-dependent, channel conformation-specific manner to block HCh function and physiologically significant intercellular Ca2+ signaling. The potency and inhibitory specificity of SRPTEKT-Hdc suggest that not only is lipidation a useful strategy for increasing the efficacy of mimetic peptides but, also, that it is possible to selectively target channels in specific conformations. Moreover, the data suggest that peptidomimetics could be designed or evaluated for their capacity to inhibit or promote communication of certain types of signals, offering a new avenue of therapeutic possibility that has not been explored.
Fig. 9.

Specificity of SRPTEKT-Hdc for pS368-Cx43 channels. Schematic representation showing the predicted mechanism underlying SRPTEKT-Hdc selectivity for pS368-Cx43 hemichannels (HChs) and gap junction channels (GJChs). Phosphorylation at S368 induces a conformational change in both GJChs and HChs that not only influences channel permselectivity (e.g., enhancing Ca2+-wave propagation but reducing conductivity) but, also, enhances peptide interaction with the channel. The result of the induced conformational change is specific and potent inhibition of pS368-Cx43 channels with little to no inhibition of channels in other conformations (e.g., Cx43 not phosphorylated at S368). dpS368, dephosphorylated S368.
Channel conformation-specific signaling.
We show here that phosphorylation at S368 leads to enhanced GJCh-mediated Ca2+-wave propagation, with waves two to three times larger in cells expressing the form of Cx43 that mimicked or favored the pS368 state than in cells with wild-type Cx43 or Cx43 in the dephosphorylated state. We also show that pS368 channels exhibit greater capacity for dye coupling than their dephosphorylated counterparts. It has been well described that connexin channels display multiple open configurations, varying between fully open and less open substates (and ≥1 closed state), with differing permeabilities for ions and metabolites. Such heterogeneity is evident at the single-channel level, with multiple open states clearly distinguished by their differences in transition conductances in electrophysiological records (26). For Cx43 channels, preference for these different open states can be altered or regulated through phosphorylation (or voltage)-dependent changes in channel conformation. As a result, a gap junction can contain multiple differentially phosphorylated channels and, consequently, macroscopic permeation properties that reflect the combined individual channel permeation properties. Importantly, Cx43 gap junctions do not show a linear relationship between junctional conductance and dye permeation rates (i.e., ionic vs. dye permeance) (23–26, 38). Indeed, some low-conductance junctions are well permeated by dyes, and some high-conductance junctions are poorly permeated by the same dyes. Differential phosphorylation of channel proteins accounts for variable permselectivity, and while it remains unclear which phosphorylation sites contribute to variable junctional permselectivity, available data indicate that inability to phosphorylate Cx43 at S368 results in junctions with increased permeation by current-carrying ions (e.g., K+ and Cl−) but reduced permeation by some larger molecules [e.g., NBD-MTMA, as demonstrated here and elsewhere (26)]. For the first time, we show that the conformation conferred by mimicking phosphorylation of Cx43 at S368 enhances GJCh-mediated Ca2+-wave propagation and increases the susceptibility of GJCh and HCh to closure by extracellular loop mimetic peptides. These data reinforce the findings from other published studies (26, 47, 66, 70) by providing additional evidence for channel conformation-specific signaling.
Traditionally, PKC-induced phosphorylation at S368 has been thought of as a beneficial response of cells and tissues to injury (e.g., wounding or ischemic insult). In the skin, for example, pS368-Cx43 levels increase substantially in basal keratinocytes in the wound region, despite overall downregulation of Cx43 expression (26, 32, 64). Similarly, acute response of the heart to ischemic insult involves increased phosphorylation at S368 of the Cx43 remaining at the intercalated disk, which may be necessary for continued conduction of the electrical impulse (52) in the face of dephosphorylation at other sites and relocation of Cx43 to lateral borders (26, 39). Other contradictory studies showed that phosphorylation at S368 decreases as a result of injury or stress (3, 42, 51, 52). These apparently disparate results may reflect differences in type and severity of ischemia, the cell systems and methods used to detect phosphorylation, and spatiotemporal aspects of the ischemic insult and assessment of effects. Several studies have shown that phosphorylation at S368 increases during the initial, acute phase of ischemia, but this phosphorylation is lost over time (3, 60). Still other studies have shown that PKC activation occurs during ischemic preconditioning, which has been demonstrated to protect against cardiac arrhythmias and cell injury (2, 11, 30). However, although PKC phosphorylates S368, it also modifies the activity of other kinases that target Cx43, including AKT (21) and MAPK (19, 21, 65, 66), which, together with PKC, target serine residues S262, S279, S282, and S373. This interaction of kinases and their multiple targets in Cx43 complicates our understanding of the impact of phosphorylation on Cx43 function. While it is not entirely clear how phosphorylation of Cx43 modulates the wound healing response, changes in Cx43 phosphorylation, localization, and expression occur as a natural cellular and tissue response to injury and are thought to alter communication in the injury region in a way that might influence repair. For instance, alterations in junctional selective permeability (as a result of preferred channel conformation and/or change in channel number) may influence the type of signals (harmful or beneficial) that are communicated, with both electrical and metabolic signaling consequences (26, 60). Interestingly, our data show enhanced Ca2+-wave propagation with phosphorylation at S368, but the physiological in vivo importance of this finding and relevance to the wound healing process are unresolved and require further study. In particular, it will be interesting to determine if the propagation of IP3 in the form of Ca2+-waves is beneficial or detrimental in a wound setting. The intercellular signals involved in preventing or enhancing the progression of injury remain unknown; however, it has been postulated that GJCh-mediated communication of IP3 and Ca2+ during injury may contribute to the spread of injury (12, 14, 15, 57). If this were the case, then inhibiting this type of communication (apparently enhanced by pS368) may turn out to be beneficial.
Selective inhibition by SRPTEKT-Hdc of pS368-Cx43 HChs and GJChs.
We showed previously that SRPTEKT-Hdc inhibition of HCh-mediated dye uptake and GJCh-mediated Ca2+-wave propagation occurred at concentrations and incubation times that minimally affected dye coupling (9). Since the SRPTEKT sequence is present in all Cx43 HChs and GJChs formed by our cells, this selective inhibition of some signals (Ca2+-waves) and not others (dye and electrical) suggested a conformation-specific susceptibility to inhibition by SRPTEKT-Hdc. Although available data support the likelihood of selective inhibition by mimetic peptides, they are not explicitly addressed as such. Our evidence (9) suggests that not all conformations of HChs (and, likely, GJChs) display similar susceptibility to inhibition by ECL mimetic peptides. Briefly, a number of reports have demonstrated that certain voltage-induced substates or subsets of Cx43 HChs vary in their sensitivity to inhibition by the ECL mimetic peptides Gap26, Gap27, and Peptide5 (8, 33, 34, 45, 73) and that these differences in mimetic peptide efficacy may reflect differences in the phosphorylation state of Cx43 (58). Conformation-dependent susceptibility to inhibition by mimetic peptides is further suggested in studies by Jiang, Gourdie, and colleagues, who showed that the CT-derived mimetic peptide αCT1 induces phosphorylation of S368 through its interaction with two positively charged lysine residues (K344 and K345) in an α-helical domain of the Cx43 CT (41). Importantly, αCT1-induced phosphorylation of S368 only occurs following injury (e.g., cryoinjury of the heart or scratch wound), as uninjured cells and tissue do not show elevated pS368-Cx43 levels in response to αCT1 treatment (55, 56). This indicates that the interaction of αCT1 with Cx43 may be affected by injury-induced changes in Cx43 channel conformation. The lack of additional studies demonstrating conformation-specific inhibition of GJCh-mediated signaling is likely due to the fact that most studies are limited to looking at a single form of intercellular communication, most often dye coupling (40, 45), Ca2+-wave propagation (40), or electrical coupling (17, 75; reviewed in Refs. 29 and 49). The variability in inhibitory effects of mimetic peptides such as Gap27 ranges from no block to complete block of these different forms of intercellular signaling, further suggesting the possibility of conformation-specific inhibition. Therefore, given the many phosphorylation sites and consequent GJCh conformations of Cx43, we set out in this study to determine which conformations are susceptible versus resistant to SRPTEKT-Hdc inhibition.
Our data show that SRPTEKT-Hdc selectively inhibits HChs and GJChs containing Cx43 phosphorylated at S368. This result is very intriguing, as the peptide shows preference for a phosphorylated form of Cx43 that may be a predominant mediator of Ca2+-waves, especially in settings of injury. This may explain the discordance in inhibitory effect we previously observed for wild-type Cx43 GJChs, where SRPTEKT-Hdc potently inhibited Ca2+-wave propagation while having no significant effect on dye coupling (9). Interestingly, the potency of inhibition by SRPTEKT-Hdc was five orders of magnitude greater for the phosphorylated form of GJChs that favored or mimicked phosphorylation at S368 than for the dephosphorylated form, and this level of potency was similar to that reported by us previously for the inhibition of Ca2+-wave propagation through wild-type Cx43 GJChs (9). Despite obtaining this important insight into phosphorylation-dependent susceptibility to peptide inhibition from the phosphomimetic mutant cells, we were unable to assess peptide specificity for physiological phosphorylation of S368 in wild-type channels using TPA alone, as TPA has off-target consequences that may result in phosphorylation of Cx43 at additional sites, removal/internalization of Cx43 from the plasma membrane, and, ultimately, abolition of Ca2+-wave propagation. This effect was best highlighted by the S368A and S368D mutants, which, despite having an S368 site unavailable for phosphorylation by PKC, showed no propagation of Ca2+-waves in the presence of TPA. Thus, TPA may close or prime the Cx43 GJChs mediating Ca2+-wave propagation for internalization through mechanisms other than S368 phosphorylation, most likely by altering phosphorylation at S262, S279, S282, or S373 (21, 65, 66, 69). To counteract these consequences of TPA, cells expressing wild-type Cx43 were preexposed to the MAPK kinase (MEK1/2) inhibitor PD98059 or the proteasome inhibitor (and AKT activator) MG132 before exposure to TPA. MAPK inhibition only marginally improved the propagation of Ca2+-waves in the presence of TPA, suggesting that the inhibitory effects of TPA on Ca2+-wave propagation may be only partially linked to MAPK phosphorylation of Cx43 and are more likely occurring through a separate mechanism. This result is in agreement with a study by Rivedal and Opsahl (61), which suggested that TPA induces inhibition of communication only partly through the MAPK pathway, as PD98059 had little effect on TPA-mediated block of communication. The small Ca2+-waves that were observed under these conditions in our current study, however, were significantly inhibited by SPRTEKT-Hdc, providing initial evidence of peptide specificity for Cx43 channels with TPA-induced S368 phosphorylation. As an alternative to MAPK inhibition, proteasome inhibition by MG132 has been linked to increased AKT activation and, ultimately, increased accretion and stability of Cx43 in gap junction plaques, even in the presence of TPA, and this is likely due to an AKT-induced disruption of the Cx43-zonula occludens-1 interaction (21, 22). Despite its effects on gap junction size, treatment with MG132 alone resulted in Ca2+-wave propagation on a scale similar to that in untreated MDCK43 cells. On the other hand, treatment with MG132 in conjunction with TPA resulted in enhanced Ca2+-waves similar to that observed in the S368D and S365A mutants, supporting the notion that pS368-Cx43 channels are important conformation-specific mediators of Ca2+ coupling. Importantly, these MG132 + TPA-treated channels were also potently inhibited by SRPTEKT-Hdc. Finally, treatment with BIM was also highly relevant, as the prevention of phosphorylation at S368 (and, possibly, other serine residues) in wild-type channels eliminated the inhibitory effect of the peptide, providing further support for the importance of the S368 site in the peptide’s interaction with Cx43.
Here we also show that SRPTEKT-Hdc preferentially inhibits the passage of dye through pS368-Cx43 channels. Despite having no significant effect on wild-type Cx43 channels, SRPTEKT-Hdc strongly inhibited dye coupling mediated by GJChs with phosphorylation at S368. This result differs from that observed for Ca2+-wave propagation, in which both wild-type and pS368-Cx43 GJChs were inhibited. A likely explanation for this difference could be the existence of other Cx43 channel configurations that continue to support a robust level of dye coupling in wild-type junctions even when pS368 channels are inhibited by SRPTEKT-Hdc. This possibility is supported by peptide-resistant Ca2+-wave propagation and dye coupling in S368A cells. In addition, we also saw inhibition of HCh-mediated dye uptake with specificity for HChs containing pS368-Cx43, and although the effect may have been slightly less for pS368 HChs than for wild-type HChs, it is evident that the peptide is interacting with HChs in a conformation-specific manner. Whether the inhibitory mechanism of pS368-Cx43 channels involves SRPTEKT-Hdc interacting independently with HChs and GJChs to alter gating or open probability or the effect is, instead, mediated through interactions with HChs to prevent the docking and formation of GJChs remains to be determined, but our previous data for wild-type Cx43 channels suggest that both mechanisms may be involved. Overall, these data indicate that SRPTEKT-Hdc (and, likely, other loop-mimetic peptides) interacts with Cx43 channels in a phosphorylation-dependent, channel conformation-specific manner.
Because wild-type Cx43 channels can contain differentially phosphorylated subunits, including pS368 in the MDCK43 cells, and because SRPTEKT-Hdc completely blocks Ca2+-wave propagation in mutants that favor or mimic phosphorylation at S368, it is tempting to conclude that SRPTEKT-Hdc preferentially inhibits channels containing the pS368 phosphorylated form of Cx43 while minimally affecting channels not containing this phosphorylated form. This conclusion is strengthened by the similar IC50 values for inhibition of Ca2+-wave propagation between cells expressing wild-type Cx43 and pS368-mimicking or -favoring mutants, as well as the failure of the peptide to block Ca2+-wave propagation in BIM-treated wild-type channels. Additional support can be gleaned from our dye-coupling studies, where the lack of inhibition of wild-type Cx43 GJChs could indicate that the peptide, while targeting pS368 channels, may be ignoring or minimally affecting other channel conformations that also support dye coupling.
We do not know all the physiologically relevant phosphorylation states that support Ca2+-wave propagation and dye coupling; presumably, other Cx43 phosphorylated forms will be implicated in mediating these types of intercellular signaling. While it may indeed be the case that phosphorylation at S368 plays a major role in Ca2+-wave propagation, we do not know the extent of phosphorylation at this site that is needed to promote this enhanced communication. Unlike the mutant channels, which homogenously contain aspartate residues at S368, such that all six Cx subunits in a HCh or all twelve subunits in the GJCh mimic phosphorylation at S368, wild-type channels may have any number of subunits phosphorylated or not at this site. Furthermore, we do not know which sites, in addition to S365 or S368, may be phosphorylated in the mutants or wild-type Cx43, with different combinations differentially regulating HCh and GJCh conformation and function. These matters raise some interesting questions, including how many Cx43 subunits must be phosphorylated for SRPTEKT-Hdc to interact and inhibit GJCh-mediated Ca2+-wave propagation and dye coupling or HCh-mediated dye uptake? Are there any specific patterns of phosphorylation that include phosphorylation at S368 in conjunction with other sites that may be linked to the channel’s inhibitory susceptibility? Are there other phosphorylation sites independent of S365 or S368 promoting a similar level of sensitivity to SRPTEKT-Hdc (that could also be involved in the strong inhibition we see for wild-type Cx43 channels)? Given the results for Cx43 channels, it will also be intriguing to evaluate inhibition of channels composed of other connexin types. Because the SRPTEKT sequence is highly conserved in 13 of 19 mouse connexins (only Cx29, Cx30.3, Cx31, Cx31.1, Cx39, and Cx40 lack the identical sequence, and Cx39 and Cx40 differ only in the last residue), it will be interesting to determine which are susceptible to inhibition by SRPTEKT-Hdc and whether a similar conformation specificity is present for these other connexin types.
In summary, it is clear that modulation of Cx43 channels can influence tissue recovery from injury, with downregulation of Cx43 and phosphorylation of S368 at the wound site occurring as a natural response. Furthermore, pharmacological agents that target Cx43 channels, especially through knockdown or inhibition, have been shown to enhance wound healing by augmenting repair processes, reducing inflammation, and, ultimately, restoring tissue homeostasis. Connexin mimetic peptides that specifically target Cx43 and impact these responses are emerging as viable therapeutic tools that can promote wound healing and are becoming increasingly prevalent in clinical trials [e.g., αCT1 in the treatment of diabetic foot ulcers (31, 36)]. Both HChs and GJChs are involved in the spread of the injury response. HCh-mediated ATP release is an important trigger of inflammatory signaling (45), and GJCh-mediated intercellular IP3 and Ca2+ signaling has been implicated in propagation of the injury (12, 14, 15, 57). To the extent that the pS368 phosphorylated form exacerbates these responses in a wound setting, particularly in increasing intercellular Ca2+ communication, our data show that SRPTEKT-Hdc potently and specifically inhibits pS368-Cx43 HChs and GJChs in a conformation-specific manner. Overall, these results not only confirm the importance of broadening the evaluation of current mimetic peptides to include signal-specific inhibition but, also, present a paradigm for the development of HCh, GJCh, Cx isotype-specific, and Cx conformation-specific inhibitory drugs.
GRANTS
These studies were funded by National Institutes of Health Grants HL-058732 and HL-131712 (J. M. Burt), HL-007249 (J. M. Burt and M. L. Cotter), NS-073664 (S. Boitano and J. Vagner), and GM-055632 (P. D. Lampe).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.L.C., S.B., P.D.L., J.L.S., J.V., J.F.E.-V., and J.M.B. conceived and designed research; M.L.C. and J.F.E.-V. performed experiments; M.L.C. analyzed data; M.L.C., S.B., and J.M.B. interpreted results of experiments; M.L.C. prepared figures; M.L.C. drafted manuscript; M.L.C., S.B., P.D.L., J.L.S., J.V., J.F.E.-V., and J.M.B. edited and revised manuscript; M.L.C., S.B., P.D.L., J.L.S., J.V., J.F.E.-V., and J.M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the members of the Burt, Boitano, Lampe, and Vagner laboratories for assistance and helpful discussions during the course of this work. NBD-MTMA was a gift from Stephen Wright (University of Arizona, Tucson, AZ).
REFERENCES
- 1.Alexander DB, Goldberg GS. Transfer of biologically important molecules between cells through gap junction channels. Curr Med Chem 10: 2045–2058, 2003. doi: 10.2174/0929867033456927. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res 61: 427–436, 2004. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 3.Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein-Rathlou NH, Andersen S, Jensen ON, Hennan JK, Kjølbye AL. Identification of ischemia-regulated phosphorylation sites in connexin43: a possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J Mol Cell Cardiol 40: 790–798, 2006. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Bednarczyk D, Mash EA, Aavula BR, Wright SH. NBD-TMA: a novel fluorescent substrate of the peritubular organic cation transporter of renal proximal tubules. Pflugers Arch 440: 184–192, 2000. doi: 10.1007/s004240000283. [DOI] [PubMed] [Google Scholar]
- 5.Blumberg PM. Protein kinase C as the receptor for the phorbol ester tumor promoters: Sixth Rhoads Memorial Award Lecture. Cancer Res 48: 1–8, 1988. [PubMed] [Google Scholar]
- 6.Boitano S, Evans WH. Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am J Physiol Lung Cell Mol Physiol 279: L623–L630, 2000. doi: 10.1152/ajplung.2000.279.4.L623. [DOI] [PubMed] [Google Scholar]
- 7.Burt JM, Nelson TK, Simon AM, Fang JS. Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am J Physiol Cell Physiol 295: C1103–C1112, 2008. doi: 10.1152/ajpcell.299.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen YS, Toth I, Danesh-Meyer HV, Green CR, Rupenthal ID. Cytotoxicity and vitreous stability of chemically modified connexin43 mimetic peptides for the treatment of optic neuropathy. J Pharm Sci 102: 2322–2331, 2013. doi: 10.1002/jps.23617. [DOI] [PubMed] [Google Scholar]
- 9.Cotter ML, Boitano S, Vagner J, Burt JM. Lipidated connexin mimetic peptides potently inhibit gap junction-mediated Ca2+-wave propagation. Am J Physiol Cell Physiol 315: C141–C154, 2018. doi: 10.1152/ajpcell.00156.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol 284: C511–C520, 2003. doi: 10.1152/ajpcell.00214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross HR, Murphy E, Bolli R, Ping P, Steenbergen C. Expression of activated PKC-ε (PKCε) protects the ischemic heart, without attenuating ischemic H+ production. J Mol Cell Cardiol 34: 361–367, 2002. doi: 10.1006/jmcc.2001.1518. [DOI] [PubMed] [Google Scholar]
- 12.Cusato K, Ripps H, Zakevicius J, Spray DC. Gap junctions remain open during cytochrome c-induced cell death: relationship of conductance to “bystander” cell killing. Cell Death Differ 13: 1707–1714, 2006. doi: 10.1038/sj.cdd.4401876. [DOI] [PubMed] [Google Scholar]
- 13.Dbouk HA, Mroue RM, El-Sabban ME, Talhouk RS. Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Commun Signal 7: 4, 2009. doi: 10.1186/1478-811X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decrock E, Krysko DV, Vinken M, Kaczmarek A, Crispino G, Bol M, Wang N, De Bock M, De Vuyst E, Naus CC, Rogiers V, Vandenabeele P, Erneux C, Mammano F, Bultynck G, Leybaert L. Transfer of IP3 through gap junctions is critical, but not sufficient, for the spread of apoptosis. Cell Death Differ 19: 947–957, 2012. doi: 10.1038/cdd.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decrock E, Vinken M, Bol M, D’Herde K, Rogiers V, Vandenabeele P, Krysko DV, Bultynck G, Leybaert L. Calcium and connexin-based intercellular communication, a deadly catch? Cell Calcium 50: 310–321, 2011. doi: 10.1016/j.ceca.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Decrock E, Vinken M, De Vuyst E, Krysko DV, D’Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. Connexin-related signaling in cell death: to live or let die? Cell Death Differ 16: 524–536, 2009. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- 17.Desplantez T, Verma V, Leybaert L, Evans WH, Weingart R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol Res 65: 546–552, 2012. doi: 10.1016/j.phrs.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Deutsch DE, Williams JA, Yule DI. Halothane and octanol block Ca2+ oscillations in pancreatic acini by multiple mechanisms. Am J Physiol Gastrointest Liver Physiol 269: G779–G788, 1995. doi: 10.1152/ajpgi.1995.269.5.G779. [DOI] [PubMed] [Google Scholar]
- 19.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci 117: 507–514, 2004. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 20.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA 92: 7686–7689, 1995. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunn CA, Lampe PD. Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J Cell Sci 127: 455–464, 2014. doi: 10.1242/jcs.142497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn CA, Su V, Lau AF, Lampe PD. Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J Biol Chem 287: 2600–2607, 2012. doi: 10.1074/jbc.M111.276261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckert R. Gap-junctional single-channel permeability for fluorescent tracers in mammalian cell cultures. Biophys J 91: 565–579, 2006. doi: 10.1529/biophysj.105.072306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ek-Vitorín JF, Burt JM. Quantification of gap junction selectivity. Am J Physiol Cell Physiol 289: C1535–C1546, 2005. doi: 10.1152/ajpcell.00182.2005. [DOI] [PubMed] [Google Scholar]
- 25.Ek-Vitorin JF, Burt JM. Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim Biophys Acta 1828: 51–68, 2013. doi: 10.1016/j.bbamem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res 98: 1498–1505, 2006. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbadawy HM, Mirabelli P, Xeroudaki M, Parekh M, Bertolin M, Breda C, Cagini C, Ponzin D, Lagali N, Ferrari S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br J Pharmacol 173: 2880–2893, 2016. doi: 10.1111/bph.13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans WH, Boitano S. Connexin mimetic peptides: specific inhibitors of gap-junctional intercellular communication. Biochem Soc Trans 29: 606–612, 2001. doi: 10.1042/bst0290606. [DOI] [PubMed] [Google Scholar]
- 29.Evans WH, Bultynck G, Leybaert L. Manipulating connexin communication channels: use of peptidomimetics and the translational outputs. J Membr Biol 245: 437–449, 2012. doi: 10.1007/s00232-012-9488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Dorado D, Ruiz-Meana M, Padilla F, Rodriguez-Sinovas A, Mirabet M. Gap junction-mediated intercellular communication in ischemic preconditioning. Cardiovasc Res 55: 456–465, 2002. doi: 10.1016/S0008-6363(02)00441-8. [DOI] [PubMed] [Google Scholar]
- 31.Ghatnekar GS, Grek CL, Armstrong DG, Desai SC, Gourdie RG. The effect of a connexin43-based peptide on the healing of chronic venous leg ulcers: a multicenter, randomized trial. J Invest Dermatol 135: 289–298, 2015. doi: 10.1038/jid.2014.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol Biol Cell 6: 1491–1501, 1995. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci 46: 1208–1218, 2005. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- 34.Gomes P, Srinivas SP, Vereecke J, Himpens B. Gap junctional intercellular communication in bovine corneal endothelial cells. Exp Eye Res 83: 1225–1237, 2006. doi: 10.1016/j.exer.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 35.Good ME, Ek-Vitorín JF, Burt JM. Extracellular loop cysteine mutant of Cx37 fails to suppress proliferation of rat insulinoma cells. J Membr Biol 245: 369–380, 2012. doi: 10.1007/s00232-012-9459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grek CL, Prasad GM, Viswanathan V, Armstrong DG, Gourdie RG, Ghatnekar GS. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: a multicenter, randomized trial. Wound Repair Regen 23: 203–212, 2015. doi: 10.1111/wrr.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 1985. [PubMed] [Google Scholar]
- 38.Heyman NS, Burt JM. Hindered diffusion through an aqueous pore describes invariant dye selectivity of Cx43 junctions. Biophys J 94: 840–854, 2008. doi: 10.1529/biophysj.107.115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hund TJ, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Protein kinase Cε mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm 4: 1183–1193, 2007. doi: 10.1016/j.hrthm.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isakson BE, Seedorf GJ, Lubman RL, Evans WH, Boitano S. Cell-cell communication in heterocellular cultures of alveolar epithelial cells. Am J Respir Cell Mol Biol 29: 552–561, 2003. doi: 10.1165/rcmb.2002-0281OC. [DOI] [PubMed] [Google Scholar]
- 41.Jiang J, Palatinus JA, He H, Iyyathurai J, Jordan J, McGowan FX, Schey K, Bultynck G, Zhang Z, Gourdie RG. Phosphorylation of connexin43 at serine 368 is necessary for induction of cardioprotection by a connexin43 carboxyl-terminal mimetic (Abstract) Circulation 134: A16380, 2018. [Google Scholar]
- 42.Johansen D, Cruciani V, Sundset R, Ytrehus K, Mikalsen SO. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol Biochem 28: 103–114, 2011. doi: 10.1159/000331719. [DOI] [PubMed] [Google Scholar]
- 43.Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry 33: 181–198, 2009. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 44.Kameritsch P, Khandoga N, Pohl U, Pogoda K. Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis 4: e584, 2013. doi: 10.1038/cddis.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y, Griffin JM, Harris PW, Chan SH, Nicholson LF, Brimble MA, O’Carroll SJ, Green CR. Characterizing the mode of action of extracellular connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim Biophys Acta, Gen Subj 1861: 68–78, 2017. doi: 10.1016/j.bbagen.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Krysko DV, Leybaert L, Vandenabeele P, D’Herde K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis 10: 459–469, 2005. doi: 10.1007/s10495-005-1875-2. [DOI] [PubMed] [Google Scholar]
- 47.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 149: 1503–1512, 2000. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem 279: 50089–50096, 2004. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- 49.Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, Laird DW, Naus CC, Green CR, Schulz R. Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol Rev 69: 396–478, 2017. doi: 10.1124/pr.115.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Dürst M, Alonso A. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci 119: 3634–3642, 2006. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- 51.Matsushita S, Kurihara H, Watanabe M, Okada T, Sakai T, Amano A. Alterations of phosphorylation state of connexin 43 during hypoxia and reoxygenation are associated with cardiac function. J Histochem Cytochem 54: 343–353, 2006. doi: 10.1369/jhc.4A6611.2005. [DOI] [PubMed] [Google Scholar]
- 52.Nassal MM, Werdich AA, Wan X, Hoshi M, Deschênes I, Rosenbaum DS, Donahue JK. Phosphorylation at connexin43 serine-368 is necessary for myocardial conduction during metabolic stress. J Cardiovasc Electrophysiol 27: 110–119, 2016. doi: 10.1111/jce.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Carroll SJ, Alkadhi M, Nicholson LF, Green CR. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun Adhes 15: 27–42, 2008. doi: 10.1080/15419060802014164. [DOI] [PubMed] [Google Scholar]
- 54.O’Carroll SJ, Gorrie CA, Velamoor S, Green CR, Nicholson LF. Connexin43 mimetic peptide is neuroprotective and improves function following spinal cord injury. Neurosci Res 75: 256–267, 2013. doi: 10.1016/j.neures.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 55.O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res 108: 704–715, 2011. doi: 10.1161/CIRCRESAHA.110.235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palatinus JA, Rhett JM, Gourdie RG. Enhanced PKCε mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels (Austin) 5: 236–240, 2011. doi: 10.4161/chan.5.3.15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peixoto PM, Ryu SY, Pruzansky DP, Kuriakose M, Gilmore A, Kinnally KW. Mitochondrial apoptosis is amplified through gap junctions. Biochem Biophys Res Commun 390: 38–43, 2009. doi: 10.1016/j.bbrc.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pollok S, Pfeiffer AC, Lobmann R, Wright CS, Moll I, Martin PE, Brandner JM. Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J Cell Mol Med 15: 861–873, 2011. doi: 10.1111/j.1582-4934.2010.01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, Becker DL. Targeting connexin43 expression accelerates the rate of wound repair. Curr Biol 13: 1697–1703, 2003. doi: 10.1016/j.cub.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 60.Richards TS, Dunn CA, Carter WG, Usui ML, Olerud JE, Lampe PD. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J Cell Biol 167: 555–562, 2004. doi: 10.1083/jcb.200404142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rivedal E, Opsahl H. Role of PKC and MAP kinase in EGF- and TPA-induced connexin43 phosphorylation and inhibition of gap junction intercellular communication in rat liver epithelial cells. Carcinogenesis 22: 1543–1550, 2001. doi: 10.1093/carcin/22.9.1543. [DOI] [PubMed] [Google Scholar]
- 62.Simon AM, Chen H, Jackson CL. Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun Adhes 13: 61–77, 2006. doi: 10.1080/15419060600631748. [DOI] [PubMed] [Google Scholar]
- 63.Smyth JW, Shaw RM. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep 5: 611–618, 2013. doi: 10.1016/j.celrep.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J 419: 261–272, 2009. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes 15: 75–84, 2008. doi: 10.1080/15419060802014016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol 217: 35–41, 2007. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Solan JL, Lampe PD. Kinase programs spatiotemporally regulate gap junction assembly and disassembly: effects on wound repair. Semin Cell Dev Biol 50: 40–48, 2016. doi: 10.1016/j.semcdb.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Solan JL, Lampe PD. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim Biophys Acta Biomembr 1860: 83–90, 2018. doi: 10.1016/j.bbamem.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol 179: 1301–1309, 2007. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem 279: 54695–54701, 2004. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 71.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266: 15771–15781, 1991. [PubMed] [Google Scholar]
- 72.Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, Kawashima S. Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: a novel catalytic activity in proteasome. Biochem Biophys Res Commun 196: 1195–1201, 1993. doi: 10.1006/bbrc.1993.2378. [DOI] [PubMed] [Google Scholar]
- 73.Wang N, De Bock M, Antoons G, Gadicherla AK, Bol M, Decrock E, Evans WH, Sipido KR, Bukauskas FF, Leybaert L. Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res Cardiol 107: 304, 2012. doi: 10.1007/s00395-012-0304-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang N, De Vuyst E, Ponsaerts R, Boengler K, Palacios-Prado N, Wauman J, Lai CP, De Bock M, Decrock E, Bol M, Vinken M, Rogiers V, Tavernier J, Evans WH, Naus CC, Bukauskas FF, Sipido KR, Heusch G, Schulz R, Bultynck G, Leybaert L. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res Cardiol 108: 309, 2013. doi: 10.1007/s00395-012-0309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Warner A, Clements DK, Parikh S, Evans WH, DeHaan RL. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J Physiol 488: 721–728, 1995. doi: 10.1113/jphysiol.1995.sp021003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wright CS, van Steensel MA, Hodgins MB, Martin PE. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen 17: 240–249, 2009. doi: 10.1111/j.1524-475X.2009.00471.x. [DOI] [PubMed] [Google Scholar]