

Abstract
The existence and regenerative potential of resident stem and progenitor cells in the adult pancreas are controversial topics. A question that has been only minimally addressed is the capacity of a progenitor cell to self-renew, a key attribute that defines stem cells. Previously, our laboratory has identified putative stem and progenitor cells from the adult murine pancreas. Using an ex vivo colony/organoid culture system, we demonstrated that these stem/progenitor-like cells have self-renewal and multilineage differentiation potential. We have named these cells pancreatic colony-forming units (PCFUs) because they can give rise to three-dimensional colonies. However, the molecular mechanisms by which PCFUs self-renew have remained largely unknown. Here, we tested the hypothesis that PCFU self-renewal requires GLIS family zinc finger 3 (GLIS3), a zinc-finger transcription factor important in pancreas development. Pancreata from 2- to 4-month-old mice were dissociated, sorted for CD133highCD71low ductal cells, known to be enriched for PCFUs, and virally transduced with shRNAs to knock down GLIS3 and other proteins. We then plated these cells into our colony assays and analyzed the resulting colonies for protein and gene expression. Our results revealed a previously unknown GLIS3–to–CD133–to–WNT signaling axis in which GLIS3 and CD133 act as factors necessary for maintaining WNT receptors and signaling molecules in colonies, allowing responses to WNT ligands. Additionally, we found that CD133, but not GLIS3 or WNT, is required for phosphoinositide 3-kinase (PI3K)/AKT Ser/Thr kinase (AKT)-mediated PCFU survival. Collectively, our results uncover a molecular pathway that maintains self-renewal of adult murine PCFUs.
Keywords: pancreas, endocrinology, stem cells, cell therapy, Wnt signaling, β-catenin, cell replacement, GLIS family zinc finger 3, progenitor cell, self-renewal
Introduction
Many adult tissues harbor resident stem or progenitor cells that maintain tissue integrity and homeostasis (1). Whether the adult pancreas also harbors progenitor cells, however, is controversial, with evidence both for (2–4) and against (5–7) their existence in vivo. Adult pancreatic progenitor cells could be a potential source of β-like cells for endogenous neogenesis or for cell replacement therapy of type 1 diabetes.
Although the in vivo evidence is controversial, our laboratory has provided data that a rare population of cells in the adult murine pancreas has stem cell-like activities in vitro: they self-renew long-term and give rise to cells that resemble all three major pancreatic lineages (i.e. duct, acinar, and endocrine), including insulin-producing β-like cells (8, 9). We have named these rare tri-potent progenitors pancreatic colony-forming units (PCFUs),3 because they, as single cells, are able to form colonies (also known as organoids) in a methylcellulose-containing semisolid medium. Methylcellulose is a biologically-inert material that increases the viscosity of the medium. The semisolid medium prevents single cells from moving and aggregating, yet it is soft enough to allow a single cell to form a colony of cells in a three-dimensional space. Using the ability to study the in vitro functions of adult murine PCFUs, this study aimed to investigate the molecular mechanism by which adult PCFUs self-renew.
GLIS family zinc finger 3 (Glis3) is a member of the Krüppel-like zinc finger transcription factor family (10). In postnatal mice, Glis3 plays an important role in the self-renewal of sperm stem cells (11). Also, when expressed in conjunction with the “Yamanaka factors” Sox2, Klf4, c-Myc, and Oct4, Glis3 enhances the reprogramming of adult human adipose–derived stromal cells into induced pluripotent stem cells (12), suggesting an important role of Glis3 in the self-renewal and pluripotency of various stem/progenitor cells. In humans, Glis3 is important for proper endocrine cell development in the pancreas. Mutations in Glis3 are linked to neonatal diabetes (13), a higher risk of type 2 diabetes in several genome-wide association studies (14), beta-cell apoptosis (15), and in some cases defects in the formation of both the exocrine and endocrine pancreas (16, 17). Glis3 is also necessary for proper pancreas development in mice, and mutations in Glis3 lead to decreased numbers of beta-cells, smaller islet volume, and subsequent neonatal diabetes (18, 19). Adult mice continually express Glis3 in the beta and duct cells of the pancreas (20). In both adult and embryonic mice, mutations or deletion of Glis3 leads to beta-cell apoptosis and a cystic duct phenotype (18, 19, 21). Using global gene expression analysis in one of our prior studies, Glis3 was found to be expressed to a higher degree in the pancreatic CD133highCD71low ductal cell population, which is highly enriched for adult murine PCFUs (9), than in other populations. Given the pleiotropic roles of Glis3 in multiple biological processes and organs of different ages, we hypothesized that Glis3 was also important in the in vitro self-renewal of adult murine PCFUs.
To test our hypothesis, lentiviral vectors carrying short-hairpin interfering RNAs (shRNAs) were used to knock down Glis3 expression, and the effects on adult murine PCFUs were measured. We found that Glis3 was required for the long-term self-renewal of PCFUs in vitro. Further analyses on the role of Glis3 in self-renewal revealed that Glis3 affected Wnt signaling molecules and, surprisingly, Prominin-1 (CD133). Follow-up studies using shRNAs against CD133 and Wnt effector β-catenin demonstrated a previously unknown Glis3–to–CD133–to-Wnt-signaling pathway that is required for maintaining the responsiveness to self-renewing signals of adult murine PCFUs.
Results
Glis3 is expressed in CD133highCD71low ductal cells from the adult murine pancreas
We previously reported that CD133highCD71low ductal cells in the adult murine pancreas are enriched for PCFUs (9). To confirm this finding, pancreata from 8- to 12-week-old C57Bl/6 mice were dissociated into single-cell suspension, stained with anti-CD133 and anti-CD71 antibodies, and sorted into CD133+CD71− (region (R)1), CD133highCD71low (R2), CD133lowCD71+ (R3), and CD133−CD71+ (R4) cell populations (Fig. S1A). Sorted cells were plated into our standard colony assay containing 5% (v/v) Matrigel (Fig. S2) (22). The CD133highCD71low (R2) population formed colonies most efficiently (Fig. S1B), consistent with our previous report (9). Quantitative (q) RT-PCR analyses confirmed the expression of ductal markers (keratin19 (CK19) and Sox9) in R1 and R2 cells (Fig. S1C) (9). Importantly, Glis3 was detected in the CD133+CD71− (R1) and CD133hiCD71low (R2) ductal cell fractions, with a higher expression in the R2 cells. These results were consistent with our prior RNA-seq study (9), and demonstrate that Glis3 is expressed in the CD133highCD71low ductal cell fraction.
Glis3 affects the genetic profile and growth of “endocrine/acinar” colonies in vitro
A functional mutation in Glis3 in the developing murine pancreas leads to decreased expression of the Ins2 gene (19, 23). We first tested whether Glis3 knockdown affected the expression of Ins2 in adult PCFU-derived colonies in vitro. Sorted CD133highCD71low cells were transduced overnight with a pool of five shRNAs directed specifically at Glis3 mRNA (shGlis3s) or an shRNA control targeting a nonmammalian gene (shControl). Subsequently, cells were plated into our Matrigel-containing colony assay with exogenous R-spondin1 (RSPO1), a Wnt agonist (24, 25), to expand PCFUs (8). After 3 weeks, colonies were pooled, dissociated into a single cell suspension, and replated into our colony assay containing laminin hydrogel (in the absence of Matrigel and RSPO1) to differentiate PCFUs into endocrine/acinar (E/A) colonies (Fig. S3A). We have previously determined that E/A colonies grown in laminin hydrogel express higher levels of endocrine and acinar cell markers, but lower levels of ductal markers, compared with colonies grown in the presence of Matrigel (8, 9). Puromycin was added throughout the culture to select for transduced cells.
Individual 10-day-old E/A colonies were hand-picked and analyzed by microfluidic qRT-PCR analysis. Microfluidic qRT-PCR allows gene expression analysis on limited material, as little as one colony (8). As expected, the expression of Glis3 and Ins2 was reduced in individual E/A colonies receiving shGlis3s compared with controls. In contrast, the expression of acinar (amylase 2A and elastase1) and ductal (mucin-1, CK7, CK19, and Sox9) cell markers was unchanged (Fig. S3B).
A PCFU has to survive before forming a colony; therefore, the number of colonies grown in a culture well was used as a proxy to indicate the survival of PCFUs that give rise to E/A colonies. After 10 days of growth in laminin hydrogel, equal numbers of E/A colonies formed from cells transduced with either shControl or shGlis3s (Fig. S3C), demonstrating that knockdown of Glis3 does not affect the survival of the PCFUs that give rise to E/A colonies. However, E/A colonies that were treated with shGlis3s had smaller diameters compared with shControl (Fig. S3D). We have previously determined that the diameter of a colony may be used as a proxy for cell number (26). To determine the reasons that growth was affected, E/A colonies were grown in the presence of EdU, handpicked, pooled, fixed, and immunostained with EdU or TUNEL to assess proliferation and apoptosis levels, respectively (Fig. S3E). The nuclei were stained with Hoechst. Compared with shControl, shGlis3s-treated E/A colonies had significantly lower EdU+Hoechst+ but higher TUNEL+Hoechst+ double-positive cells (Fig. S3F). These results demonstrate that Glis3 knockdown negatively affects the proliferation and the survival of cells within E/A colonies. Together, these results are consistent with previous reports in that Glis3 affects Ins2 expression (18, 27) and beta-cell survival (28) and confirm the effectiveness of Glis3 knockdown in our colony system.
Glis3 is required for self-renewal of adult PCFU in vitro
To determine whether knockdown of Glis3 affected the self-renewal of PCFUs, sorted CD133highCD71low cells were transduced with shGlis3s or shControl. Subsequently, cells were plated in a Matrigel-containing colony assay known to support robust PCFU self-renewal in the presence of the Wnt agonist RSPO1 (8). The resulting colonies were serially dissociated and replated (passaged) for a total of four generations in Matrigel and RSPO1-containing cultures (Fig. 1A). Puromycin was again added throughout the cultures to select for transduced cells. Treatment of PCFUs with shGlis3s, compared with shControl, reduced the number of PCFUs by the 4th generation (Fig. 1B). The reduction of PCFUs was not due to survival inhibition of shGlis3s, as there was no difference between the number of first generation colonies derived from the freshly-sorted cells that received shGlis3s or shControl (Fig. 1C). Together, these results demonstrate that, in the presence of Wnt signaling, Glis3 is required for long-term self-renewal, but not survival, of adult PCFUs.
Figure 1.

Knockdown of Glis3 reduces Wnt targets/signaling transducers and CD133 in short-term culture and inhibits long-term self-renewal of PCFUs. A, experimental scheme to assess self-renewal by serial dissociation and replating of cystic colonies grown in standard (Matrigel-containing) colony assay containing a Wnt agonist RSPO1. B, knockdown of Glis3 reduced the total number of PCFUs at the 4th generation, suggesting that Glis3 was required for long-term self-renewal of PCFUs in vitro. Data are from five pooled biological samples run in four independent experiments with four technical replicates each and are represented as mean ± S.E. C, Glis3 knockdown did not affect the number of first generation cystic colonies in the primary culture, suggesting that Glis3 was not required for the survival of PCFUs that gave rise to cystic colonies. Data are from five pooled biological samples run in nine independent experiments with four technical replicates each and are represented as mean ± S.E. D, conventional qRT-PCR analyses of pooled colonies showed that, compared with the “no RSPO1” control, addition of exogenous RSPO1 increased Wnt genes, Lgr5 and Axin2, but not Glis3 or CD133, whereas the addition of a porcupine inhibitor “PORCNi” did not change the expression of Lgr5 or Axin2. Results are shown as fold change of ΔΔCt values from PORCNi or RSPO1 versus no RSPO1-treated colonies. β-Actin was used to calculate ΔΔCt values of each gene. Data represent the mean ± S.E. of three independent experiments. E, microfluidic qRT-PCR analyses showed that expression of Wnt genes and CD133 in individual cystic colonies was reduced by Glis3 knockdown. None of the genes displayed were up-regulated. Data represent mean ± S.E. of a total of 28 colonies gathered from three independent experiments. F, decreased protein expression of Lgr5, total Lrp6, phospho-Lrp6 (Ser-1490), and CD133 in cystic colonies treated with shGlis3s was confirmed by Western blotting analyses. β-Actin was used as a loading control. Images are representatives of three independent experiments showing similar trends. G and H, IPA revealed top down-regulated pathways or genes affected by Glis3 knockdown in modules of “diseases” (G) and “predicted transcription factors” (H). Data are from five pooled biological samples run in two independent experiments and are represented as mean ± S.E. *, p value <0.05; **, p value <0.01; ****, p value <0.0001; n.s., not significant.
Glis3 lies upstream of Wnt signaling
To further investigate the relationship between Glis3 and the Wnt pathway, we tested the hypothesis that Glis3 was downstream of Wnt signaling. Pooled 3-week-old colonies (first generation) grown in Matrigel with or without RSPO1 were examined for Glis3 mRNA expression, which was not affected by exogenous RSPO1 (Fig. 1D). This suggests that Glis3 may lie upstream of, or in parallel to, the Wnt-signaling pathway.
To test the hypothesis that Glis3 lies upstream of Wnt signaling, individual 3-week-old colonies, grown in Matrigel and RSPO1, treated with shGlis3s or shControl were hand-picked and analyzed by microfluidic qRT-PCR for Wnt gene expression. Compared with shControl, shGlis3s-treated colonies had decreased expression of the canonical Wnt effector β-catenin (29), the target genes Axin2 (30) and Lgr5 (31), and the receptor Lrp6 (Fig. 1E). The decreased Lgr5 and Lrp6 proteins were confirmed by Western blot analysis (Fig. 1F).
To determine whether Glis3 affected the activation of the Lrp6 receptor, we examined the levels of phosphorylated Lrp6 (p-Lrp6; Ser-1490), which is known to increase when Wnt ligand binds to Lrp6 on the cell surface (32). Glis3 knockdown did not affect the protein ratio of p-Lrp6 to total Lrp6 (Fig. 1F), suggesting that Glis3 did not affect the activation of Lrp6. Due to the lack of a useful anti-Glis3 antibody for Western blotting analyses, the effectiveness of shGlis3s was confirmed by the reduced Glis3 gene expression in individual colonies, compared with shControl (Fig. 1E). Together, these results suggest that, in 3-week-old colonies grown in Matrigel and stimulated with RSPO1, Glis3 affects the transcription of Wnt genes, but not the initiation of the Wnt signal transduction into the cells, and that Glis3 lies upstream of the Wnt pathway.
Gene expression analysis in long-term cultures initiated with Glis3 knockdown
Germ-line whole-body knockout of Glis3 results in hyperproliferative ducts in the newborn pancreas of mice (18). We therefore sought to determine the long-term consequence of Glis3 knockdown on ductal gene expression at the 4th generation colonies grown in the presence of RSPO1 in Matrigel-containing assay as in Fig. 1A. Despite continuous selection pressure with puromycin, Glis3 was no longer significantly down-regulated in the 4th culture (Fig. S3G), potentially due to promoter silencing or compensatory effects (33). The former possibility was unlikely because shCD133s, which we used for the next series of experiments, maintained long-term knockdown of CD133 in the 4th culture (data not shown), suggesting that there were compensatory effects resulting in an increased expression of Glis3. Ductal markers, including CAII, CK19, Hnf1b, and Sox9, were significantly increased in colonies initiated with Glis3 knockdown compared with shControl. The Wnt gene Axin2, as well as β-catenin, that were initially down-regulated in the first generation (Fig. 1E) was now up-regulated in the 4th generation colonies (Fig. S3G). Additionally, c-myc, a Wnt/β-catenin target gene important for proliferation (34), was significantly enhanced in 4th generation colonies initiated with Glis3 knockdown. These results suggest that long-term Glis3 knockdown may result in a switch from PCFUs still capable of self-renewal in the early culture into the nonself-renewing ductal cells with activated Wnt signaling in the 4th generation. These results are therefore consistent with the phenotype of hyperproliferative ducts in Glis3 knockout mice (18) and may explain the minor effects of Glis3 knockdown in the number of total PCFUs in the 2nd and 3rd generations (Fig. 1B).
Glis3 knockdown affects multiple gene pathways
To further investigate the overall effects of Glis3 knockdown, we performed global gene expression analysis using RNA-seq on RSPO1-stimulated 3-week-old colonies grown in Matrigel transduced with shControl or shGlis3s. Next, the Ingenuity Pathway Analysis (IPA) software was used to analyze and categorize the differentially-expressed genes. Under the function “disease annotations” in IPA, decreased cellular movement and migration were found to be top-ranked in the shGlis3-decreased gene category (Fig. 1G). We employed another tool from IPA, the “upstream regulators,” that predicts affected genes that lie upstream of the down-regulated genes affected by shGlis3s. To our gratification, β-catenin (CTNNB1) was ranked in the top 10 transcriptional regulators affected by the knockdown of Glis3 (Fig. 1H) which confirmed that Glis3 lies upstream of Wnt signaling.
Glis3 lies upstream of CD133 signaling
Because CD133 is expressed by various types of progenitor cells (35–37) and is used to enrich PCFUs in our laboratory (8, 9, 22, 26, 38–40), the expression of CD133 is routinely monitored in colonies. Serendipitously, we found that whenever Glis3 was knocked down, CD133 was significantly down-regulated on both mRNA (Fig. 1E) and protein levels (Fig. 1F), suggesting that CD133 is also downstream of Glis3. Previous reports suggest that CD133 networks with the Wnt pathway (41, 42). To test whether CD133 was interacting with Wnt in our system, the expression of CD133 in colonies grown in Matrigel with or without RSPO1 was examined. CD133 expression did not change between colonies treated with or without RSPO1 (Fig. 1D), suggesting that CD133 is not downstream but may lie either upstream or in parallel to the Wnt pathway.
CD133 lies upstream of Wnt signaling
We next tested the hypothesis that CD133 was upstream of Wnt signaling in adult PCFUs. Sorted CD133highCD71low cells were transduced with a pool of five lentiviral vectors carrying shRNAs against CD133 (shCD133s) or shControl, plated into Matrigel colony assay in the presence of exogenous RSPO1, and the resulting colonies were analyzed by single-colony qRT-PCR for Wnt genes. As expected, individual 3-week-old colonies treated with shCD133s, compared with shControl, had decreased expression of CD133 both in mRNA (Fig. 2A) and protein (Fig. 2B). β-Catenin, Axin2, and Lgr5 were also significantly decreased in shCD133-treated colonies, compared with control (Fig. 2A). The reduced protein expression of Lgr5 was confirmed by Western blot analysis (Fig. 2B). In contrast to Glis3 knockdown, CD133 knockdown did not change mRNA or protein expression of Lrp6 (Fig. 2, A and B), suggesting a divergent effect of Glis3 and CD133 on this Wnt receptor. Similar to Glis3 knockdown, the ratio of p-Lrp6 to total Lrp6 did not change in response to CD133 knockdown, suggesting a lack of involvement of CD133 in Lrp6 activation. Finally, knockdown of CD133 did not affect Glis3 expression (Fig. 2A), demonstrating that CD133 is downstream of Glis3 but upstream of Wnt.
Figure 2.

Knockdown of CD133 decreases Wnt targets/signaling transducers and phospho-AKT, but not Glis3, in 3-week-old cystic colonies grown in Matrigel and RSPO1. A, single-colony microfluidic qRT-PCR analysis showed that knockdown of CD133 reduced the expression of Wnt genes but not Glis3, suggesting CD133 lies downstream of Glis3 but upstream of Wnt signaling. None of the displayed genes were significantly up-regulated. Data represent mean ± S.E. of a total of 37 cystic colonies from three independent experiments. B, Western blot analysis showed, in colonies treated with shCD133s, decreased protein expression of CD133, Lgr5, active β-catenin, and phospho-AKT (Ser-473), but not total Lrp6, phospho-Lrp6 (Ser-1490), total AKT, total Stat3, and phospho-Stat3 (Tyr-705). β-Actin was used as a loading control. Images are representatives of three independent experiments showing similar trends. C and D, IPA revealed top down-regulated pathways or genes affected by CD133 knockdown in modules of “diseases” (C) and “predicted transcription factors” (D). Data are from five pooled biological samples run in two independent experiments. *, p value <0.05; **, p value <0.01; N/A, not applicable.
CD133 knockdown affects multiple gene pathways
Using RNA-seq and IPA analyses, differentially expressed genes in 3-week-old colonies grown in Matrigel and RSPO1 and treated with shCD133s or shControl had shocking similarity to the results obtained by Glis3 knockdown. The “disease annotation” showed reduced cell movement and migration as the first and third most affected cellular functions (Fig. 2C). β-Catenin (CTNNB1) was again in the top 10 predicted decreased transcriptional regulators following shCD133s treatment (Fig. 2D). These results support the finding that CD133 lies upstream of Wnt signaling.
CD133 signaling is required for in vitro self-renewal and survival of adult PCFUs
To determine whether CD133 may also be involved in PCFU self-renewal, sorted CD133highCD71low cells were transduced with shCD133s or shControl lentiviruses and serially replated in the Matrigel colony assay in the presence of exogenous RSPO1 for four generations. Compared with control, CD133 knockdown significantly decreased the number of PCFUs in all generations (Fig. 3A). Unlike the results from shGlis3s (Fig. 1C), knockdown of CD133 in freshly-sorted cells reduced the total number of 1st-generation colonies grown in Matrigel and RSPO1 (Fig. 3B), suggesting that CD133 was necessary to maintain PCFU survival in vitro. To adjust for the effect of CD133 in PCFU survival, the self-renewal data were re-analyzed by accommodating for the 40% reduction of PCFUs from shCD133-treated cells (Fig. 3B). As expected, there was no longer a difference in colony numbers in the first generation between shControl and shCD133-treated groups after adjustments; however, the total PCFUs in the 2nd generation remained significantly different (Fig. 3C), demonstrating that PCFU self-renewal is indeed affected by CD133 knockdown.
Figure 3.

Knockdown of CD133 decreases both self-renewal and survival of PCFUs grown in Matrigel and RSPO1. A, number of PCFUs, assessed by a serial replating strategy, was reduced by knockdown of CD133. Data are from five pooled biological samples run in four independent experiments with four technical replicates each and are represented as mean ± S.E. B, number of primary (first generation) cystic colonies grown in standard (Matrigel-containing) colony assay plus RSPO1 was reduced by CD133 knockdown, suggesting that CD133 is required for the survival of PCFUs. Data represent mean ± S.E. from four experiments. C, data from A were re-analyzed after adjusting for the 40% reduction of PCFU survival caused by CD133 knockdown, which showed that self-renewal of PCFUs indeed requires CD133. D and E, pharmacological activation of PI3K by 740 Y-P and AKT by SC79 rescued the number of primary cystic colonies treated with shCD133s, demonstrating the survival effects of CD133 were mediated by the PI3K/AKT pathway. Data are from five pooled biological samples run in three independent experiments with four technical replicates from each molecule and are represented as mean ± S.E. *, p value <0.05; **, p value <0.01; ***, p value <0.001; ****, p value <0.0001; n.s., not significant.
CD133 affects PCFU survival via PIK/AKT pathway
CD133 affects the survival and tumorigenicity of glioma and colon cancer stem cells through interactions with the PI3K/AKT complex (43, 44). PI3K/AKT signaling is required for the survival of many progenitor cell types (45, 46). To determine whether PI3K/AKT was affected by CD133 knockdown in our system, we employed the “upstream complexes” tool in IPA to predict the protein complexes affected by shCD133s (Fig. S4). The PI3K complex was ranked the 2nd most significantly–down-regulated complexes in the CD133 knockdown.
To further confirm the involvement of the PI3K/AKT pathway, pooled 3-week-old colonies grown in Matrigel treated with shCD133s or shControl were examined by Western blotting for the presence of phosphorylated AKT (p-AKT; Ser-473), an indicator of AKT activation (47). Knockdown of CD133 resulted in a reduced level of p-AKT, although the total AKT protein was unchanged (Fig. 2B). These results demonstrate that CD133 is required for AKT activation in PCFU-derived colonies.
Next, we sought to determine whether the survival inhibition of PCFUs induced by shCD133s could be rescued by activating PI3K/AKT signaling. To test this, freshly sorted CD133highCD71low cells were transduced with shCD133s or shControl and then plated in the presence of pharmacological activators of PI3K (740 Y-P) or AKT (SC79). Both 740 Y-P (Fig. 3D) and SC79 (Fig. 3E), compared with vehicle control, significantly increased colony formation from shCD133s-transduced PCFUs. In contrast, 740 Y-P or SC79 had no effect on total colonies formed from shControl-transduced PCFUs (Fig. 3, D and E). These results demonstrate that the requirement of CD133 for PCFU survival is mediated, at least in part, through PI3K/AKT signaling.
Stat3 is also known to affect survival of cells (48), and it was ranked in the IPA analysis as the 3rd most significantly–down-regulated complexes in the CD133 knockdown (Fig. S4). In contrast to PI3K/AKT, knockdown of CD133 did not change the levels of total Stat3 proteins nor phospho-Stat3 (p-Stat3; Tyr-705) in Western blot analysis (Fig. 2B).
RSPO1 partially rescues inhibition of PCFU self-renewal mediated by Glis3 and CD133 knockdowns
To confirm that the effect of Glis3 or CD133 on PCFU self-renewal was indeed mediated through Wnt signaling, we attempted to perform rescue experiments (Fig. 4A). Sorted CD133highCD71low cells were transduced with shGlis3s, shCD133s, or shControl lentiviruses and serially-replated in the Matrigel colony assay without exogenous RSPO1. In parallel, exogenous RSPO1 was added to each group. Without RSPO1, the shControl-treated cells had a base-line level of self-renewal (Fig. 4B, blue dotted line), consistent with our previous reports (8). Also consistent with our previous finding (8), addition of RSPO1 to shControl-treated cells increased self-renewal (Fig. 4B, comparing blue dotted versus solid lines), confirming the bioactivity of the RSPO1 used. In the presence of RSPO1, shGlis3s and shCDS133s reduced self-renewal at the 4th generation, compared with shControl (Fig. 4B; comparing the three solid lines), which confirmed the results obtained in Figs. 1B and 3A.
Figure 4.

Glis3 and CD133 affect self-renewal of PCFUs through Wnt signaling. A, experimental scheme for PCFUs transduced with lentivirus carrying shGlis3s, shCD133s, or shControl followed by serial replating in Matrigel-containing culture with or without exogenous RSPO1. B, in the absence of exogenous RSPO1, knockdown of Glis3 (red dotted line) and CD133 (green dotted line) stopped self-renewal at the second passage. Addition of exogenous RSPO1 to PCFUs that were treated with shGlis3s (red solid line) or shCD133s (green solid line) partially rescued the self-renewal to the same levels as the PCFUs that received shControl (blue dotted line). The biological activity of the exogenous RSPO1 was further confirmed by the increase of self-renewal in PCFUs that received shControl and RSPO1 (blue solid line), compared with shControl and no RSPO1 (blue dotted line). Results are represented as mean ± S.E. from four independent experiments. *, p value <0.05.
Knockdowns of Glis3 and CD133 stopped PCFU self-renewal in the absence of RSPO1 after the second passaging (Fig. 4B; comparing red and green dotted lines with the blue dotted line), suggesting that Glis3 and CD133 were needed to maintain self-renewal even in the absence of exogenous RSPO1. This could be due to endogenous production of Wnt ligands, or effects from alternative signaling pathways such as EGF and nicotinamide, which are known to support self-renewal of adult murine PCFUs (40).
When RSPO1 was added, the self-renewal of PCFUs with respect to Glis3 and CD133 knockdowns was restored to the basal level of the shControl-treated cells (blue dotted line) (Fig. 4B, comparing red and green dotted lines with red and green solid lines), demonstrating partial rescues. This result also demonstrated that exogenous RSPO1 did not bypass Glis3 and CD133 to fully activate self-renewal. This is expected because Glis3 and CD133 were needed for the optimal expression of Lgr5 and Lrp6 (Figs. 1F and 2B), two molecules required to initiate RSPO1-mediated signaling (49). Together, these data demonstrate that the effects of Glis3 and CD133 on PCFU self-renewal are mediated through Wnt signaling.
Next, we sought to exclude the possibility that cells in colonies could produce Wnt ligands, which can act as autocrine or paracrine factors to influence Wnt signaling. Porcupine is known to mediate the secretion of Wnt ligands (50). Therefore, sorted CD133highCD71low cells were treated with 1 μm of a porcupine inhibitor, LGK-974 (PORCNi) (51), and plated in Matrigel culture in the absence of exogenous RSPO1 for 3 weeks (Fig. 1D). Gene expression analysis demonstrated that porcupine inhibitor, compared with the vehicle control, did not significantly alter the expression of Wnt target genes Lgr5 or Axin2 in colonies. These results demonstrate that cells in colonies grown in Matrigel without exogenous RSPO1 produce minimal amounts of Wnt ligands. Therefore, these results preclude the possibility that knockdown of Glis3 or CD133 reduces Wnt signaling by inhibiting endogenous Wnt production or secretion.
β-Catenin is necessary for in vitro self-renewal of PCFUs
Canonical Wnt signals through its effector β-catenin (52). Previously, we showed that inhibiting canonical Wnt signaling using a Wnt receptor antagonist, Dkk1, resulted in a decreased PCFU self-renewal (8). To confirm the effect of canonical Wnt on the self-renewal of PCFUs, sorted CD133highCD71low cells were transduced with a pool of five lentiviral vectors carrying shRNAs against β-catenin or shControl and serially-replated as described above. As expected, knockdown of β-catenin, compared with control, reduced the self-renewal of adult PCFUs in vitro (Fig. 5A). Knockdown of β-catenin did not affect PCFU survival (Fig. 5B), similar to the results shown by Glis3 knockdown (Fig. 1C).
Figure 5.
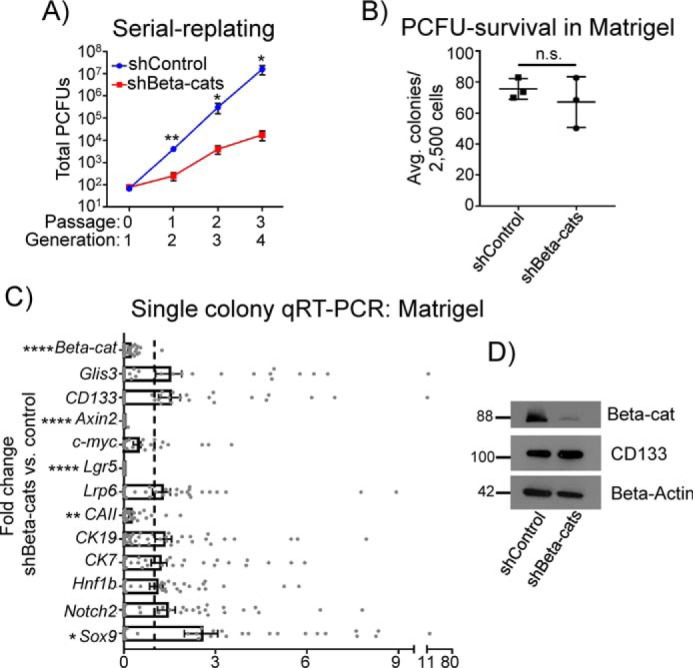
β-Catenin knockdown reduces PCFU self-renewal and Wnt genes, but not CD133 or Glis3. A, self-renewal of PCFUs, assessed by a serial replating strategy, was reduced by knockdown of β-catenin, a canonical Wnt effector. Data are from five pooled biological samples run in three independent experiments with four technical replicates each and are represented as mean ± S.E. B, number of primary (first generation) cystic colonies grown in Matrigel and RSPO1 was not reduced by β-catenin knockdown, suggesting that β-catenin is not required for the survival of PCFUs. Data are from five pooled biological samples run in three independent experiments with four technical replicates each and are represented as mean ± S.E. C, microfluidic qRT-PCR analyses showed that knockdown of β-catenin reduced gene expression in individual cystic colonies of Wnt genes but not Glis3 or CD133. None of the genes displayed were significantly up-regulated. Data represent mean ± S.E. of a total of 48 colonies gathered from three independent experiments each consisting of five pooled biological replicates. D, Western blotting analyses confirmed decreased β-catenin, but not CD133, proteins in cystic colonies treated with shβ-cats, compared with control. β-Actin was used as a loading control. Images are representative of two independent experiments showing similar trends. Molecular weights are indicated. *, p value <0.05; **, p value <0.01; ****, p value <0.0001; n.s., not significant.
The effectiveness of the shRNAs was confirmed using single-colony microfluidic qRT-PCR, which showed the reduced expression of β-catenin in shβ-cats–treated first generation colonies compared with shControl (Fig. 5C). Reduced protein expression of β-catenin was confirmed by Western blot analysis, again confirming knockdown (Fig. 5D). Expression of Axin2 and Lgr5 was reduced in response to β-catenin knockdown (Fig. 5C); however, neither the gene expression of Glis3 or CD133 (Fig. 5C) nor the protein expression of CD133 (Fig. 5D) was changed in colonies treated with shβ-cats, again confirming that Wnt lies downstream of both Glis3 and CD133.
CD133 regulates β-catenin activation
To further interrogate the relationship between CD133 and β-catenin, we tested whether activation of β-catenin was able to rescue the inhibited expression of Lgr5 by CD133 knockdown. Freshly-sorted cells were transduced with shCD133s and grown in the presence of Matrigel and RSPO1 for 3 weeks. A GSK-3β inhibitor, CHIR99021 (53), which inhibits the destruction complex of β-catenin thereby activating Wnt signaling, was added to 3-week-old colonies for 1 day. As expected, CD133 knockdown reduced the gene expression of Lgr5 in colonies grown without CHIR99021 (Fig. 6A, comparing 1st and 3rd columns). Addition of CHIR99021 rescued Lgr5 expression (Fig. 6A, comparing 3rd and 4th columns), suggesting CD133 mediates its effect through β-catenin activation caused by GSK-3β inactivation.
Figure 6.

CD133 co-localizes with β-catenin via E-cadherin and directs activation of β-catenin via inhibiting GSK-3β in 3-week-old cystic colonies grown in Matrigel and RSPO1. A, inhibiting GSK-3β rescued the gene expression of Lrg5 inhibited by shCD133s. PCFUs were transduced with shControl or shCD133s and grown in the presence of Matrigel and RSPO1 for 3 weeks. The 3-week-old colonies were then treated with the GSK3 inhibitor CHIR99021 (5 μm) or DMSO vehicle (0.02%) for 1 day before being pooled and analyzed for gene expression for Lgr5 and CD133 by conventional qRT-PCR. Data represent mean ± S.E. from three experiments. B, CD133 maintained levels of an active form of β-catenin. Western blot analysis showed that shCD133s decreased protein expression of active (nonphosphorylated) β-catenin, unchanged levels of total β-catenin, and unchanged levels of E-cadherin. β-Actin was used as a loading control. Images are representatives of two to three independent experiments showing similar trends. C, co-immunoprecipitation with anti-E-cadherin antibody showed precipitation of β-catenin and CD133, demonstrating co-localization of the three molecules in 3-week-old colonies grown in Matrigel and RSPO1. The 5% input of total sample and mouse IgG1, the isotype of the anti-E-cadherin antibody, were used as controls. Images representative of two independent experiments showing similar trends. *, p value<0.05; **, p value<0.01; n.s., not significant.
To further confirm, we performed Western blotting on total and activated β-catenin in 3-week-old colonies grown in Matrigel and RSPO1. Compared with controls, knockdown of CD133 led to decreased protein levels of β-catenin in the activated form (nonphosphorylated at Ser-33/Ser-37/Thr-41) (Fig. 6B). In contrast, total β-catenin protein levels appeared to be unaffected, although the mRNA level was consistently yet minimally down-regulated (Fig. 2A). These results demonstrate that CD133 is required for β-catenin activation at the protein level.
To test whether CD133 may bind to β-catenin directly, we performed co-immunoprecipitation using antibody against β-catenin. However, CD133 was not present in the complexes being pulled down by anti-β-catenin (data not shown). Previous reports show that β-catenin is capable of binding to E-cadherin at the cell membrane (54). Additionally, CD133 can interact with E-cadherin in expanding primary human renal progenitor cells and function as a permissive factor for canonical Wnt signaling (55). We therefore tested the potential interactions of these molecules. Three-week-old colonies grown in the presence of Matrigel and RSPO1 were lysed, and the protein complexes were pulled down by anti-E-cadherin antibodies followed by Western blot analysis. Both β-catenin and CD133 were found to be associated with E-cadherin (Fig. 6C), demonstrating that E-cadherin, CD133, and β-catenin form a complex. These results demonstrate that, although not in direct contact, CD133 is in close physical proximity to β-catenin via E-cadherin in the 3-week-old colonies.
Discussion
Using FACS-sorted PCFUs from adult murine pancreata and our unique colony assays, we demonstrate here that a Glis3–CD133–Wnt signaling axis regulates PCFU self-renewal. Glis3 (11), CD133 (43), and Wnt (31) have individually been implicated in regulating the self-renewal of stem and progenitor cells in tissues other than the pancreas. The current results, to the best of our knowledge, appear to be the first to delineate the sequence of signaling hierarchy among these molecules in regulating the self-renewal of progenitors. However, we caution that due to technical difficulties, we could not rescue Glis3 knockdown with overexpression of CD133; therefore, the specificity of the effect of Glis3 mediated through CD133 requires further investigation.
This study confirmed the necessity of canonical Wnt signaling for PCFU self-renewal in vitro (Fig. 5A). Previously, the increase of self-renewal by exogenous Wnt ligand (RSPO1) in adult murine PCFUs has been demonstrated in our published studies (8), and the same conclusion was corroborated by other studies (56, 57). In addition to the Wnt findings, the new findings from this study are the roles of Glis3 and CD133 for in vitro self-renewal of adult murine PCFUs.
Our data add to a growing body of literature demonstrating the importance of Glis3 in the self-renewal and maintenance of progenitor cells. As mentioned, a mutation of Glis3 in murine sperm stem cells results in a decreased expression of genes important for self-renewal (i.e. Lhx1, Sall4, and Id4), leading to a severe lack of mature sperm cells (11). Overexpression of Glis3 increases reprogramming efficiency of canonical Yamanaka factors (12), presumably through the up-regulation of FoxA2, Wnt, and the induction of pluripotency genes, similar to those induced by the family member Glis1 (58), which has 94% homology to Glis3 (10, 59).
Here, we show that Glis3 regulates self-renewal of adult murine PCFUs in organoids by changing the gene and protein expression of certain Wnt genes (Figs. 1 and 4). Interestingly, the negative effect of Glis3 knockdown on self-renewal occurred later at the 4th generation, compared with CD133 and β-catenin knockdowns at the 2nd generation. Knockdown of Glis3 down-regulates and up-regulates Wnt-related genes in the 1st and 4th generations, respectively. These contrasting results are likely due to the known pleiotropic effects of Glis3 impacting multiple pathways (60, 61). We speculate that Glis3 fine-tunes its effects over time in a cell-dependent manner: in the earlier PCFUs to sustain the expression of Wnt receptors (e.g. Lgr5 and Lrp6) as well as CD133, and, once the PCFUs are exhausted or in the differentiated ductal cells, inhibiting proliferation via a currently unknown mechanism.
While our manuscript was in revision, Jeon et al. (62) showed that overexpression of Glis3 in human embryonic stem cells (hESCs) causes up-regulation of Wnt ligand genes, such as Wnt3A. This increase was caused by a direct binding of Glis3 to the Wnt3A promoter, demonstrated by ChIP-seq analysis using FLAG-tagged Glis3. In silico analysis utilizing JASPAR motifs and the eukaryotic promoter database demonstrated potential Glis3 consensus-binding sites in promoter regions of CD133, Axin2, Lgr5, and β-catenin (data not shown), genes not reported by Jeon et al. (62). Again, it is possible that the effects of Glis3 are dependent on cellular context, and further analysis will be necessary to compare how Glis3 may differentially affect Wnt genes in our PCFUs versus hESCs.
The role of CD133 in self-renewal and proliferation has been well-documented in tumor-initiating cells (63). Overexpression of CD133 in carcinoma cell lines originating from the mouth, head, and neck increases the expression of stem cell-specific genes, Oct4, Sox2, and Nanog (64, 65). CD133 has also been shown to be necessary for the proliferation and self-renewal of various tumor cells and human embryonic kidney cells through interacting with HDAC6 to influence Wnt signaling (41). Finally, knockdown of CD133 in glioblastoma tumor cells caused a significant decrease in self-renewal (63) demonstrating that in tumor cells CD133 is required for stemness properties.
Despite the wealth of the above-mentioned research in tumor cells, the functional role of CD133 has not been extensively explored in normal murine tissues. In the mouse embryo, CD133 affects neural stem cell (NSC) maintenance through changes to asymmetrical division where NSCs retain and differentiated young neurons lack CD133 (66). This may suggest a positive role for CD133 in self-renewal of embryonic NSCs. In adult hematopoietic stem cells (HSCs), however, CD133 knockdown does not affect the self-renewal of HSCs but leads to an enhanced differentiation of HSCs toward the myeloid, as opposed to the lymphoid, lineage (67). These studies, together with our current results (discussed more below), suggest that the roles of CD133 are likely cell type–specific.
In contrast to hESCs (68), the colonies grown in our standard Matrigel assay (without exogenous RSPO1) do not appear to secrete endogenous Wnt ligands (Fig. 1D), which should prevent a positive feed-forward loop. Importantly, the ratio of phosphorylated Lrp6 to total Lrp6 protein was not changed by either Glis3 or CD133 knockdown, suggesting that Wnt signaling initiation may be triggered without Glis3 or CD133. Additionally, we found that CD133 was required for optimal Lgr5 expression (Figs. 2, A and B, and 6A), as well as maintaining some active form of β-catenin (Fig. 6B), which is known to activate Lgr5 transcription (31). Glis3 maintained the expression of Lrp6 in addition to Lgr5 (Fig. 1, E and F), highlighting overlapping yet divergent effects of Glis3 and CD133 in maintaining the expression of Wnt receptors. Because RSPO1 depends on Lgr5 and Lrp6 for signal transmission (49), these results suggest that Glis3 and CD133 collectively endow PCFUs the ability to respond to external Wnt stimuli by maintaining the expression of these Wnt receptors.
To the best of our knowledge, our study is the first to demonstrate the physical association of CD133 with β-catenin via E-cadherin in primary murine cells. This association may suggest functional interactions between CD133 and β-catenin, although we could not detect a direct physical interaction between these two molecules by co-immunoprecipitation analysis (data not shown). Previously, it has been shown that E-cadherin and β-catenin interact at adherens junctions on the cell membrane to increase cell–cell adhesion (54). When bound to E-cadherin, β-catenin is typically considered “sequestered” and not able to stimulate Wnt signaling (69). Knockdown of CD133 did not affect E-cadherin levels in our cells (Fig. 6B), suggesting that the sequestering mechanism of β-catenin by E-cadherin is in place. However, a switch in β-catenin function from adhesion to Wnt signaling may be regulated by phosphorylating β-catenin at specific tyrosine residues (70, 71). CD133 can act as a scaffold for other serine/threonine kinases, including AKT (43), which has been shown to directly activate β-catenin (43, 72). Additionally, CD133 can directly interact with the tyrosine kinase Src in vitro (73), which has been shown to promote activation of focal adhesion kinase (FAK), leading to β-catenin nuclear localization and murine epidermal cell migration (74). We therefore speculate that certain tyrosine kinases associated with CD133 can play a direct role in activating β-catenin.
Alternatively, CD133-mediated β-catenin activation may be controlled through GSK-3β inhibition. All activities of GSK-3β, including that in the destruction complex for β-catenin, are inhibited by CHIR99021 (53, 75). The observation that CHIR99021, when added to colonies with CD133 knockdown, rescued Wnt target gene expression (Fig. 6A) suggests that CD133 may inhibit GSK-3β, but this hypothesis requires further investigation.
In this study CD133, but not Glis3 or β-catenin, is required for both self-renewal (Fig. 3, A and C) and PI3K/AKT-mediated survival (Fig. 3, B, D, and E), demonstrating the functional importance of CD133 for adult murine PCFUs. However, the fact that only cells that express high levels of CD133 on their cell surface can form colonies, but not every single CD133high cell is capable of forming a colony (Fig. S1, A and B) (26), suggests that CD133 is necessary but not sufficient to confer a PCFU status. Whether Glis3 is a molecule that enables CD133high cells to form colonies needs further investigation.
IPA analyses of Glis3- and CD133-affected genes in 3-week-old colonies grown in Matrigel and RSPO1 revealed interesting biological pathways and upstream regulators (Figs. 1, G and H, and 2, C and D). Cell movement, migration, and NF-κB, a key regulator in immune response (76), are affected by the knockdowns of both Glis3 and CD133, which may implicate the involvement in these processes by their common downstream signal, Wnt. These biological processes are not typically associated with stemness of other organs. However, prior findings did show roles of Wnt in cytoskeletal rearrangement and chemokine expression, which directly affected cell movement and migration in various cell types (77), as well as cross-talk in the inflammatory responses (78). The upstream complex analyses in the CD133-knockdown genes identified the possible involvement of both PI3K/AKT and Stat3 signaling (Fig. S4). However, only the activation of AKT but not Stat3 was affected by CD133 knockdown in colonies grown in Matrigel (Fig. 2B). This result suggests that, although dependent on CD133, AKT signaling affects downstream genes independent of Stat3. It is known that Stat3 can directly affect AKT expression (79), and therefore, the IPA analysis may have indiscriminately identified Stat3 by association. Regardless, the other pathways identified in the IPA analyses will open new avenues to further decipher the biology of adult PCFUs.
In summary, this study reveals a Glis3–CD133–Wnt molecular pathway and its functions in in vitro self-renewal and survival of adult murine PCFUs (Fig. 7). These results may have implications for targeting this signaling axis for maintenance and expansion of adult PCFUs for endogenous neogenesis or cell-replacement therapy of type 1 diabetes, in which insulin-producing beta cells are destroyed by an autoimmune attack (80).
Figure 7.

Proposed model for the Glis3–CD133–Wnt-signaling pathway in adult murine PCFU-derived 3-week-old cystic colonies grown in Matrigel and RSPO1. A, at the resting state, freshly-sorted PCFUs express Glis3 and CD133. Glis3 maintains the levels of Wnt receptors Lgr5 and Lrp6 in colonies, making the cells permissive to further receive and respond to RSPO1. Glis3 also maintains the expression of CD133 in colonies. CD133, E-cadherin, and β-catenin form a complex, potentially at the adherens junction (54). CD133 sustains the levels of the activated AKT (p-AKT), thereby conserving the activated form of β-catenin. In the literature, p-AKT, a serine/threonine kinase, is known to be capable of directly activating β-catenin (72). Literature also suggests that CD133 may directly bind to certain tyrosine kinases, such as Src in in vitro binding assays (73). Src indirectly activates β-catenin at tyrosine residues via FAK that allows β-catenin to switch roles from adhesion to a transcriptionally-active form (74). CD133 may also inhibit the activities of GSK-3β, a molecule in the destruction complex for β-catenin, thereby activating β-catenin. The activated β-catenin translocates into the nucleus (52), which maintains the gene expression of Wnt-signaling molecules, such as Lrg5, and eventually self-renewal of PCFUs. B, for PCFU survival, CD133 sustains the activation of AKT, most likely through its interactions with PI3K (43). Literature has shown that AKT activation leads to increased expression of survival-associated genes (45, 46), which in turn should allow PCFUs to survive and form colonies.
Experimental procedures
Mice
C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) aged 8–12 weeks old from both sexes were used in this study. All mice were maintained under specific pathogen-free conditions, and animal experiments were conducted according to the Institutional Animal Care and Use Committee at the City of Hope. All procedures and protocols were approved by the Institutional Animal Care and Use Committee at the City of Hope.
Dissociation of pancreas into single cells
Murine pancreata were dissected, cleared of fat tissue under a dissecting microscope, and rinsed three times in cold Dulbecco's PBS (DPBS) containing 0.1% bovine serum albumin (BSA), 100 units/ml penicillin, and 100 μg/ml streptomycin; this wash solution is referred to as PBS/BSA. Pancreata were minced in a dry Petri dish placed on ice using spring scissors for 3 min or until finely minced. The tissue was transferred to a 50-ml conical tube and resuspended in PBS/BSA containing collagenase B (2–4 mg/ml) (Roche Applied Science, Mannheim, Germany) and DNase I (2000 units/ml) (Calbiochem, Darmstadt, Germany). Tissue was incubated at 37 °C for 16 min with swirling every 2–3 min and gently passed through a 16-gauge syringe needle every 8 min. Cells were then washed in cold PBS/BSA supplemented with 2000 units/ml DNase I and successively passed through a 100- and a 40-μm mesh filter (BD Biosciences) to yield a mostly single cell suspension.
Cell sorting
Sorting was performed similarly to our previous publication (9). Briefly, dissociated pancreatic cells were first incubated with anti-mouse CD16/32 (10 μg/ml) (BioLegend, San Diego) for 5 min on ice to diminish nonspecific binding. Biotin-conjugated anti-mouse CD133 (clone 13A4; 5 μg/ml; eBioscience, San Diego) and phycoerythrin/Cy7 (PECy7)-conjugated anti-mouse CD71 (clone RI7217; 5 μg/ml; BioLegend, San Diego) antibodies were added. Cells were incubated for 20 min on ice, washed twice, then treated with streptavidin-labeled allophycocyanin (2 μg/ml BioLegend, San Diego) for 15 min on ice, washed twice and resuspended in PBS/BSA/DNase I containing DAPI (0.2 μg/ml). Control antibodies were biotin-conjugated rat IgG1 (5 μg/ml; eBioscience, San Diego) and PE/Cy7-conjugated rat IgG1 (5 μg/ml; BioLegend, San Diego). Acquired flow cytometry data were analyzed with software provided by FlowJo (TreeStar, Ashland, OR). Cell sorting was performed on an Aria special-order research product (BD Biosciences). All analyses included an initial gating of forward and side scatters to exclude debris. In cell-sorting experiments, doublets were further excluded by gating out high pulse-width cells, and live cells were selected by DAPI-negative staining (Fig. S5).
Colony assay
Sorted cells were resuspended at a density of 2.5 × 103 cells/well/0.5 ml for the Matrigel assay or 2.5 × 104 cells/well/0.5 ml for the laminin-hydrogel assay as described previously (8). Culture media contained DMEM/F-12 media, 1% methylcellulose (Sinetsu Chemical, Tokyo, Japan), 50% conditioned media from mouse embryonic stem cell-derived pancreatic-like cells (81), 5% fetal calf serum (FCS), 10 mmol/liter nicotinamide (Sigma), 10 ng/ml human recombinant activin B (R&D Systems, Minneapolis, MN), 0.1 nmol/liter exendin-4 (Sigma), 1 ng/ml vascular endothelial growth factor-A (Sigma), and 750 ng/ml R-spondin-1 (R&D Systems, Minneapolis, MN). When indicated, either 5% (v/v) Matrigel (referred to as the standard assay in this study) or 100 μg/ml laminin hydrogel (8) was added to the media for the generation of cystic or endocrine/acinar colonies, respectively. Cells were plated in 24-well ultra-low protein-binding plates (Corning, New York) and incubated in a humidified 5% CO2 atmosphere at 37 °C. Colonies grown in Matrigel or laminin hydrogel were counted 3 weeks or 10 days after plating, respectively.
Colony dissociation and replating
Warmed PBS/BSA (1 ml per well) was added to each well of a 24-well plate containing colonies. Cystic colonies were collected in a 50-ml conical tube, washed, resuspended in 10 ml of 2–4 mg/ml collagenase B, incubated for 15 min at 37 °C with mixing every 5 min, and washed in PBS/BSA. Subsequently, colonies were treated with 20 ml of 0.25% (w/v) trypsin-EDTA, incubated for 3 min at 37 °C, and pipetted thoroughly to generate mostly single cells. Warmed FCS (4 ml) was added to the cells to stop the trypsin digestion. Cells were washed in PBS/BSA and kept at room temperature. A portion was mixed with 0.02% (w/v) trypan blue, and the concentration of live cells (i.e. the trypan blue–negative cells) was determined by a hemocytometer. For replating experiments, the final single cell suspension was mixed with medium containing Matrigel or laminin hydrogel as indicated.
Lentivirus transduction
Lentiviral particles containing shRNAs against control (catalog no. SHC002V), Glis3 (SHCLNV-NM_175459), Prominin-1 (CD133) (SHCLNV-NM_008935), or CTNNB1 (β-catenin) (SHCLNV-NM_007614) were purchased (Sigma). Five independent clones of shRNAs were pooled with equal ratios. Freshly-sorted cells were seeded at a concentration of 100,000 cells/96-well plate/100 μl in a liquid medium (i.e. colony assay medium minus Matrigel, laminin hydrogel, RSPO1, and methylcellulose) in a flat-bottom low-attachment plate (Thermo Fisher Scientific, Waltham, MA) and incubated at 37 °C for 4 h. Viral particles were added at a mode of infection of 20. The final volumes were brought to 300 μl/well using DMEM/F-12 containing penicillin/streptomycin and 4 μg/ml hexadimethrine bromide (polybrene) (Sigma). Cells were incubated overnight at 37 °C, washed twice with warmed PBS/BSA, counted, and plated into a colony assay medium. Puromycin (2 μg/ml) was added to each well to select for infected cells.
Conventional or microfluidic quantitative (q)RT-PCR
For conventional and microfluidic qRT-PCR analyses, the same procedures were employed as reported previously (8). Duplicate samples were used in all analyses. Microfluidic qRT-PCR was performed using the BioMarkTM 48.48 Dynamic Array system (Fluidigm, South San Francisco, CA). Single colonies were lifted one by one from the methylcellulose-containing medium under direct microscopic visualization by using a 10-μl Eppendorf pipette, collected in reaction buffer (10 μl), and followed by preamplification (12 cycles for Matrigel-grown cystic colonies and 18 cycles for E/A colonies) according to the manufacturer's instructions (Fluidigm). Amplified cDNA was loaded onto a 48.48 Dynamic Array system using the NanoFlex integrated fluidic circuit controller (Fluidigm). Threshold cycle (Ct), as a measure of fluorescence intensity, was determined by the BioMark PCR analysis software (Fluidigm) and expressed as ΔCt. All experiments were performed with negative (water) and positive (adult C57BL/6J pancreatic cells) controls. TaqMan probes used in this study are listed in Table S1.
Western blotting
Cells were lysed on ice in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) containing 50 mm Tris, pH 7.4, 150 mm NaCl, 0.1 mm EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, phosphatase inhibitor mixture III (Calbiochem/Merck, Bad Soden, Germany), and complete protease inhibitor (Roche Applied Science, Mannheim, Germany). Extracts were sedimented at 10,000 × g for 15 min at 4 °C to remove insoluble material. Protein concentrations were determined using the Pierce® BCA protein assay (Thermo Fisher Scientific). Samples of proteins (10 μg) were boiled in 1× sample buffer, separated by SDS-PAGE on a 10% Laemmli gel, transferred to a polyvinylidene difluoride membrane (Bio-Rad), incubated with primary and secondary antibodies, and developed using enhanced luminol chemiluminescence (ECL-kit; Bio-Rad). Images were detected using Crystal Blue X-ray film (Bio-Rad). Antibodies used in Western blotting are listed in Table S2.
Co-immunoprecipitation
Three-week-old colonies were hand-picked, pooled, and lysed on ice in Pierce® IP lysis buffer (Thermo Fisher Scientific) containing phosphatase inhibitor mixture III (Calbiochem/Merck, Bad Soden, Germany) and complete protease inhibitor (Roche Applied Science, Mannheim, Germany) for 30 min with gentle pipetting every 10 min. Protein concentration was measured using the Pierce® BCA protein assay (Thermo Fisher Scientific). Lysates were pre-cleared using protein G magnetic beads (Cell Signaling Technology, Danvers, MA) for 1 h at 4 °C with rotation. The beads were pelleted via magnetic separation rack and the lysate was removed. Primary antibodies were added to the pre-cleared lysates overnight with rotation at 4 °C. The next day, protein G magnetic beads were added for 1 h at 4 °C with rotation, and the beads were pelleted using a magnetic separation rack. Beads containing pulled down proteins were then washed five times with lysis buffer and boiled at 95 °C for 15 min in 3× sample buffer. Samples were brought to 1× sample buffer and then run on SDS-PAGE gel for subsequent Western blot analysis.
Proliferation and apoptosis analyses
For proliferation assay, Click-iT EdU (Thermo Fisher Scientific) was added to cells grown in laminin hydrogel-containing colony assay, and 10 days after plating, individual E/A colonies were hand-picked, pooled, and fixed with 4% paraformaldehyde for 20 min at room temperature. Colonies were washed and stored at 4 °C in PBS containing 0.01% Triton X-100. The Alexa Fluor 594 imagine kit (Thermo Fisher Scientific) was used to visualize EdU+ cells. For apoptosis assay, fixed 10-day-old E/A colonies were stained using the Click-iT TUNEL Alexa Fluor 488 imagine kit (Thermo Fisher Scientific) according to the manufacturer's instructions. For imaging, E/A colonies were resuspended in Hoechst solution, placed in 35-mm glass-bottom dishes, and serially imaged using the z-stack function on a Zeiss 700 LSM. Individual nuclei positive for Hoechst were analyzed for the presence of EdU or TUNEL, and the fraction of positive nuclei was recorded.
mRNA sequencing
Each biological sample submitted for RNA-Seq was collected from a total of five pancreata that had previously been dissociated into single cell suspension, sorted for CD133highCD71low cells, infected with lentivirus carrying either shControl, shGlis3s, or shCD133s, and plated into a standard Matrigel-containing colony assay in the presence of exogenous RSPO1. The resulting 3-week-old cystic colonies were pooled and prepared for sequencing. Two biological replicates were used from each knockdown. cDNA libraries were prepared with Kapa Stranded mRNA-seq kit (KAPA Biosystems, Wilmington, MA) according to the manufacturer's protocol with minor modifications. Briefly, polyadenylated RNAs were enriched from 500 ng of total RNAs using oligo(dT) magnetic beads, fragmented with divalent cations under elevated temperature, and reverse-transcribed into the first-strand followed by the second-strand cDNAs. Subsequently, the double-stranded cDNAs underwent end repair and 3′-end adenylation, ligated with barcoded adaptors, and amplified by PCR (12 cycles). The cDNA libraries were validated by the Agilent Bioanalyzer (Agilent) and prepared for sequencing using cBot cluster generation system (Illumina) with HiSeq SR Cluster Version 4 kit (Illumina). Using the HiSeq 2500 platform with HiSeq SBS Version 4 kits (Illumina), the sequencing run was performed in the single read mode with 51 cycles of “read1” and seven cycles of “index read.” Real-time analysis 2.2.38 software (Illumina) was used to process the image analysis and base calling.
Data analysis for mRNA sequencing
Raw sequence reads were mapped to the mouse genome (mm10) using TopHat (82), and the frequency of Refseq genes was counted using HTseq. The raw counts were then normalized using the method of trimmed mean of M values (TMM) and compared using Bioconductor package “edgeR”. Reads per kilobase per million (RPKM) were also calculated from the raw counts. Differentially expressed genes were identified if the RPKM were ≥1 in at least one sample, fold-change of ≥2, and p ≤ 0.05. These differential genes were then imported into Ingenuity Pathway Analysis for functional pathway analysis. Raw RNA-Seq results were deposited on the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) under the ID code GSE124944.
Statistical analysis
All values shown are represented as either mean ± S.D. or mean ± S.E. p values were calculated using Student's two-tailed t test; p < 0.05 was considered significant.
Author contributions
J. R. T. and H. T. K. conceptualization; J. R. T. and K. L. data curation; J. R. T. and H. T. K. formal analysis; J. R. T. methodology; J. R. T. writing-original draft; J. R. T. and H. T. K. writing-review and editing; K. L. validation; H. T. K. supervision; H. T. K. funding acquisition.
Supplementary Material
Acknowledgments
We thank Lucy Brown and Alex Spalla for cell-sorting assistance and the integrative genomics core for RNA-seq analysis. We thank Dr. Yisheng Yang, Dr. Janice Huss, Dr. Rama Natarajan, and Dr. Arthur Riggs for scientific discussion.
This work was supported in part by National Institutes of Health Grant R01DK099734 and a grant from Wanek Family Project of Type 1 Diabetes (to H. T. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5, Tables S1 and S2, and supporting Ref. 1.
RNA-Seq results were deposited on the Gene Expression Omnibus (GEO) database under code GSE124944.
- PCFU
- pancreatic colony-forming unit
- PI3K
- phosphoinositide 3-kinase
- AKT
- Ser/Thr kinase
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- qRT
- quantitative PCR
- EdU
- 5-ethynyl-2′-deoxyuridine
- TUNEL
- deoxynucleotidyltransferase-mediated dUTP nick end-labeling
- FAK
- focal adhesion kinase
- DAPI
- 4′,6-diamidino-2-phenylindole
- E/A
- endocrine/acinar
- IPA
- Ingenuity Pathway Analysis
- shβ-cats
- shRNAs against β-catenin
- NSC
- neural stem cell
- HSC
- hematopoietic stem cell
- hESC
- human embryonic stem cell
- RPKM
- reads per kilobase per million
- PE
- phycoerythrin.
References
- 1. Blanpain C., Horsley V., and Fuchs E. (2007) Epithelial stem cells: turning over new leaves. Cell 128, 445–458 10.1016/j.cell.2007.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sancho R., Gruber R., Gu G., and Behrens A. (2014) Loss of Fbw7 reprograms adult pancreatic ductal cells into α, δ, and β cells. Cell Stem Cell 15, 139–153 10.1016/j.stem.2014.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu X., D'Hoker J., Stangé G., Bonné S., De Leu N., Xiao X., Van de Casteele M., Mellitzer G., Ling Z., Pipeleers D., Bouwens L., Scharfmann R., Gradwohl G., and Heimberg H. (2008) Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197–207 10.1016/j.cell.2007.12.015 [DOI] [PubMed] [Google Scholar]
- 4. Criscimanna A., Speicher J. A., Houshmand G., Shiota C., Prasadan K., Ji B., Logsdon C. D., Gittes G. K., and Esni F. (2011) Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 141, 1451–1462, 1462.e1–6 10.1053/j.gastro.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kopp J. L., Dubois C. L., Schaffer A. E., Hao E., Shih H. P., Seymour P. A., Ma J., and Sander M. (2011) Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138, 653–665 10.1242/dev.056499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dor Y., Brown J., Martinez O. I., and Melton D. A. (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 10.1038/nature02520 [DOI] [PubMed] [Google Scholar]
- 7. Xiao X., Chen Z., Shiota C., Prasadan K., Guo P., El-Gohary Y., Paredes J., Welsh C., Wiersch J., and Gittes G. K. (2013) No evidence for beta cell neogenesis in murine adult pancreas. J. Clin. Invest. 123, 2207–2217 10.1172/JCI66323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin L., Feng T., Shih H. P., Zerda R., Luo A., Hsu J., Mahdavi A., Sander M., Tirrell D. A., Riggs A. D., and Ku H. T. (2013) Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc. Natl. Acad. Sci. U.S.A. 110, 3907–3912 10.1073/pnas.1301889110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin L., Gao D., Feng T., Tremblay J. R., Ghazalli N., Luo A., Rawson J., Quijano J. C., Chai J., Wedeken L., Hsu J., LeBon J., Walker S., Shih H. P., Mahdavi A., et al. (2016) Cells with surface expression of CD133(high)CD71(low) are enriched for tripotent colony-forming progenitor cells in the adult murine pancreas. Stem Cell Res. 16, 40–53 10.1016/j.scr.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim Y. S., Nakanishi G., Lewandoski M., and Jetten A. M. (2003) GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 31, 5513–5525 10.1093/nar/gkg776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang H. S., Chen L. Y., Lichti-Kaiser K., Liao G., Gerrish K., Bortner C. D., Yao H. H., Eddy E. M., and Jetten A. M. (2016) Transcription factor GLIS3: a new and critical regulator of postnatal stages of mouse spermatogenesis. Stem Cells 34, 2772–2783 10.1002/stem.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee S. Y., Noh H. B., Kim H. T., Lee K. I., and Hwang D. Y. (2017) Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget 8, 77041–77049 10.18632/oncotarget.20334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senée V., Chelala C., Duchatelet S., Feng D., Blanc H., Cossec J. C., Charon C., Nicolino M., Boileau P., Cavener D. R., Bougnères P., Taha D., and Julier C. (2006) Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 38, 682–687 10.1038/ng1802 [DOI] [PubMed] [Google Scholar]
- 14. Wen X., and Yang Y. (2017) Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J. Mol. Endocrinol. 58, R73–R85 10.1530/JME-16-0232 [DOI] [PubMed] [Google Scholar]
- 15. Nogueira T. C., Paula F. M., Villate O., Colli M. L., Moura R. F., Cunha D. A., Marselli L., Marchetti P., Cnop M., Julier C., and Eizirik D. L. (2013) GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 9, e1003532 10.1371/journal.pgen.1003532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dimitri P., Habeb A. M., Garbuz F., Millward A., Wallis S., Moussa K., Akcay T., Taha D., Hogue J., Slavotinek A., Wales J. K., Shetty A., Hawkes D., Hattersley A. T., Ellard S., and De Franco E. (2015) Expanding the clinical spectrum associated with GLIS3 mutations. J. Clin. Endocrinol. Metab. 100, E1362–E1369 10.1210/jc.2015-1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dimitri P., Warner J. T., Minton J. A., Patch A. M., Ellard S., Hattersley A. T., Barr S., Hawkes D., Wales J. K., and Gregory J. W. (2011) Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur. J. Endocrinol. 164, 437–443 10.1530/EJE-10-0893 [DOI] [PubMed] [Google Scholar]
- 18. Kang H. S., Kim Y. S., ZeRuth G., Beak J. Y., Gerrish K., Kilic G., Sosa-Pineda B., Jensen J., Pierreux C. E., Lemaigre F. P., Foley J., and Jetten A. M. (2009) Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 29, 6366–6379 10.1128/MCB.01259-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watanabe N., Hiramatsu K., Miyamoto R., Yasuda K., Suzuki N., Oshima N., Kiyonari H., Shiba D., Nishio S., Mochizuki T., Yokoyama T., Maruyama S., Matsuo S., Wakamatsu Y., and Hashimoto H. (2009) A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 583, 2108–2113 10.1016/j.febslet.2009.05.039 [DOI] [PubMed] [Google Scholar]
- 20. Kang H. S., Takeda Y., Jeon K., and Jetten A. M. (2016) The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta, PP and ductal cells. PloS One 11, e0157138 10.1371/journal.pone.0157138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim Y. S., Kang H. S., Takeda Y., Hom L., Song H. Y., Jensen J., and Jetten A. M. (2012) Glis3 regulates neurogenin 3 expression in pancreatic beta-cells and interacts with its activator, Hnf6. Mol. Cells 34, 193–200 10.1007/s10059-012-0109-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tremblay J. R., LeBon J. M., Luo A., Quijano J. C., Wedeken L., Jou K., Riggs A. D., Tirrell D. A., and Ku H. T. (2016) In vitro colony assays for characterizing tri-potent progenitor cells isolated from the adult murine pancreas. J. Vis. Exp. 112 10.3791/54016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Y., Chang B. H., Yechoor V., Chen W., Li L., Tsai M. J., and Chan L. (2011) The Kruppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia 54, 2595–2605 10.1007/s00125-011-2255-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Lau W., Peng W. C., Gros P., and Clevers H. (2014) The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 28, 305–316 10.1101/gad.235473.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim K. A., Zhao J., Andarmani S., Kakitani M., Oshima T., Binnerts M. E., Abo A., Tomizuka K., and Funk W. D. (2006) R-Spondin proteins: a novel link to β-catenin activation. Cell Cycle 5, 23–26 10.4161/cc.5.1.2305 [DOI] [PubMed] [Google Scholar]
- 26. Jin L., Feng T., Zerda R., Chen C. C., Riggs A. D., and Ku H. T. (2014) In vitro multilineage differentiation and self-renewal of single pancreatic colony-forming cells from adult C57Bl/6 mice. Stem Cells Dev. 23, 899–909 10.1089/scd.2013.0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. ZeRuth G. T., Takeda Y., and Jetten A. M. (2013) The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol. 27, 1692–1705 10.1210/me.2013-1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Y., Chang B. H., and Chan L. (2013) Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol. Med. 5, 92–104 10.1002/emmm.201201398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Behrens J., von Kries J. P., Kühl M., Bruhn L., Wedlich D., Grosschedl R., and Birchmeier W. (1996) Functional interaction of β-catenin with the transcription factor LEF-1. Nature 382, 638–642 10.1038/382638a0 [DOI] [PubMed] [Google Scholar]
- 30. Jho E. H., Zhang T., Domon C., Joo C. K., Freund J. N., and Costantini F. (2002) Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 10.1128/MCB.22.4.1172-1183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barker N., van Es J. H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P. J., and Clevers H. (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 10.1038/nature06196 [DOI] [PubMed] [Google Scholar]
- 32. Liu G., Bafico A., Harris V. K., and Aaronson S. A. (2003) A novel mechanism for Wnt activation of canonical signaling through the LRP6 receptor. Mol. Cell. Biol. 23, 5825–5835 10.1128/MCB.23.16.5825-5835.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Herbst F., Ball C. R., Tuorto F., Nowrouzi A., Wang W., Zavidij O., Dieter S. M., Fessler S., van der Hoeven F., Kloz U., Lyko F., Schmidt M., von Kalle C., and Glimm H. (2012) Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol. Ther. 20, 1014–1021 10.1038/mt.2012.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang S., Li Y., Wu Y., Shi K., Bing L., and Hao J. (2012) Wnt/β-catenin signaling pathway upregulates c-Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat. Rec. 295, 2104–2113 10.1002/ar.22592 [DOI] [PubMed] [Google Scholar]
- 35. Richardson G. D., Robson C. N., Lang S. H., Neal D. E., Maitland N. J., and Collins A. T. (2004) CD133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 117, 3539–3545 10.1242/jcs.01222 [DOI] [PubMed] [Google Scholar]
- 36. Horn P. A., Tesch H., Staib P., Kube D., Diehl V., and Voliotis D. (1999) Expression of AC133, a novel hematopoietic precursor antigen, on acute myeloid leukemia cells. Blood 93, 1435–1437 [PubMed] [Google Scholar]
- 37. Sanai N., Alvarez-Buylla A., and Berger M. S. (2005) Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811–822 10.1056/NEJMra043666 [DOI] [PubMed] [Google Scholar]
- 38. Ghazalli N., Mahdavi A., Feng T., Jin L., Kozlowski M. T., Hsu J., Riggs A. D., Tirrell D. A., and Ku H. T. (2015) Postnatal pancreas of mice contains tripotent progenitors capable of giving rise to duct, acinar, and endocrine cells in vitro. Stem Cells Dev. 24, 1995–2008 10.1089/scd.2015.0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jin L., Feng T., Chai J., Ghazalli N., Gao D., Zerda R., Li Z., Hsu J., Mahdavi A., Tirrell D. A., Riggs A. D., and Ku H. T. (2014) Colony-forming progenitor cells in the postnatal mouse liver and pancreas give rise to morphologically distinct insulin-expressing colonies in 3D cultures. Rev. Diabet. Stud. 11, 35–50 10.1900/RDS.2014.11.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wedeken L., Luo A., Tremblay J. R., Rawson J., Jin L., Gao D., Quijano J., and Ku H. T. (2017) Adult murine pancreatic progenitors require epidermal growth factor and nicotinamide for self-renewal and differentiation in a serum- and conditioned medium-free culture. Stem Cells Dev. 26, 599–607 10.1089/scd.2016.0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mak A. B., Nixon A. M., Kittanakom S., Stewart J. M., Chen G. I., Curak J., Gingras A. C., Mazitschek R., Neel B. G., Stagljar I., and Moffat J. (2012) Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2, 951–963 10.1016/j.celrep.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rappa G., Mercapide J., Anzanello F., Le T. T., Johlfs M. G., Fiscus R. R., Wilsch-Bräuninger M., Corbeil D., and Lorico A. (2013) Wnt interaction and extracellular release of prominin-1/CD133 in human malignant melanoma cells. Exp. Cell Res. 319, 810–819 10.1016/j.yexcr.2013.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wei Y., Jiang Y., Zou F., Liu Y., Wang S., Xu N., Xu W., Cui C., Xing Y., Liu Y., Cao B., Liu C., Wu G., Ao H., Zhang X., and Jiang J. (2013) Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc. Natl. Acad. Sci. U.S.A. 110, 6829–6834 10.1073/pnas.1217002110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shimozato O., Waraya M., Nakashima K., Souda H., Takiguchi N., Yamamoto H., Takenobu H., Uehara H., Ikeda E., Matsushita S., Kubo N., Nakagawara A., Ozaki T., and Kamijo T. (2015) Receptor-type protein tyrosine phosphatase κ directly dephosphorylates CD133 and regulates downstream AKT activation. Oncogene 34, 1949–1960 10.1038/onc.2014.141 [DOI] [PubMed] [Google Scholar]
- 45. Hossini A. M., Quast A. S., Plötz M., Grauel K., Exner T., Küchler J., Stachelscheid H., Eberle J., Rabien A., Makrantonaki E., and Zouboulis C. C. (2016) PI3K/AKT signaling pathway is essential for survival of induced pluripotent stem cells. PLoS One 11, e0154770 10.1371/journal.pone.0154770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Ito T., Udaka N., Okudela K., Yazawa T., and Kitamura H. (2005) PI3K-AKT pathway mediates growth and survival signals during development of fetal mouse lung. Tissue Cell 37, 25–35 10.1016/j.tice.2004.09.002 [DOI] [PubMed] [Google Scholar]
- 47. Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., and Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 10.1016/S0960-9822(06)00122-9 [DOI] [PubMed] [Google Scholar]
- 48. Hirano T., Ishihara K., and Hibi M. (2000) Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 19, 2548–2556 10.1038/sj.onc.1203551 [DOI] [PubMed] [Google Scholar]
- 49. Pinson K. I., Brennan J., Monkley S., Avery B. J., and Skarnes W. C. (2000) An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 407, 535–538 10.1038/35035124 [DOI] [PubMed] [Google Scholar]
- 50. Biechele S., Cox B. J., and Rossant J. (2011) Porcupine homolog is required for canonical Wnt signaling and gastrulation in mouse embryos. Dev. Biol. 355, 275–285 10.1016/j.ydbio.2011.04.029 [DOI] [PubMed] [Google Scholar]
- 51. Han T., Schatoff E. M., Murphy C., Zafra M. P., Wilkinson J. E., Elemento O., and Dow L. E. (2017) R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 8, 15945 10.1038/ncomms15945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Molenaar M., van de Wetering M., Oosterwegel M., Peterson-Maduro J., Godsave S., Korinek V., Roose J., Destrée O., and Clevers H. (1996) XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399 10.1016/S0092-8674(00)80112-9 [DOI] [PubMed] [Google Scholar]
- 53. Ring D. B., Johnson K. W., Henriksen E. J., Nuss J. M., Goff D., Kinnick T. R., Ma S. T., Reeder J. W., Samuels I., Slabiak T., Wagman A. S., Hammond M. E., and Harrison S. D. (2003) Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 52, 588–595 10.2337/diabetes.52.3.588 [DOI] [PubMed] [Google Scholar]
- 54. Nelson W. J., and Nusse R. (2004) Convergence of Wnt, β-catenin, and cadherin pathways. Science 303, 1483–1487 10.1126/science.1094291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brossa A., Papadimitriou E., Collino F., Incarnato D., Oliviero S., Camussi G., and Bussolati B. (2018) Role of CD133 molecule in Wnt response and renal repair. Stem Cells Transl. Med. 7, 283–294 10.1002/sctm.17-0158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huch M., Bonfanti P., Boj S. F., Sato T., Loomans C. J., van de Wetering M., Sojoodi M., Li V. S., Schuijers J., Gracanin A., Ringnalda F., Begthel H., Hamer K., Mulder J., van Es J. H., et al. (2013) Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 32, 2708–2721 10.1038/emboj.2013.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dorrell C., Tarlow B., Wang Y., Canaday P. S., Haft A., Schug J., Streeter P. R., Finegold M. J., Shenje L. T., Kaestner K. H., and Grompe M. (2014) The organoid-initiating cells in mouse pancreas and liver are phenotypically and functionally similar. Stem Cell Res. 13, 275–283 10.1016/j.scr.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maekawa M., Yamaguchi K., Nakamura T., Shibukawa R., Kodanaka I., Ichisaka T., Kawamura Y., Mochizuki H., Goshima N., and Yamanaka S. (2011) Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474, 225–229 10.1038/nature10106 [DOI] [PubMed] [Google Scholar]
- 59. Kim Y. S., Lewandoski M., Perantoni A. O., Kurebayashi S., Nakanishi G., and Jetten A. M. (2002) Identification of Glis1, a novel Gli-related, Kruppel-like zinc finger protein containing transactivation and repressor functions. J. Biol. Chem. 277, 30901–30913 10.1074/jbc.M203563200 [DOI] [PubMed] [Google Scholar]
- 60. Jetten A. M. (2018) GLIS1–3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell. Mol. Life Sci. 75, 3473–3494 10.1007/s00018-018-2841-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rurale G., Persani L., and Marelli F. (2018) GLIS3 and thyroid: a pleiotropic candidate gene for congenital hypothyroidism. Front. Endocrinol. 9, 730 10.3389/fendo.2018.00730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jeon K., Kumar D., Conway A. E., Park K., Jothi R., and Jetten A. M. (2019) GLIS3 transcriptionally activates WNT genes to promote differentiation of human embryonic stem cells into posterior neural progenitors. Stem Cells 37, 202–215 10.1002/stem.2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brescia P., Ortensi B., Fornasari L., Levi D., Broggi G., and Pelicci G. (2013) CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 31, 857–869 10.1002/stem.1317 [DOI] [PubMed] [Google Scholar]
- 64. Moon Y., Kim D., Sohn H., and Lim W. (2016) Effect of CD133 overexpression on the epithelial–to-mesenchymal transition in oral cancer cell lines. Clin. Exp. Metastasis 33, 487–496 10.1007/s10585-016-9793-y [DOI] [PubMed] [Google Scholar]
- 65. Lee J., Park M., Ko Y., Kim B., Kim O., Hyun H., Kim D., Sohn H., Moon Y. L., and Lim W. (2017) Ectopic overexpression of CD133 in HNSCC makes it resistant to commonly used chemotherapeutics. Tumour Biol. 39, 1010428317695534 10.1177/1010428317695534 [DOI] [PubMed] [Google Scholar]
- 66. Kosodo Y., Röper K., Haubensak W., Marzesco A. M., Corbeil D., and Huttner W. B. (2004) Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 23, 2314–2324 10.1038/sj.emboj.7600223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arndt K., Grinenko T., Mende N., Reichert D., Portz M., Ripich T., Carmeliet P., Corbeil D., and Waskow C. (2013) CD133 is a modifier of hematopoietic progenitor frequencies but is dispensable for the maintenance of mouse hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 110, 5582–5587 10.1073/pnas.1215438110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. ten Berge D., Kurek D., Blauwkamp T., Koole W., Maas A., Eroglu E., Siu R. K., and Nusse R. (2011) Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat. Cell Biol. 13, 1070–1075 10.1038/ncb2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tang Y., Liu Z., Zhao L., Clemens T. L., and Cao X. (2008) Smad7 stabilizes β-catenin binding to E-cadherin complex and promotes cell-cell adhesion. J. Biol. Chem. 283, 23956–23963 10.1074/jbc.M800351200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brembeck F. H., Schwarz-Romond T., Bakkers J., Wilhelm S., Hammerschmidt M., and Birchmeier W. (2004) Essential role of BCL9–2 in the switch between β-catenin's adhesive and transcriptional functions. Genes Dev. 18, 2225–2230 10.1101/gad.317604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Taddei M. L., Chiarugi P., Cirri P., Buricchi F., Fiaschi T., Giannoni E., Talini D., Cozzi G., Formigli L., Raugei G., and Ramponi G. (2002) β-Catenin interacts with low-molecular-weight protein tyrosine phosphatase leading to cadherin-mediated cell-cell adhesion increase. Cancer Res. 62, 6489–6499 [PubMed] [Google Scholar]
- 72. Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G. B., Kobayashi R., Hunter T., and Lu Z. (2007) Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 10.1074/jbc.M611871200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu C., Li Y., Xing Y., Cao B., Yang F., Yang T., Ai Z., Wei Y., and Jiang J. (2016) The interaction between cancer stem cell marker CD133 and Src protein promotes focal adhesion kinase (FAK) phosphorylation and cell migration. J. Biol. Chem. 291, 15540–15550 10.1074/jbc.M115.712976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ridgway R. A., Serrels B., Mason S., Kinnaird A., Muir M., Patel H., Muller W. J., Sansom O. J., and Brunton V. G. (2012) Focal adhesion kinase is required for β-catenin-induced mobilization of epidermal stem cells. Carcinogenesis 33, 2369–2376 10.1093/carcin/bgs284 [DOI] [PubMed] [Google Scholar]
- 75. Chen E. Y., DeRan M. T., Ignatius M. S., Grandinetti K. B., Clagg R., McCarthy K. M., Lobbardi R. M., Brockmann J., Keller C., Wu X., and Langenau D. M. (2014) Glycogen synthase kinase 3 inhibitors induce the canonical WNT/β-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 111, 5349–5354 10.1073/pnas.1317731111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lawrence T. (2009) The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651 10.1101/cshperspect.a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sedgwick A. E., and D'Souza-Schorey C. (2016) Wnt signaling in cell motility and invasion: drawing parallels between development and cancer. Cancers 8, E80 10.3390/cancers8090080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma B., and Hottiger M. O. (2016) Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front. Immunol. 7, 378 10.3389/fimmu.2016.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carpenter R. L., and Lo H. W. (2014) STAT3 target genes relevant to human cancers. Cancers 6, 897–925 10.3390/cancers6020897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Atkinson M. A. (2012) The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2, a007641 10.1101/cshperspect.a007641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Winkler M., Trieu N., Feng T., Jin L., Walker S., Singh L., and Ku H. T. (2011) A quantitative assay for insulin-expressing colony-forming progenitors. J. Vis. Exp. 2011, e3148 10.3791/3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Trapnell C., Pachter L., and Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.