

Abstract
Epithelial Na+ channel (ENaC)-mediated Na+ transport has a key role in the regulation of extracellular fluid volume, blood pressure, and extracellular [K+]. Among the thousands of human ENaC variants, only a few exist whose functional consequences have been experimentally tested. Here, we used the Xenopus oocyte expression system to investigate the functional roles of four nonsynonymous human ENaC variants located within the β7-strand and its adjacent loop of the α-subunit extracellular β-ball domain. αR350Wβγ and αG355Rβγ channels exhibited 2.5- and 1.8-fold greater amiloride-sensitive currents than WT αβγ human ENaCs, respectively, whereas αV351Aβγ channels conducted significantly less current than WT. Currents in αH354Rβγ-expressing oocytes were similar to those expressing WT. Surface expression levels of three mutants (αR350Wβγ, αV351Aβγ, and αG355Rβγ) were similar to that of WT. However, three mutant channels (αR350Wβγ, αH354Rβγ, and αG355Rβγ) exhibited a reduced Na+ self-inhibition response. Open probability of αR350Wβγ was significantly greater than that of WT. Moreover, other Arg-350 variants, including αR350G, αR350L, and αR350Q, also had significantly increased channel activity. A direct comparison of αR350W and two previously reported gain–of–function variants revealed that αR350W increases ENaC activity similarly to αW493R, but to a much greater degree than does αC479R. Our results indicate that αR350W along with αR350G, αR350L, and αR350Q, and αG355R are novel gain–of–function variants that function as gating modifiers. The location of these multiple functional variants suggests that the αENaC β-ball domain portion that interfaces with the palm domain of βENaC critically regulates ENaC gating.
Keywords: epithelial sodium channel (ENaC), genetic polymorphism, acid-sensing ion channel (ASIC), patch clamp, Xenopus, amiloride, blood pressure, degenerin, Na+ self-inhibition, nonvoltage-gated ion channel
Introduction
The epithelial Na+ channel (ENaC)2 is a member of the ENaC/degenerin family of nonvoltage-gated ion channels. ENaCs are expressed in the apical plasma membranes of specific epithelia and, in parallel with the basolateral Na+,K+-ATPase, mediate the absorption of Na+ from the lumen of the aldosterone-sensitive distal nephron (ASDN), the distal colon, and the airway and alveolae. ENaC-mediated Na+ absorption plays significant roles in the regulation of extracellular fluid volume and blood pressure and fluid volume in airways and alveolae (1–4). ENaC-mediated Na+ absorption is also tightly linked to K+ secretion in the ASDN, and changes in extracellular [K+] influence activity of the Na+–Cl− cotransporter in the distal convoluted tubule and blood pressure (5, 6). Inherited forms of human hypertension or hypotension are largely associated mutations in specific genes that encode either renal tubular Na+ transporters or their regulators (7, 8). Other than well-defined Liddle syndrome mutations that disrupt or result in a loss of a Pro-Tyr (PY) motif in the C terminus of the β- or γ-subunit, correlating nonsynonymous ENaC variants that alter channel activity with predicted changes in blood pressure in humans has been challenging. This may reflect that fact that only two of the common ENaC nonsynonymous variants alter ENaC function in heterologous expression systems, and these have not been clearly linked to changes in blood pressure in humans (9–14). Other functional ENaC nonsynonymous variants that we and others have described are rare or of low-frequency (15–19). It is difficult to show that these rare functional human ENaC variants affect blood pressure in epidemiological studies, and the effects of specific ENaC variants on blood pressure in animal models have not yet been addressed. Another barrier is that the vast majority of ENaC variants, including synonymous and nonsynonymous ones, have no defined functional roles.
The resolved crystal structure of an acid-sensing ion channel 1 (ASIC1), a member of the ENaC/degenerin family, has provided important insights regarding the highly-organized structure of the extracellular domains of ENaC-subunits and has recently been confirmed by a resolved structure of αβγENaC (20, 21). A central core is formed by multiple β-strands that form the β-ball (β2, β4, β5, β7, and β8) and palm domains. The β-ball domain contributes to an acidic pocket in ASIC1 that has a role in fine-tuning acid activation of the channel (20). Its functional role in ENaC has not been clearly defined. A recently resolved cryo-EM structure of the extracellular domain of human ENaC revealed that it is strikingly similar to ASIC1, with a few notable differences. The α- and γ-subunits have an embedded inhibitory track in their finger domains, and the region encompassing this track is formed by β-strands and is not present in ASICs (21). We identified several nonsynonymous variants in the β7-strand and its following loop of the human α-subunit that is part of the β-ball (αR350W, rs181065138; αV351A, rs139861603; αH354R, rs753035419; and αG355R, rs189376498). We found that all these variants except αH354R alter human ENaC activity when expressed in Xenopus oocytes. Among these variants, αR350W exhibited the most robust effect on enhancing channel activity and open probability (Po).
Results
Location of the β7-strand in the extracellular domain of the resolved ENaC and ASIC1 structures
In the structures of ASIC1 and ENaC, five short β-strands form the β-ball domain (20, 21). When we examined human gene variant databases (NCBI dbSNP, 1000 Genome Project, TopMed (22) and ExAC (Exome Aggregation Consortium (23)), we noted that several nonsynonymous human SCNN1A (encoding αENaC) variants (αR350W, αV351A, αH354R, and αG355R) are present in the β7-strand and its following loop that form part of the β-ball of the α-subunit (Fig. 1, A and B). Sequence alignments show αArg-350, αVal-351, and αHis-354 are well-conserved among the ENaC/degenerin family, but αGly-355 is only conserved among α-subunits from different species (Fig. 1C).
Figure 1.
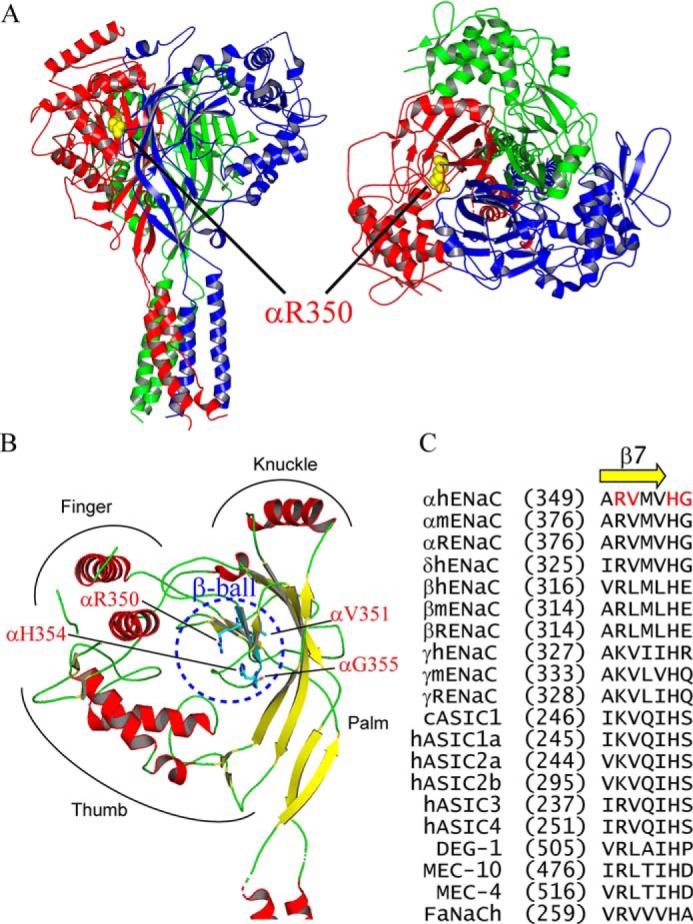
Location of variants in the human ENaC structure. A, location of αArg-350 in a trimeric human ENaC model (PDB 6BQN (21)). The α, β, and γ-subunits are shown as red, blue, and green ribbons, respectively, using PyMOL 2.0 (60). Side chain of αArg-350 is shown as yellow spheres. Left, side view, and right, top view. Both views show that αArg-350 is near the palm domain of βENaC. B, locations of αArg-350, αVal-351, αHis-354, and αGly-355 in the extracellular β-ball domain of αENaC. Helical domains are displayed in red and β-strands in yellow. All four residues are shown as sticks with carbons in cyan, oxygen in red, and nitrogen in blue. C, sequence alignments of ENaC/degenerin members. Alignments were performed using Vector NTI 11 (Thermo Fisher Scientific). Only sequences of the β7-strand and its following residues are shown. Amino acid numbers of the first residue in all sequences are shown in parentheses. Four residues where variants of this study reside are shown in red letters.
ENaC variants alter channel activity
To investigate the effects of these β7-strand human ENaC variants, we generated the αR350W, αV351A, αH354R, and αG355R mutations and co-expressed wildtype (WT) or mutant human α-subunit with WT human β- and γ-subunits in Xenopus oocytes. Amiloride-sensitive whole-cell currents were assessed by two-electrode voltage clamp. Representative current recordings from oocytes expressing WT or mutant ENaCs are shown in Fig. 2, A, C, E, and G. Oocytes expressing the αR350W variant had 2.51 ± 1.37-fold greater amiloride-sensitive currents than WT (Fig. 2B, n = 82–89, p < 0.0001 versus WT), whereas oocytes expressing the αG355R variant had 1.78 ± 1.11-fold greater amiloride-sensitive currents than WT (Fig. 2H, n = 58–59, p < 0.0001 versus WT). In contrast, oocytes expressing αV351A had 0.45 ± 0.36-fold reduced currents when compared with WT (Fig. 2D, n = 76–77, p < 0.0001). The αH354R mutant currents were similar to WT (Fig. 2F, n = 50–51, p > 0.05 versus WT).
Figure 2.

Three variants of human αENaC changed channel activity in Xenopus oocytes. Oocytes expressing WT and mutant human ENaCs were clamped at −100 mV (membrane potential), and whole-cell currents were recorded in a bath solution (NaCl-110, containing 110 mm NaCl) in the absence and presence of 10 μm amiloride. A, C, E, and G, representative recordings of WT and mutant ENaCs. Traces were superimposed with the same time and current scales. Negative values reflect inward Na+ currents. B, D, F, and H, normalized currents, representing amiloride-sensitive currents in all cells that were divided by the mean of the WT group in the same batch of oocytes. Data were pooled from three to five batches of oocytes. Dot plots were overlaid with mean ± S.D. The p values were from Student's t test.
Functional variants do not alter levels of ENaC surface expression
We examined whether the differences in whole-cell currents we observed with the gain–of–function variants (αR350W and αG355R) or loss–of–function variant (αV351A) in oocytes reflected changes in numbers of channels at the plasma membrane. Levels of surface expression of WT and mutant human ENaCs in oocytes were determined using a chemiluminescence assay, using a human β-subunit construct with an extracellular epitope FLAG tag (17). As shown in Fig. 3, oocytes expressing either WT or mutant ENaCs had similar levels of surface expression.
Figure 3.

Functional variants do not alter ENaC surface expression in oocytes. A, surface expression levels in oocytes injected with cRNAs for WT αβFγ (βF for βFLAG), αR350WβFγ or WT αβγ (no FLAG control) ENaCs. B, surface expression levels in oocytes injected with WT αβFγ, αV351AβFγ, αG355RβFγ, or WT αβγENaC. Levels of surface expression were assessed using a chemiluminescence assay 48 h after cRNA injection. Relative light units measured from individual oocytes were normalized to the mean relative light units of the same batch of oocytes expressing WT (αβFγ). Data were combined from three batches of oocytes. Similar relative surface expression levels of WT (αβFγ) and mutants (αR350WβFγ in A and V351AβFγ and αG355RβFγ in B) were observed (NS, not significant). However, levels of FLAG-tagged WT (αβFγ, positive control) were significantly greater than nontagged WT (αβγ, negative control, p < 0.0001, one-way ANOVA with Dunnett's post hoc test).
ENaC trafficking and expression may vary in different cells, and the three subunits may traffic to the cell surface in a noncoordinate fashion (24). To confirm our observations in oocytes, we examined ENaC surface expression in a mammalian cell line (Fisher rat thyroid, FRT cells) by co-transfection of nontagged human β- and γENaCs, and WT or mutant human αENaC with N-terminal HA tag and C-terminal V5 tag (25). Biotin-labeled surface proteins were purified and immunoblotted with anti-HA antibody. ENaC-subunit maturation in the biosynthetic pathway involves furin cleavages of both α- and γ-subunit (26–28). As shown in Fig. 4, all three mutants (αR350W, αV351A, and αG355R) and WT showed similar surface levels of both full-length (90 kDa) and cleaved (22 kDa) forms of αENaC (Fig. 4, A, C, E, and G). All four groups also had similar levels of expression of the full-length and cleaved forms of αENaC in whole-cell lysates (Fig. 4, B, D, F, and G).
Figure 4.

Function variants do not alter ENaC surface expression in FRT cells. Cells were transfected with a WT or mutant (αR350W, αV351A, or αG355R) α-subunit with an N-terminal HA and C-terminal V5 epitope tag, together with nontagged WT β- and γ-subunits. Surface proteins were labeled with NHS-SS-biotin and following cell lysis were isolated with avidin beads. Following SDS-PAGE, proteins were immunoblotted with a horseradish peroxidase-conjugated anti-HA antibody. Chemiluminescence was quantified with Bio-Rad ChemiDocTM system and then normalized to GAPDH expression. Resultant values were normalized to WT to obtain the relative expression levels for each experiment. Relative αENaC surface expression levels are shown in A (full-length, 90 kDa), C (cleaved, 22 kDa), and E (full-length+ cleaved, 90 + 22 kDa). Relative total αENaC expression levels (5% of the cell lysate) are shown in B (full-length, 90 kDa), D (cleaved, 22 kDa), and F (full-length + cleaved, 90 kDa + 22 kDa). G, representative blots for surface and total (5% of the cell lysate) expression of αENaC. Negative control (NC) represents results with cells transfected with nontagged human α, β, and γ cDNAs. WT, 350, 351, and 355 represent the cells transfected with cDNAs for HA-α-V5, HA-αR350W-V5, HA-αV351A-V5, or HA-αG355R-V5, respectively, accompanied with nontagged β- and γENaCs. Bars are mean ± S.D. All values in A–F were not significantly different (p > 0.05, n = 3–4, one-way ANOVA).
These results suggest that the increases in whole-cell currents seen with αR350Wβγ and αG355Rβγ, as well as the reduction in current seen with αV351Aβγ, compared with WT, likely reflected a change in channel open probability and/or single channel conductance.
Gain–of–function variants αR350W and αG355R suppress the Na+ self-inhibition response
In addition to transporting Na+, ENaC open probability is suppressed by extracellular Na+, a process referred to as Na+ self-inhibition (4, 29, 30). We examined whether the increase in current seen with the αR350W variant reflected a loss of the inhibitory effect of extracellular Na+. A typical Na+ self-inhibition current trace recorded in oocytes expressing WT ENaC is shown in Fig. 5A. An increase in bath [Na+] from 1 to 100 mm was associated with a rapid increase in inward Na+ current reaching a peak value (Ipeak), followed by a slower reduction in inward Na+ current that reflects Na+ self-inhibition, with the current reaching a steady state (Iss). We used the ratio of Iss to Ipeak as measure of the magnitude of the Na+ self-inhibition response. Oocytes expressing αR350Wβγ had a blunted Na+ self-inhibition response, with an Iss/Ipeak of 0.86 ± 0.04 (n = 19, p < 0.0001 versus 0.52 ± 0.05, n = 20 for WT, Fig. 5, A and B). Another gain–of–function αG355R variant also reduced Na+ self-inhibition response (Fig. 5, G and H). These results suggest that the increases in current seen with αR350Wβγ and αG355Rβγ reflect an increase in ENaC open probability. Unexpectedly, the loss–of–function αV351A variant did not show an enhanced Na+ self-inhibition (Fig. 5, C and D). The “silent” αH354R variant modestly, but significantly, reduced Na+ self-inhibition (Fig. 5, E and F).
Figure 5.

αR350W, αH354R, and αG355R reduce the Na+ self-inhibition response. A, C, E, and G, representative recordings in oocytes expressing WT or mutant ENaCs to show the Na+ self-inhibition response. Oocytes were clamped at −100 mV, whereas bath Na+ concentration was increased from 1 mm (NaCl-1, gray bar) to 110 mm (NaCl-110, black bar). Traces were superimposed with the same time and current scales. B, D, F, and H, Iss/Ipeak represents the magnitude of Na+ self-inhibition. Values were obtained from amiloride-sensitive Iss and Ipeak. Horizontal bars are mean ± S.D. Data were collected from three batches of oocytes. The p values were from Student's t tests, NS, not significant.
αR350W increases ENaC open probability
If a mutation is considered as a gating modifier, it should cause a change in channel open probability in a predicted manner. We used a cell-attached path-clamp technique to determine the open probability of WT αβγ, αR350Wβγ, and αV351Aβγ human ENaCs in oocytes. The open probability of αR350Wβγ channels was 0.35 ± 0.12 (n = 11), significantly greater than that of WT (0.23 ± 0.09, p < 0.05, n = 10; Fig. 6, A and E). In contrast, the open probability of αV351Aβγ channels was 0.19 ± 0.13, similar to that of WT (p > 0.05, n = 7; Fig. 6, A and E). Both NPo and N (number of channels within patches) in αR350Wβγ-expressing oocytes were moderately greater than in WT-expressing cells (Fig. 6, C and D). Single channel conductances measured with 110 mm LiCl in patch pipettes were similar between WT, αR350Wβγ, and αV351Aβγ (Fig. 6B).
Figure 6.

αR350W has an increased channel open probability. A, representative traces of single channel recordings from oocytes expressing WT αβγ, αV351Aβγ, and αR350Wβγ. Cell-attached patches were performed at −80 mV (opposite to pipette potential) with a NaCl-110 bath solution and a pipette solution containing 110 mm LiCl. C and O represent closed and open states. Recordings were further filtered at 20 Hz with a low-pass Gaussian following 100 Hz of low-pass Gaussian algorithm by ClampFit 10 (Molecular Devices) for display. The filtering did not eliminate transitions, as judged by visual inspection. B, single channel conductances of WT and the two variants, determined by linear regression of unitary currents and clamping voltages in the range of −20 to −100 mV. Values were not significantly different (p > 0.05, one-way ANOVA). C, NPo as the product of N (channel number in patches) and Po (open probability) of WT and the mutant channels. D, N (channel numbers) of WT and mutant channels. E, Po of WT and mutant channels. B–E, data are shown as dot plots with mean and S.D. as bars. Significantly different values in C–E are noted (p < 0.05, one-way ANOVA followed with a Dunnett's post hoc test). NS, not significant.
Other αArg-350 variants also increase ENaC activity
During our study, additional variants at αArg-350 were revealed in NCBI dbSNP. We examined the other variants to further explore the function role of αArg-350. All three variants (αR350G, αR350Q, and αR350L) showed significantly increased amiloride-sensitive currents (Fig. 7). Although the relative increases in currents seen with αR350G (1.37 ± 0.83, n = 41, p < 0.05 versus WT) and αR350Q (1.47 ± 0.82, n = 40, p < 0.01 versus WT) were modest, a robust increase was seen with αR350L (2.63 ± 1.54, n = 38, p < 0.001 versus WT). Like αR350W, all three mutations significantly reduced Na+ self-inhibition response as evidenced by greater Iss/Ipeak values (αR350G, 0.81 ± 0.05, n = 10, p < 0.001 versus WT; αR350Q, 0.84 ± 0.07, n = 9, p < 0.001 versus WT; and αR350L, 0.69 ± 0.07, n = 9, p < 0.001 versus WT, from Student's t tests).
Figure 7.

Other variants at αArg-350 increase ENaC activity. Normalized currents obtained in the same batches of oocytes expressing WT and mutant channels are shown in A (αR350G and WT), B (αR350Q and WT), and C (αR350L and WT). Normalized currents (measured at −100 mV) were obtained as described in Fig. 2 legend. Data were pooled from three batches of oocytes for each WT and mutant pair. Bars are mean ± S.D. The p values were from Student's t tests.
Comparison of gain–of–function variants located in the α-subunit
Besides the well-known gain–of–function mutations in β- and γENaCs that cause Liddle syndrome, emerging evidence indicates that gain–of–function mutations/variants in the α-subunit present novel causes of salt-related clinical disorders (15, 16, 31). The point mutation αC479R was reported in a family with Liddle syndrome (31). In addition, αW493R variant was found in a group of patients with a cystic fibrosis phenotype and with either a single mutant cystic fibrosis transmembrane conductance regulator (CFTR) allele or no CFTR mutation (15, 16). We compared αR350W, αC479R, and αW493R for functional changes in ENaC activity, normalized to WT. Relative amiloride-sensitive currents of αR350Wβγ (3.0 ± 1.5, n = 49) were similar to that of αW493Rβγ (3.2 ± 1.9, n = 44, p > 0.05), and both were significantly greater than that of both αC479Rβγ (1.4 ± 0.7, n = 46, p < 0.0001) and WT (1.0 ± 0.4, n = 43, p < 0.0001, Fig. 8A). As shown in Fig. 8B, Na+ self-inhibition response was dramatically reduced in all three variants (n = 17–19, p < 0.0001 versus WT). The increases in Iss/Ipeak seen with αW493R were modestly greater than that seen with αR350W, and both were significantly greater than that observed with αC479R.
Figure 8.
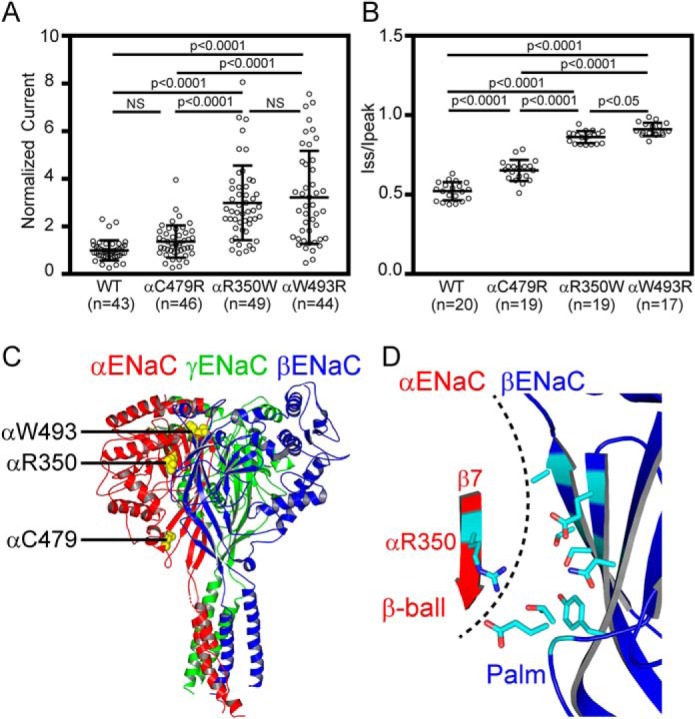
Comparisons of the effects of αR350W and other known gain–of–function human ENaC variants. ENaC activities and Na+ self-inhibition responses were examined in the same batches of oocytes expressing human ENaC αβγ (WT), αR350Wβγ, αC479Rβγ, or αW493Rβγ. A, normalized currents of WT, αC479R, αR350W, and αW493R mutants, obtained in three batches of oocytes with numbers of cells in parentheses. B, Iss/Ipeak of WT and the three variants, obtained in three batches of oocytes. Significantly different values in both A and B are noted (p < 0.0001, one-way ANOVA followed by Tukey's post hoc test). NS, not significant. C, locations of αArg-350, αTrp-493, and αCys-479 in a trimeric model of human ENaC. Three subunits (α, β, and γ) are shown as three colored ribbons, from PDB 6BQN (21), using PyMOL 2.0 (60). Three residues where the gain–of–function variants reside are shown as yellow spheres. D, zoomed-in view of the same model as C. For clarity, only αArg-350 in β7 of α-subunit and a partial palm domain of β-subunit are shown. Eight βENaC residues within 8 Å of the side chain of αArg-350, predicted by PyMOL, are shown. The dashed line identifies α/β-subunit interface.
DISCUSSION
The increasing number of sequenced human genomes has been accompanied with the identification of an increasing number of human ENaC missense variants. We and others have found human ENaC variants located in the extracellular domains of ENaC subunits that affect channel activity when expressed in oocytes, primarily though changes in open probability that are associated with changes in Na+ self-inhibition (16–19). Although functional human ENaC variants have been described in the thumb, finger, knuckle, and palm domains, the only functional variant in the β-ball previously described was a loss–of–function variant associated with pseudohypoaldosteronism type 1 (αC133Y (7, 32)). We found that the human ENaC variants αR350W, αV351A, αG355R, located in β7 and the succeeding loop of the β-ball domain, significantly altered ENaC activity when expressed in oocytes. αR350W and αG355R are novel gain–of–function variants, and for αR350W, this gain–of–function was associated with a reduction in Na+ self-inhibition along with an increase in channel open probability. The changes in ENaC activity seen with αR350W, αV351A, and αG355R were not accompanied by a change in channel surface expression as assessed by a chemiluminescence-based assay in oocytes and a surface biotinylation assay in FRT cells, although patch-clamp analysis suggested that the channel number in patches of αR350W was modestly greater than WT. These differing results may reflect differences in the sensitivities of these assays to detect changes in ENaC surface expression. Although we did not perform single channel recordings of αG355Rβγ channels, the increase in current and the accompanying decrease in Na+ self-inhibition suggest that channels with the αG355R variant have an increase in open probability when compared with WT (30). Although the αV351A variant significantly reduced ENaC currents, it did not affect surface expression level, Na+ self-inhibition response, nor open probability. At present, it is unclear what caused the current reduction.
We used a β-subunit with an extracellular epitope tag to determine surface expression of αβγENaCs. As β-subunits alone do not transit to the plasma membrane at a measurable level in Xenopus oocytes (33), β-subunits at the surface level largely reflect αβγ channels (34–36). We observed similar levels of surface expression of WT and mutant ENaCs in Xenopus oocytes and FRT cells, using either an epitope-tagged β-subunit or an epitope-tagged α-subunit.
The identification of multiple functional variants in the β-ball domain of αENaC strongly suggests that this domain plays an important role in ENaC-gating regulation. The β-ball is a structure formed by five β-strands and is surrounded by the helical finger, thumb, and knuckle domains and the β-sheet palm domains (Fig. 1) (20, 21). In ASICs, Arg-191 in the β-ball interacts with protonable residues at the acidic cavity, potentially contributing to acid sensing (20). Extracellular protons in the physiological range selectively activate human ENaCs by presumably interacting with multiple β- and γENaC residues (37, 38). It would be interesting to examine whether αArg-350 plays any role in the pH regulation of ENaC. In MEC-4, touch-disrupting mutations were identified in the β-ball domain (39). However, there have been few studies examining the specific roles of residues in ENaC β-ball domains (32, 40–42). Mutations of a pair of Cys residues within the rat ENaC β-ball (first and sixth extracellular Cys forming a sulfide bridge) greatly hampered channel delivery to plasma membrane (32), suggesting that the structural integrity of the β-ball domains in all three-subunits is essential for efficient functional expression of ENaCs. We previously observed that mutations of the same pair of Cys residues of mouse αENaC suppressed the Na+ self-inhibition response (40), which could reflect some degree of misfolding. Edelheit et al. (41) reported that αR350A significantly reduced whole-cell current that was associated with a moderately reduced surface expression. Although the magnitude of the Na+ self-inhibition response was not altered, the rate of the decrease in Na+ current in response to a rapid increase in [Na+] was enhanced (41). The homologous mutation in γ-subunit (γK328A) led to a reduced channel current, associated with reduced channel surface expression (42). The authors also noted a reduced Na+ self-inhibition response, which should increase ENaC activity. We found that four substitutions (Trp, Gly, Gln, and Leu) at αArg-350 led to increased channel currents, associated with a suppressed Na+ self-inhibition response. Taken together, our results and previous studies suggest that the β-ball domains have important roles in the regulation of ENaC gating.
The increases in current seen with channels expressing the αR350W and αG355R variants were similar in magnitude to that seen with the αW493R variant and significantly greater that that seen with the αC479R variant (Figs. 2 and 8) (16, 31). The observation that the αC479R variant was present on one allele of a sibling pair with a Liddle syndrome phenotype (31) suggests that individuals with an αR350W, αG355R, or αW493R variant are at risk for hypertension presenting as Liddle syndrome.
Low-frequency and rare variants likely influence the heritability of complex disorders (43–45). Furthermore, normal physiological processes may be modified by rare variants (45). All functional variants in this study are rare. The αArg-350 variants (Trp, Gly, Gln, and Leu) have reported allele frequencies of less than 0.001 (dbSNP Build 152). αR350Q was reported as a de novo mutation in an individual with nonfamilial Brugada syndrome together with a KCNB2 mutation (46), and SCNN1A has been included in the current list of genes associated with Brugada syndrome (47). The αR350Q variant was reported in an individual with Dent disease (48), although its contribution to the disease is unclear. As sequencing efforts increase, we expect to see reports of additional associations of ENaC variants with human diseases.
The variants αR350W, αW493R, and αC479R suppress the Na+ self-inhibition response (Fig. 7) (16). All three residues are located at an intersubunit interface (Fig. 8C), highlighting the important role of intersubunit interfaces in ENaC gating (17, 21, 31, 41, 42, 49–52). The αArg-350 side chain in the resolved ENaC structure projects toward the β-subunit palm domain (21). Examination of the structure in the vicinity of αArg-350 indicates that most βENaC residues near αArg-350 are polar (Fig. 8D). We speculate that αArg-350 and nearby α-subunit polar residues form a hydrophilic patch interacting with their counterparts in the β-subunit palm domain, facilitating the Na+ self-inhibition response. Replacing αArg-350 with Trp or Leu would interfere with these hydrophilic interactions between the α- and β-subunits and with the Na+ self-inhibition response.
Based on structural information from ASIC1 (53), we previously suggested αTrp-520 in mouse ENaC (equivalent to human αTrp-493) and nearby residues in the α-subunit form a hydrophobic patch that facilitates interactions with neighboring structures within ENaC (54). The orientation of αTrp-493 in the resolved human ENaC structure (21) is consistent with this notion. Shobair et al. (55) suggested a different mechanism of ENaC activation by αW493R based on a heterotetrameric αβαγ model, where W493R on one α-subunit interfaces with γGlu-348, and W493R on the other α-subunit interfaces with residues on the β-subunit. The resolved structure of ENaC does not support their model, as ENaC is an αβγ trimer (21), where αTrp-493 is in proximity to hydrophobic and aromatic residues, including residues in loops connecting the β6 and β7 and the β8 and β9 in the γ-subunit. Multiple substitutions at αTrp-493 (Ala, Cys, or Glu) result in an ∼2.5-fold increase in amiloride-sensitive current (16). This observation suggests that it is the loss of αTrp-493 interactions with neighboring residues when other amino acids are placed at this site that leads to the loss of Na+ self-inhibition, rather than interactions between αW493R and γGlu-348 (55).
In summary, αR350W, αG350G, αR350Q, αR350L, and αG355R are novel gain–of–function human ENaC variants, whereas αV531A is a loss–of–function variant. When expressed in Xenopus oocytes, αR350W shares similar features with the αW493R and γL511Q variants (16, 17) as well as αC479R mutation implicated in Liddle syndrome (31). These variants increase whole-cell Na+ currents and open probability and suppress the Na+ self-inhibition response. When examined, little or no effect on the number of channels expressed at the plasma membrane has been observed. These variants form a new class of ENaC extracellular gain–of–function variants with properties that are distinct from classic Liddle mutations targeting the PY motif. Further studies are needed to determine the contributions of these variants to human disorders.
Experimental procedures
Materials
All reagents were purchased from Sigma unless otherwise noted.
Site-directed mutagenesis
Point mutations in human αENaC cDNA were generated using QuikChange II XL site-directed mutagenesis kit (Agilent, Santa Clara, CA). Target mutations were verified by direct DNA sequencing. Mutant and WT human ENaC cRNAs were synthesized with either SP6 or T7 RNA polymerase (Thermo Fisher Scientific, Waltham, MA), using linearized plasmids as templates. Synthesized cRNAs were purified with an RNA purification kit (Qiagen, Germantown, MD), and concentrations were quantified by spectrophotometry.
ENaC expression
For functional expression of human ENaCs, cRNAs for α-, β-, and γENaC subunits (2 ng/subunit) were co-injected into oocytes obtained from female Xenopus laevis. The University of Pittsburgh Institutional Animal Care and Use Committee approved the animal protocol. Injected oocytes were incubated for 20–30 h at 18 °C in modified Barth's saline (MBS: 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca (NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 10 μg/ml streptomycin sulfate, 100 μg/ml gentamycin sulfate, and 10 μg/ml sodium penicillin, pH 7.4).
Two-electrode voltage clamp
Two-electrode voltage clamp was performed 24 h after cRNA injection at room temperature (20–24 °C), using either an TEV200A amplifier (Dagan, Minneapolis, MN) with a DigiData 1550 interface (Molecular Devices, Sunnyvale, CA) or an Axoclamp 900A amplifier (Molecular Devices) with a DigiData 1440A interface, controlled by pClamp 10 (Molecular Devices), as reported previously (17, 56). Pipettes filled with 3 m KCl had a resistance of 0.2–2.0 megohms. Oocytes were continuously voltage-clamped at −100 mV (membrane potential).
Na+ self-inhibition
Na+ self-inhibition was performed as reported previously (17, 57). Na+ self-inhibition responses were recorded following a rapid transition from 1 mm Na+ bath solution (NaCl-1: 1 mm NaCl, 109 mm N-methyl-d-glucamine, 2 mm KCl, 2 mm CaCl2, and 10 mm HEPES, pH 7.4) to 110 mm Na+ solution (NaCl-110: 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, and 10 mm HEPES, pH 7.4). Oocytes were then perfused with 110 mm Na+ solution containing 10 μm amiloride to determine the amiloride-insensitive component of the whole-cell current. The ratio of steady-state amiloride-sensitive current (Iss) in 110 mm Na+ solution, obtained 40 s after transition to the 110 mm Na+ solution, to the peak amiloride-sensitive current (Ipeak) observed following the transition to 110 mm Na+, reflects the magnitude of Na+ self-inhibition.
Patch clamp
Cell-attached patch clamp was performed in oocytes expressing WT αβγ, αR350Wβγ, or αV351Aβγ human ENaC. Bath solution contained 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4. Pipette solution contained 110 mm LiCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4. Patch clamp was carried out using a PC-One patch-clamp amplifier (Dagan Corp.) and a DigiData 1440A interface connected to a PC. Cell-attached patches were clamped at −80 or −100 mV (negative value of pipette potential). pClamp 10 software (Molecular Devices) was used for data acquisition and analyses. Single-channel recordings were acquired at 5 kHz, filtered at 1 kHz with a built-in Bessel filter. Channel open probability was estimated with the single-channel search function of pClamp 10 from recordings that were a minimum of 5 min in length. Unitary currents at clamping voltages (i.e. membrane potentials) of 20, −20, −40, −60, −80, −100, and −120 mV were determined by cursor measurements and used to generate a current-voltage plot yielding single channel slope conductance.
Surface expression in Xenopus oocytes
ENaC surface expression was determined using a chemiluminescence assay and a human βENaC construct with an extracellular epitope FLAG tag (58), as described previously (17). Oocytes were injected with 2 ng/subunit cRNAs for WT or mutant human α-subunit, WT human γ-subunit, and human β-subunit with an extracellular FLAG epitope tag (DYKDDDDK) that was inserted between residues Thr-137 and Arg-138. Oocytes injected with a WT β-subunit cRNA without the FLAG tag and WT α- and γ-subunit cRNAs were used as a negative control group. Surface expression was assayed 48 h after cRNA injection. All steps were performed on ice, except for the last step that was performed at room temperature. Briefly, following a 30-min incubation with MBS (without antibiotics) supplemented with 1% BSA (MBS/BSA), oocytes were incubated with MBS/BSA supplemented with 1 μg/ml of a human anti-FLAG M2 mAb (Sigma) for 1.5 h. Oocytes were then washed six times for 5 min in MBS/BSA and incubated in MBS/BSA supplemented with 1 μg/ml secondary antibody (peroxidase-conjugated AffiniPure F(ab′)2 fragment goat anti-mouse IgG; Jackson ImmunoResearch, West Grove, PA) for 1 h. Cells were extensively washed six times for 5 min in MBS/BSA and finally washed six times for 5 min in MBS without BSA. Individual oocytes were transferred into a white U-bottom 96-well plate, and 100 μl of SuperSignal ELISA Femto Maximum Sensitivity Substrates (Thermo Fisher Scientific, Rockford, IL) was added to each well. Following a 1-min incubation at room temperature, chemiluminescence was quantified with a GloMax-Multi+ detection system (Promega, Madison, WI). Results are presented in relative light units.
Surface expression in FRT cells
FRT cells were maintained in DMEM/F-12 with 8% fetal bovine serum (Life Technologies, Inc.) at 37 °C incubation. Human αENaC with an N-terminal HA tag and a C-terminal V5 tag (HA-αENaC-V5), β- and γ-subunit DNAs were cloned into pcDNA3.1 (25). Mutations (αR350W, αV351A, and αG355R) were introduced into HA-αENaC-V5. FRT cells were grown on plastic wells (6-well size from Costar, Corning, NY) and transfected with 2 μg of plasmid DNA per well using Lipofectamine 3000 transfection kit (Invitrogen), as described previously (36).
Surface biotinylation was performed 24 h following transfection, as described previously (25, 59). Confluent FRT cells were washed four times with cold Dulbecco's PBS with 1.0 mm CaCl2 and 0.5 mm MgCl2 (PBS, Corning Life Sciences). Cells were biotinylated with 1 mg/ml sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3′-dithiopropionate (sulfo-NHS-SS-biotin, Thermo Fisher Scientific, Rockford, IL) in a buffer containing 137 mm NaCl, 15 mm sodium borate, pH 9.0. After quenching biotin with 8% fetal bovine serum in DMEM/F1–2, cells were washed twice with PBS. Cells were then lysed in a detergent solution (100 mm NaCl, 40 mm KCl, 1 mm EDTA, 10% glycerol, 1% Nonidet P-40, 0.4% deoxycholate, 20 mm HEPES, pH 7.4) supplemented with protease inhibitor mixture III (Calbiochem) for 20 min. 5% of the cell lysate was saved for analysis of total protein expression. The remaining cell lysate was incubated with 50 μl of immobilized avidin-coated beads (Thermo Fisher Scientific) overnight at 4 °C. Precipitated proteins were heated for 3 min in Laemmli buffer containing 5% β-mercaptoethanol at 95 °C, then resolved by SDS-PAGE on a 4–15% polyacrylamide gel, and immunoblotted with either a horseradish peroxidase-conjugated anti-HA antibody (0.05 μg/ml, 3F10, Sigma) or a horseradish peroxidase-conjugated anti-GAPDH antibody (0.2 μg/ml, ProteinTech, Rosemont, IL). Immunoblots were developed with a chemiluminescence reagent (PierceTM ECL Western blotting substrate, Thermo Fisher Scientific). A Bio-Rad ChemiDocTM system was used to image blots. Experiments were repeated in four batches of FRT cells.
Statistical analyses
Data are presented as either mean ± S.D. alone or together with dot plots from individual datum points. Statistical significance was examined by the Student's t test for two group data and one-way ANOVA followed by Dunnett's (for comparison between a mutant and WT) or Tukey's post hoc test for multiple group data, using Prism 8 (GraphPad Software, San Diego). A p value of < 0.05 was considered statistically significant.
Author contributions
X. W., J .C., S. Shi, and S. Sheng data curation; X. W., J. C., and S. Sheng formal analysis; X. W., J. C., S. Sheng, and T. R. K. investigation; X. W. writing-original draft; X. W., S. Sheng, and T. R. K. writing-review and editing; S. Shi methodology; S. Sheng and T. R. K. conceptualization; T. R. K. resources; T. R. K. supervision; T. R. K. funding acquisition; T. R. K. project administration.
This work was supported by National Institutes of Health Grants R01 HL147818, R03 DK119752, and P30 DK079307 and the Xiangya Scholar Fund (to X. W.) from The Third Xiangya Hospital, Central South University, China. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial Na+ channel
- ASDN
- aldosterone-sensitive distal nephron
- PDB
- Protein Data Bank
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ANOVA
- analysis of variance
- DMEM
- Dulbecco's modified Eagle's medium
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
References
- 1. Eaton D. C., Helms M. N., Koval M., Bao H. F., and Jain L. (2009) The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 71, 403–423 10.1146/annurev.physiol.010908.163250 [DOI] [PubMed] [Google Scholar]
- 2. Soundararajan R., Pearce D., Hughey R. P., and Kleyman T. R. (2010) Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 285, 30363–30369 10.1074/jbc.R110.155341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warnock D. G., Kusche-Vihrog K., Tarjus A., Sheng S., Oberleithner H., Kleyman T. R., and Jaisser F. (2014) Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat. Rev. Nephrol. 10, 146–157 10.1038/nrneph.2013.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleyman T. R., Kashlan O. B., and Hughey R. P. (2018) Epithelial Na(+) channel regulation by extracellular and intracellular factors. Annu. Rev. Physiol. 80, 263–281 10.1146/annurev-physiol-021317-121143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shekarabi M., Zhang J., Khanna A. R., Ellison D. H., Delpire E., and Kahle K. T. (2017) WNK kinase signaling in ion homeostasis and human disease. Cell Metab. 25, 285–299 10.1016/j.cmet.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 6. Terker A. S., Zhang C., Erspamer K. J., Gamba G., Yang C. L., and Ellison D. H. (2016) Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 89, 127–134 10.1038/ki.2015.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossier B. C., Pradervand S., Schild L., and Hummler E. (2002) Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu. Rev. Physiol. 64, 877–897 10.1146/annurev.physiol.64.082101.143243 [DOI] [PubMed] [Google Scholar]
- 8. Hanukoglu I., and Hanukoglu A. (2016) Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579, 95–132 10.1016/j.gene.2015.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baker E. H., Dong Y. B., Sagnella G. A., Rothwell M., Onipinla A. K., Markandu N. D., Cappuccio F. P., Cook D. G., Persu A., Corvol P., Jeunemaitre X., Carter N. D., and MacGregor G. A. (1998) Association of hypertension with T594M mutation in β-subunit of epithelial sodium channels in black people resident in London. Lancet 351, 1388–1392 10.1016/S0140-6736(97)07306-6 [DOI] [PubMed] [Google Scholar]
- 10. Huber R., Krueger B., Diakov A., Korbmacher J., Haerteis S., Einsiedel J., Gmeiner P., Azad A. K., Cuppens H., Cassiman J. J., Korbmacher C., and Rauh R. (2010) Functional characterization of a partial loss–of–function mutation of the epithelial sodium channel (ENaC) associated with atypical cystic fibrosis. Cell Physiol. Biochem. 25, 145–158 10.1159/000272059 [DOI] [PubMed] [Google Scholar]
- 11. Polfus L. M., Boerwinkle E., Gibbs R. A., Metcalf G., Muzny D., Veeraraghavan N., Grove M., Shete S., Wallace S., Milewicz D., Hanchard N., Lupski J. R., Hashmi S. S., and Gupta-Malhotra M. (2016) Whole-exome sequencing reveals an inherited R566X mutation of the epithelial sodium channel β-subunit in a case of early-onset phenotype of Liddle syndrome. Cold Spring Harb. Mol. Case Stud. 2, a001255 10.1101/mcs.a001255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2007) Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J. Biol. Chem. 282, 6153–6160 10.1074/jbc.M610636200 [DOI] [PubMed] [Google Scholar]
- 13. Jones E. S., Owen E. P., Davidson J. S., Van Der Merwe L., and Rayner B. L. (2011) The R563Q mutation of the epithelial sodium channel β-subunit is associated with hypertension. Cardiovasc. J. Afr. 22, 241–244 10.5830/CVJA-2010-084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhanjal M. K., Owen E. P., Anthony J. A., Davidson J. S., and Rayner B. L. (2006) Association of pre-eclampsia with the R563Q mutation of the β-subunit of the epithelial sodium channel. BJOG 113, 595–598 10.1111/j.1471-0528.2006.00899.x [DOI] [PubMed] [Google Scholar]
- 15. Azad A. K., Rauh R., Vermeulen F., Jaspers M., Korbmacher J., Boissier B., Bassinet L., Fichou Y., des Georges M., Stanke F., De Boeck K., Dupont L., Balascáková M., Hjelte L., Lebecque P., et al. (2009) Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum. Mutat. 30, 1093–1103 10.1002/humu.21011 [DOI] [PubMed] [Google Scholar]
- 16. Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J. J., Dötsch J., Sticht H., and Korbmacher C. (2010) A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self-inhibition. J. Physiol. 588, 1211–1225 10.1113/jphysiol.2009.180224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J., Kleyman T. R., and Sheng S. (2013) Gain–of–function variant of the human epithelial sodium channel. Am. J. Physiol. Renal. Physiol. 304, F207–F213 10.1152/ajprenal.00563.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rauh R., Soell D., Haerteis S., Diakov A., Nesterov V., Krueger B., Sticht H., and Korbmacher C. (2013) A mutation in the β-subunit of ENaC identified in a patient with cystic fibrosis-like symptoms has a gain–of–function effect. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L43–L55 10.1152/ajplung.00093.2012 [DOI] [PubMed] [Google Scholar]
- 19. Ray E. C., Chen J., Kelly T. N., He J., Hamm L. L., Gu D., Shimmin L. C., Hixson J. E., Rao D. C., Sheng S., and Kleyman T. R. (2016) Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am. J. Physiol. Renal Physiol. 311, F908–F914 10.1152/ajprenal.00426.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jasti J., Furukawa H., Gonzales E. B., and Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449, 316–323 10.1038/nature06163 [DOI] [PubMed] [Google Scholar]
- 21. Noreng S., Bharadwaj A., Posert R., Yoshioka C., and Baconguis I. (2018) Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 7, e39340 10.7554/eLife.39340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program. (2018) BRAVO Variant Browser. University of Michigan and NHLBI [Google Scholar]
- 23. Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O'Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weisz O. A., Wang J. M., Edinger R. S., and Johnson J. P. (2000) Noncoordinate regulation of endogenous epithelial sodium channel (ENaC)-subunit expression at the apical membrane of A6 cells in response to various transporting conditions. J. Biol. Chem. 275, 39886–39893 10.1074/jbc.M003822200 [DOI] [PubMed] [Google Scholar]
- 25. Heidrich E., Carattino M. D., Hughey R. P., Pilewski J. M., Kleyman T. R., and Myerburg M. M. (2015) Intracellular Na+ regulates epithelial Na+ channel maturation. J. Biol. Chem. 290, 11569–11577 10.1074/jbc.M115.640763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., and Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 10.1074/jbc.M307003200 [DOI] [PubMed] [Google Scholar]
- 27. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., and Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 10.1074/jbc.C400080200 [DOI] [PubMed] [Google Scholar]
- 28. Hughey R. P., Bruns J. B., Kinlough C. L., and Kleyman T. R. (2004) Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J. Biol. Chem. 279, 48491–48494 10.1074/jbc.C400460200 [DOI] [PubMed] [Google Scholar]
- 29. Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., and Kleyman T. R. (2006) Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 290, F1488–F1496 10.1152/ajprenal.00439.2005 [DOI] [PubMed] [Google Scholar]
- 30. Maarouf A. B., Sheng N., Chen J., Winarski K. L., Okumura S., Carattino M. D., Boyd C. R., Kleyman T. R., and Sheng S. (2009) Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J. Biol. Chem. 284, 7756–7765 10.1074/jbc.M807060200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salih M., Gautschi I., van Bemmelen M. X., Di Benedetto M., Brooks A. S., Lugtenberg D., Schild L., and Hoorn E. J. (2017) A missense mutation in the extracellular domain of αENaC causes Liddle syndrome. J. Am. Soc. Nephrol. 28, 3291–3299 10.1681/ASN.2016111163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Firsov D., Robert-Nicoud M., Gruender S., Schild L., and Rossier B. C. (1999) Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC). Identification of cysteines essential for channel expression at the cell surface. J. Biol. Chem. 274, 2743–2749 10.1074/jbc.274.5.2743 [DOI] [PubMed] [Google Scholar]
- 33. Harris M., Garcia-Caballero A., Stutts M. J., Firsov D., and Rossier B. C. (2008) Preferential assembly of epithelial sodium channel (ENaC)-subunits in Xenopus oocytes: role of furin-mediated endogenous proteolysis. J. Biol. Chem. 283, 7455–7463 10.1074/jbc.M707399200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chalfant M. L., Denton J. S., Langloh A. L., Karlson K. H., Loffing J., Benos D. J., and Stanton B. A. (1999) The NH2 terminus of the epithelial sodium channel contains an endocytic motif. J. Biol. Chem. 274, 32889–32896 10.1074/jbc.274.46.32889 [DOI] [PubMed] [Google Scholar]
- 35. Konstas A. A., and Korbmacher C. (2003) The γ-subunit of ENaC is more important for channel surface expression than the β-subunit. Am. J. Physiol. Cell Physiol. 284, C447–C456 10.1152/ajpcell.00385.2002 [DOI] [PubMed] [Google Scholar]
- 36. Sheng S., Chen J., Mukherjee A., Yates M. E., Buck T. M., Brodsky J. L., Tolino M. A., Hughey R. P., and Kleyman T. R. (2018) Thumb domains of the three epithelial Na+ channel-subunits have distinct functions. J. Biol. Chem. 293, 17582–17592 10.1074/jbc.RA118.003618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collier D. M., and Snyder P. M. (2009) Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J. Biol. Chem. 284, 792–798 10.1074/jbc.M806954200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collier D. M., Peterson Z. J., Blokhin I. O., Benson C. J., and Snyder P. M. (2012) Identification of extracellular domain residues required for epithelial Na+ channel activation by acidic pH. J. Biol. Chem. 287, 40907–40914 10.1074/jbc.M112.417519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eastwood A. L., and Goodman M. B. (2012) Insight into DEG/ENaC channel gating from genetics and structure. Physiology 27, 282–290 10.1152/physiol.00006.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sheng S., Maarouf A. B., Bruns J. B., Hughey R. P., and Kleyman T. R. (2007) Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J. Biol. Chem. 282, 20180–20190 10.1074/jbc.M611761200 [DOI] [PubMed] [Google Scholar]
- 41. Edelheit O., Hanukoglu I., Dascal N., and Hanukoglu A. (2011) Identification of the roles of conserved charged residues in the extracellular domain of an epithelial sodium channel (ENaC)-subunit by alanine mutagenesis. Am. J. Physiol. Renal Physiol. 300, F887–F897 10.1152/ajprenal.00648.2010 [DOI] [PubMed] [Google Scholar]
- 42. Edelheit O., Ben-Shahar R., Dascal N., Hanukoglu A., and Hanukoglu I. (2014) Conserved charged residues at the surface and interface of epithelial sodium channel-subunits–roles in cell surface expression and the sodium self-inhibition response. FEBS J. 281, 2097–2111 10.1111/febs.12765 [DOI] [PubMed] [Google Scholar]
- 43. Bandyopadhyay B., Chanda V., and Wang Y. (2017) Finding the sources of missing heritability within rare variants through simulation. Bioinform. Biol. Insights 11, 1177932217735096 10.1177/1177932217735096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bloom J. S., Ehrenreich I. M., Loo W. T., Lite T. L., and Kruglyak L. (2013) Finding the sources of missing heritability in a yeast cross. Nature 494, 234–237 10.1038/nature11867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manolio T. A., Collins F. S., Cox N. J., Goldstein D. B., Hindorff L. A., Hunter D. J., McCarthy M. I., Ramos E. M., Cardon L. R., Chakravarti A., Cho J. H., Guttmacher A. E., Kong A., Kruglyak L., Mardis E., et al. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Juang J. M., Lu T. P., Lai L. C., Ho C. C., Liu Y. B., Tsai C. T., Lin L. Y., Yu C. C., Chen W. J., Chiang F. T., Yeh S. F., Lai L. P., Chuang E. Y., and Lin J. L. (2014) Disease-targeted sequencing of ion channel genes identifies de novo mutations in patients with nonfamilial Brugada syndrome. Sci. Rep. 4, 6733 10.1038/srep06733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campuzano O., Sarquella-Brugada G., Fernandez-Falgueras A., Cesar S., Coll M., Mates J., Arbelo E., Perez-Serra A., Del Olmo B., Jordá P., Fiol V., Iglesias A., Puigmulé M., Lopez L., Pico F., et al. (2019) Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum. Mutat. 40, 749–764 10.1002/humu.23730 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Y., Fang X., Xu H., and Shen Q. (2017) Genetic analysis of Dent's disease and functional research of CLCN5 mutations. DNA Cell Biol. 36, 1151–1158 10.1089/dna.2017.3731 [DOI] [PubMed] [Google Scholar]
- 49. Chen J., Myerburg M. M., Passero C. J., Winarski K. L., and Sheng S. (2011) External Cu2+ inhibits human epithelial Na+ channels by binding at a-subunit interface of extracellular domains. J. Biol. Chem. 286, 27436–27446 10.1074/jbc.M111.232058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Collier D. M., and Snyder P. M. (2011) Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric αγβ channel architecture. J. Biol. Chem. 286, 6027–6032 10.1074/jbc.M110.198127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Collier D. M., Tomkovicz V. R., Peterson Z. J., Benson C. J., and Snyder P. M. (2014) Intersubunit conformational changes mediate epithelial sodium channel gating. J. Gen. Physiol. 144, 337–348 10.1085/jgp.201411208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kashlan O. B., Blobner B. M., Zuzek Z., Tolino M., and Kleyman T. R. (2015) Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J. Biol. Chem. 290, 568–576 10.1074/jbc.M114.606152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gonzales E. B., Kawate T., and Gouaux E. (2009) Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460, 599–604 10.1038/nature08218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen J., Ray E. C., Yates M. E., Buck T. M., Brodsky J. L., Kinlough C. L., Winarski K. L., Hughey R. P., Kleyman T. R., and Sheng S. (2015) Functional roles of clusters of hydrophobic and polar residues in the epithelial Na+ channel knuckle domain. J. Biol. Chem. 290, 25140–25150 10.1074/jbc.M115.665398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shobair M., Dagliyan O., Kota P., Dang Y. L., He H., Stutts M. J., and Dokholyan N. V. (2016) Gain–of–function mutation W493R in the epithelial sodium channel allosterically reconfigures intersubunit coupling. J. Biol. Chem. 291, 3682–3692 10.1074/jbc.M115.678052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sheng S., Perry C. J., and Kleyman T. R. (2002) External nickel inhibits epithelial sodium channel by binding to histidine residues within the extracellular domains of α and γ-subunits and reducing channel open probability. J. Biol. Chem. 277, 50098–50111 10.1074/jbc.M209975200 [DOI] [PubMed] [Google Scholar]
- 57. Chraïbi A., and Horisberger J. D. (2002) Na self-inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J. Gen. Physiol. 120, 133–145 10.1085/jgp.20028612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Firsov D., Schild L., Gautschi I., Mérillat A. M., Schneeberger E., and Rossier B. C. (1996) Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. U.S.A. 93, 15370–15375 10.1073/pnas.93.26.15370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kashlan O. B., Kinlough C. L., Myerburg M. M., Shi S., Chen J., Blobner B. M., Buck T. M., Brodsky J. L., Hughey R. P., and Kleyman T. R. (2018) N-Linked glycans are required on epithelial Na(+) channel-subunits for maturation and surface expression. Am. J. Physiol. Renal Physiol. 314, F483–F492 10.1152/ajprenal.00195.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schrödinger L. (2010) The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC, New York [Google Scholar]