

Abstract
The chemistry of protein–ligand binding is the basis of virtually every biological process. Ligand binding can be essential for a protein to function in the cell by stabilizing or altering the conformation of a protein, particularly for partially or completely unstructured proteins. However, the mechanisms by which ligand binding impacts disordered proteins or influences the role of disorder in protein folding is not clear. To gain insight into this question, the mechanism of folding induced by the binding of a Pro-rich peptide ligand to the SH3 domain of phosphatidylinositol 3-kinase unfolded in the presence of urea has been studied using kinetic methods. Under strongly denaturing conditions, folding was found to follow a conformational selection (CS) mechanism. However, under mildly denaturing conditions, a ligand concentration–dependent switch in the mechanism was observed. The folding mechanism switched from being predominantly a CS mechanism at low ligand concentrations to being predominantly an induced fit (IF) mechanism at high ligand concentrations. The switch in the mechanism manifests itself as an increase in the reaction flux along the IF pathway at high ligand concentrations. The results indicate that, in the case of intrinsically disordered proteins too, the folding mechanism is determined by the concentration of the ligand that induces structure formation.
Keywords: Src homology 3 domain (SH3 domain), protein folding, intrinsically disordered protein, ligand-binding protein, peptides, conformational selection, induced fit, reaction flux
Introduction
The binding of ligands to macromolecules is key to all processes in a living cell. Tight regulation of biological processes, including allosteric regulation (1, 2), is made possible by the binding of specific ligands. Two models were proposed more than 50 years ago to describe protein–ligand binding interactions and binding cooperativity. The sequential model (3) posited a conformational change upon ligand binding, whereas the concerted model (4) posited the pre-existence of at least two conformational states to which ligand could bind. Although these models were proposed to explain the allostery seen in the case of many multimeric proteins, the same concepts of induced fit (IF)2 and conformational selection (CS) can be used to describe structural changes induced upon ligand binding to monomeric proteins.
Importantly, proteins are stabilized when a ligand binds more strongly to the folded state than to the unfolded state. Hence, a protein that would be unfolded in the absence of a ligand in the presence of denaturant is expected to fold completely upon the addition of ligand. Consequently, it becomes possible to study the folding reaction at high denaturant concentration (5, 6). This capability enables an important question in protein folding to be addressed: is the mechanism of folding at high denaturant concentration the same as that at low denaturant concentration?
To address this question, it is first necessary to determine the mechanism of binding-induced folding. Because there are two steps, ligand binding and folding, associated with the acquisition of structure, there are two possible mechanisms for attaining the folded state (Fig. 1). The IF mechanism involves weak binding of the ligand to the unfolded state followed by folding. It posits that the binding energy, although not large, is sufficient to drive folding to the structured state. In contrast, the CS mechanism posits that the ligand binds selectively to a pre-existing, binding-competent conformation of the protein, which is in equilibrium with the unbound, structureless state. Thermodynamic coupling eventually leads to the folding of all the protein molecules.
Figure 1.
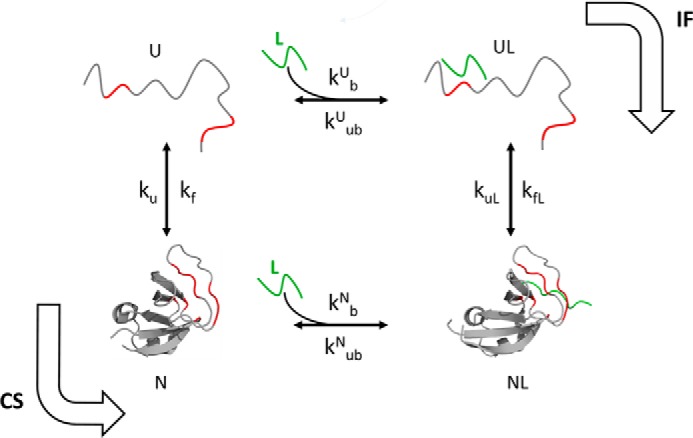
Ligand binding coupled to the folding of the PI3K SH3 domain. The binding site in the protein is highlighted in red, and the ligand is shown in green. The Protein Data Bank (PDB) codes of the structures represented as N and NL are 3I5S and 3I5R, respectively.
An understanding of binding-induced folding in the case of denaturant-unfolded proteins will provide insight into the mechanism by which an intrinsically disordered protein (IDP) gains structure upon binding to its ligand. For IDPs, it has been predicted that both the IF and CS pathways may be operative at the same time (7–9). Nevertheless, their folding has been invariably found to be describable by the IF mechanism (10–13). This is surprising given that folded protein molecules have been shown to sample many different partially unfolded (14–17) and even fully unfolded (18, 19) conformations. IDPs are therefore expected to have thermally accessible folded conformations in equilibrium with the unfolded conformation, and their folding should be describable by the CS mechanism at least under some conditions.
Distinguishing between the IF and CS mechanisms (20, 21) is important in other contexts too. Both IF and CS mechanisms have been proposed to be the underlying basis of the prion hypothesis that describes the spread of infectious protein amyloid aggregates in many neurodegenerative diseases (22–25). The ability to distinguish between the CS and IF mechanisms has a profound bearing on devising strategies to combat the spread of the disease in the brain (26).
SH3 domains are found in many proteins and are involved in diverse biological processes, including signal transduction (27–29). These biological processes are mediated by the formation of multimeric protein complexes. The formation of these complexes is facilitated by the binding of the SH3 domain to a Pro-rich motif present in its binding partner. SH3 domains recognize and bind to peptides having a PXXP (X is any amino acid residue) sequence segment (27). No significant conformational change occurs in the SH3 domain upon such binding (30, 31).
In this study, the mechanism of ligand-induced folding of the SH3 domain of phosphatidylinositol 3-kinase was characterized. The ligand used was a Pro-rich peptide having nine amino acid residues (RPLPPRPSK). This peptide had been shown to bind to the PI3K SH3 domain in earlier studies (30). Here, binding to the peptide ligand is shown to stabilize the native protein against urea-induced denaturation. Addition of the peptide ligand to the protein in unfolding conditions is shown to drive the folding of the unfolded protein. To understand the mechanism of ligand-induced folding, the kinetics of the folding process was studied using fluorescence spectroscopy. A ligand concentration–mediated switch in the mechanism was found to occur, from being dominated by CS at low ligand concentrations to being IF at high ligand concentrations, under mildly denaturing conditions. The current study showing that binding-induced folding under unfolding conditions can be tuned from following only a CS pathway to following both the CS and IF pathways suggests that IDP folding is also tuneable by varying ligand concentration.
Results
In this study, the mechanism of folding of the PI3K SH3 domain, induced by a peptide ligand, has been studied. The PI3K SH3 domain is a well-folded, globular protein that does not undergo any major structural change upon binding to peptide ligands (30, 31). Folding induced by the binding of the peptide ligand was studied under unfolding conditions where the free protein is mostly unfolded, but the protein–ligand complex is folded (Fig. 2c).
Figure 2.

Characterization of the PI3K SH3 domain in the absence and presence of the ligand. a, fluorescence emission spectra of N (solid black line), U (dashed black line), and ligand-bound N (dotted line) upon excitation at 268 nm. b, the binding curve was obtained by measuring the change in the intrinsic Trp fluorescence signal at 320 nm upon excitation at 268 nm. The data were obtained by equilibrating 0.3 μm protein with the indicated ligand concentrations. The raw data were normalized to values of 1 for the fluorescence signals of the completely unbound state of the protein. The solid line through the points is a fit to Equation 1. A value of 7 μm was obtained for KDN. c, equilibrium unfolding curves in the absence (○) and presence (▵) of the ligand (350 μm) were determined by monitoring the fluorescence at 300 and 320 nm, respectively, upon excitation at 268 nm. The data were converted to fraction unfolded (fu) values and plotted against the concentration of urea. The solid lines through the data are fits to a two-state model of unfolding.
Pro-rich peptide binds to PI3K SH3 domain and induces its folding in the presence of urea
Equilibrium binding studies were carried out at different urea concentrations by incubating a fixed concentration of protein with various concentrations of a Pro-rich peptide ligand for 5 h. The concentration of protein used in each case was at least 20-fold less than the KD value at every urea concentration so that binding did not affect the free ligand concentration. The sole Trp residue in the protein, Trp-53, is at the binding interface, and an ∼20-nm blue shift in the wavelength of the maximum fluorescence emission, accompanied by an increase in the fluorescence quantum yield, was seen upon ligand binding (Fig. 2a) upon excitation at 268 nm. Fig. 2a also shows that, in the absence of any peptide ligand, the Tyr fluorescence of the protein, which is maximum at 300 nm (32, 33), increases upon unfolding as the quenching of Tyr fluorescence by FRET occurring from Tyr residues to Trp-53 in N (native) is released upon unfolding to U (unfolded).
The equilibrium binding curve obtained at each of four different urea concentrations (Fig. 3, a–d) yielded the overall apparent dissociation constant, which was found to increase in value with an increase in urea concentration (Fig. 3, a–d). Nevertheless, binding was observed at high ligand concentration. The fluorescence emission spectrum of the ligand-bound state at any of the urea concentrations showed maximum fluorescence emission at 335 nm (Fig. 3, inset) as did the bound state under native conditions (Fig. 2a). Because folding is induced by ligand binding (Fig. 1), at equilibrium, both the ligand-bound N state and the ligand-bound U state will contribute to the fraction of protein with bound ligand. The data were fit to Equation 2 and led to the determination of KDU. The fraction of protein in the UL state was found to be negligible compared with the fraction of protein in the NL state. The logarithm of KDU was found to increase linearly with an increase in urea concentration (Fig. S1b).
Figure 3.

Binding curves of the PI3K SH3 domain obtained at different concentrations of urea. The binding curves were obtained by measuring the change in the intrinsic Trp fluorescence signal at 320 nm upon excitation at 268 nm in 6 (a), 5 (b), 3.5 (c), and 2.8 m (d) urea. The data were obtained by equilibrating 1–5 μm protein with varying ligand concentrations. To determine the fraction of the protein bound to ligand (fb), the data were normalized to values of 0 and 1 for the fluorescence signal of the completely unbound state and completely bound state, respectively. The solid lines through the points are fits to Equation 2. For fitting, the value of KDN was fixed to the value obtained from the t = 0 points of ligand-induced folding traces, and the value of KU used at each urea concentration was first determined from the equilibrium unfolding curve obtained in the absence of any ligand. The inset in each panel shows the fluorescence spectra of the bound (dashed line) and unbound (solid line) states upon excitation at 268 nm. The vertical dashed lines indicate the wavelength at which the binding curve was acquired. The values of KDU obtained are listed in Table 1.
Kinetics of ligand-induced folding of the unfolded PI3K SH3 domain
The kinetics of ligand-induced folding was monitored at various concentrations of ligand, at different urea concentrations, in manual mixing experiments. The kinetic trace at each concentration of urea was describable by a single-exponential equation (Fig. 4, a–d). At all the urea concentrations, the t = ∞ points of the kinetic folding traces fell on the equilibrium binding curve, indicating that the reactions had been monitored till completion (Fig. 5, a–d). Nevertheless, a burst phase was observed to have occurred within the dead time of manual mixing (Fig. 4, a–d) whose amplitude increased with a decrease in the concentration of urea (Fig. 5, a–d). Stopped-flow mixing (dead time, 11 ms) did not abolish the burst phase (Fig. S4).
Figure 4.

Kinetic traces of folding induced by the ligand at different urea concentrations. Ligand-induced folding in 6 (a), 5 (b), 3.5 (c), and 2.8 m (d) urea was monitored by measurement of the change in the intrinsic Trp fluorescence signal at 320 nm upon excitation at 268 nm. The protein at different concentrations of urea was diluted to different concentrations of ligand manually without changing the urea concentration. The concentrations of the peptide ligand were, from top to bottom, 3500, 1000, and 100 μm (a); 1500, 500, and 100 μm (b); 8700, 400, and 10 μm (c); 2400, 125, and 10 μm (d). Each trace was normalized to a value of 1 for the fluorescence signal of the protein in the absence of ligand. The reactions were carried out under pseudo-first-order conditions, and the solid lines through the data are fits to a single exponential equation.
Figure 5.
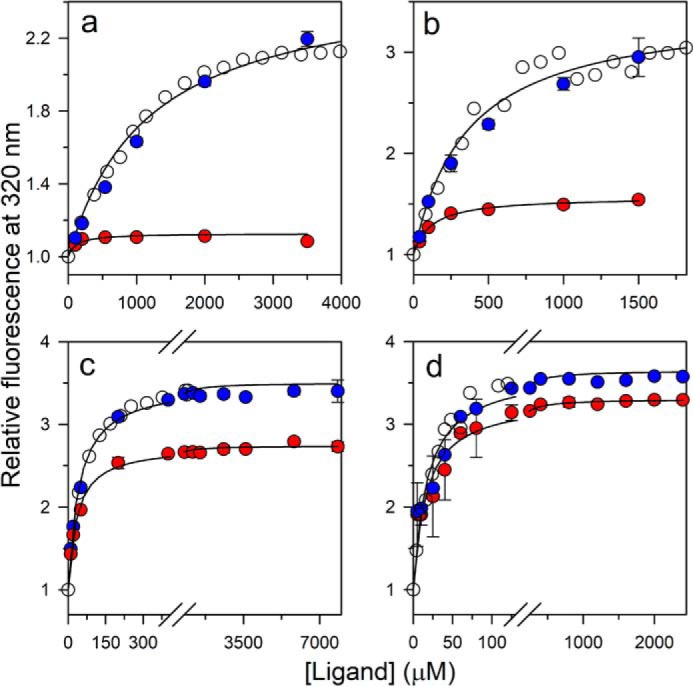
Comparison of the kinetic amplitudes with the equilibrium amplitudes of folding in 6 (a), 5 (b), 3.5 (c), and 2.8 m urea (d). Open circles, equilibrium binding curve; red filled circles, the t = 0 points; and blue filled circles, the t = ∞ points of the kinetic traces of folding induced by ligand binding. For each urea concentration, the data have been normalized to a value of 1 for the protein fluorescence signal in the absence of ligand. The solid lines through the t = 0 points (red filled circles) are fits to Equation 1. The error bars, showing the spread in the data, were obtained from two or more independent experiments.
The origin of the burst phase change in fluorescence could be understood by remembering that, at each of the four concentrations of urea, some U state is present. The percentage of molecules present in the U state, as determined from the equilibrium unfolding curve measured in the absence of ligand (Fig. 2c), is 98, 88, 30, and 15% in 6, 5. 3.5 and 2.8 m urea, respectively. Hence, it was likely that the burst phase corresponded to the very rapid binding of the ligand to the pre-existing N state population at that urea concentration and that this binding occurred on a much faster time scale than the folding reaction. Indeed, when protein and ligand were mixed under native conditions in the absence of any urea, under which essentially no U state is present, the entire change in fluorescence was observed to occur in a burst phase (Fig. S3). To confirm that the burst phase change did indeed correspond to the binding of ligand to pre-existing N, it was shown that the relative burst phase amplitude observed at a particular urea concentration corresponded to the fraction of molecules present as N at that urea concentration (Fig. 6a). Thus, the burst phase change corresponds to the binding of ligand to pre-existing N, which is not accompanied by any structural change, whereas the observable phase of fluorescence change at each of the four urea concentrations corresponds to the ligand binding–induced folding of pre-existing U.
Figure 6.

Dependence of the fraction unfolded protein and rate constants of folding and unfolding obtained from urea-induced folding and unfolding experiments and from ligand-induced folding experiments. a, the equilibrium unfolding curve was measured by monitoring the fluorescence at 300 nm upon excitation at 268 nm. The data were converted to fraction unfolded (fu) values and plotted versus urea concentration (open circles). The solid line through the data are a fit to a two-state model of unfolding. Fraction unfolded was also calculated both from the burst phase amplitude (red filled circles) and from the folding and unfolding rate constants (blue filled circles) determined from kinetic analysis of the ligand-induced folding reactions. b, observed rate constants of folding and unfolding (open circles) obtained from urea-jump experiments. The solid line through the data points is a fit to Equation 11. The folding (purple filled circles) and unfolding (green filled circles) rate constants were obtained from ligand-induced folding experiments. The error bars showing the spread in the data, were obtained from two or more independent experiments.
Hence, the dependence of the burst phase amplitude on the ligand concentration represented the binding curve for the N state at each urea concentration. The signal for unbound N was taken from the equilibrium binding curve because the N and U signals at the wavelength used for acquiring the binding curves were not very different (Fig. 2a). The value of KD for N (KDN) at each urea concentration was thus obtained; not surprisingly, the presence of urea caused the binding affinity of the ligand for N to decrease (Figs. 5 and S1a and Table 1).
Table 1.
Parameters governing the ligand-induced folding of the PI3K SH3 domain
The values of kf and ku at 6 and 5 m urea were determined by fitting the data in Fig. 7, a and b, respectively, to Equation 3. The values of kf, ku, kfL, and kuL at 3.5 and 2.8 m urea were determined by fitting the data in Fig. 7, c and d, respectively, to Equation 4. The values of KDN at all concentrations of urea were determined by fitting the data in Fig. 5, a–d, to Equation 1. The values of KDU at all concentrations of urea were determined by fitting the data in Fig. 3, a–d, to Equation 2. It should be noted that the values estimated for of KDU suggest that very little binding of ligand to the protein would occur in the range of ligand concentrations used.
| 6 m urea | 5 m urea | 3.5 m urea | 2.8 m urea | |
|---|---|---|---|---|
| kf (s−1) | (5 ± 0.2) × 10−4 | (1.5 ± 0.4) × 10−3 | (5.2 ± 0.08) × 10−3 | (1.2 ± 0.07) × 10−2 |
| ku (s−1) | (1.4 ± 0.2) × 10−2 | (7.3 ± 0.4) × 10−3 | (4.7 ± 0.4) × 10−3 | (3 ± 0.5) × 10−3 |
| kfL (s−1) | (1 ± 0.06) × 10−2 | (2 ± 0.2) × 10−2 | ||
| kuL (s−1) | (2 ± 0.4) × 10−4 | (1 ± 0.6) × 10−4 | ||
| KDN (μm) | 121 ± 11 | 118 ± 12 | 34 ± 2 | 18 ± 8 |
| KDU (μm) | 10,900 ± 1550 | 8,000 ± 1100 | 1,800 ± 320 | 774 ± 25 |
Mechanism of ligand binding to the PI3K SH3 domain in the presence of urea
It is assumed that the ligand binding steps are much faster than the conformational conversion steps. When the concentration of the ligand is much higher than the protein concentration, the observed rate constants of the two extreme mechanisms, CS and IF, are expected to decrease and increase, respectively, with an increase in ligand concentration (7).
Fig. 7 shows the dependences on ligand concentration of observed rate constants of folding induced by ligand. Fig. 7, a and b, show the observed rate constants at 6 and 5 m urea where the fraction of protein in the N state is 2 and 12%, respectively. The observed rate constants decreased asymptotically in a hyperbolic manner with an increase in the concentration of ligand. When fit to the CS model (Equation 3), the data yielded the folding and unfolding rate constants at each urea concentration (Table 1), which were in agreement with the folding and unfolding rate constants obtained from folding and unfolding experiments with the free protein in the absence of ligand (Figs. 6b and S2, a–d).
Figure 7.

Dependence of the observed rate constants of ligand-induced folding on ligand concentration. Dependence of the observed rate constants of ligand-induced folding were determined in 6 (a), 5 (b), 3.5 (c), and 2.8 m (d) urea. The solid lines through the data in a and b are fits to Equation 3, and the solid lines through the data in c and d are fits to Equation 4. The values of KDN were fixed to the values obtained from the t = 0 points of ligand-induced folding traces, and the values of KDU were fixed to the values obtained from equilibrium binding curves. The parameters obtained from fitting the data are listed in Table 1. The error bars, showing the spread in the data, were obtained from two or more independent experiments.
Fig. 7, c and d, show the dependences of the observed rate constants on ligand concentration at 3.5 and 2.8 m urea, respectively. In these cases, the rate constants decreased at low ligand concentrations and thereafter increased at high ligand concentrations. The data were explainable by a model that took into account both the mechanisms being operative simultaneously. The rate constants of folding and unfolding of the free PI3K SH3 domain and of the protein–ligand complex were obtained by fitting the data in Fig. 7, c and d, to Equation 4 (Table 1). The folding and unfolding rate constants of the free protein were in good agreement with the rate constants obtained from urea-induced folding and unfolding experiments carried out in the absence of any ligand (Figs. 6b and S2, a–d). The flux along each pathway at 3.5 and 2.8 m urea was then calculated from the knowledge of the rate constants and the population of each species on each pathway (Equations 7 and 10). The fractional flux along the IF pathway was found to increase at high ligand concentrations as the fractional flux along the CS pathway decreased (Fig. 8, a and b). It was also noted that, at 2.8 m urea, the fractional flux along the IF pathway became greater than that along the CS pathway at a lower concentration of peptide ligand than at 3.5 m urea (Fig. 8, a and b).
Figure 8.
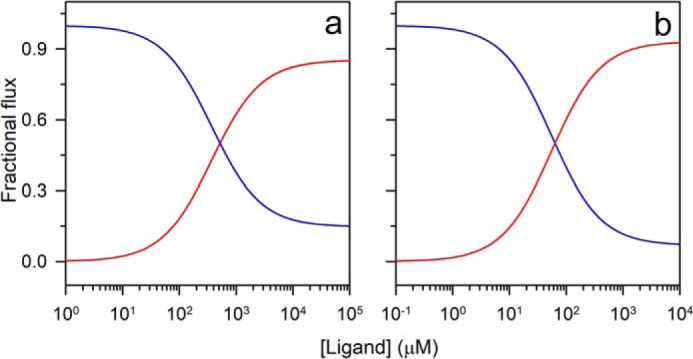
Dependence of fractional flux along CS and IF pathways of ligand concentration. The reaction flux along the CS (blue) and the IF (red) pathways at 3.5 (a) and 2.8 m (b) urea was calculated by using the values of the parameters obtained from the ligand-induced folding reaction in Equations 7 and 10. The reaction flux along each pathway was then converted to fractional flux values.
It was important to rule out any nonspecific binding of ligand to protein at very high ligand concentrations. This was done by monitoring binding by measurement of fluorescence anisotropy, whose value depends on the size of the protein–ligand complex formed. The binding curve obtained with fluorescence anisotropy as the probe was found to be coincident with that obtained using fluorescence intensity as the probe, even at high ligand concentrations (Fig. S6). This ruled out the possibility of any nonspecific binding occurring at high ligand concentrations. Moreover, previous NMR experiments carried out under native conditions had shown that chemical shift perturbation occurs only at the primary binding site (34), even at a 2 mm concentration of the same peptide ligand used in the current study.
Measurement of folding rate constants under denaturing conditions and unfolding rate constants under renaturing conditions
Direct measurement of folding rate constants under denaturing conditions and unfolding rate constants under renaturing conditions was possible because of the coupling of the folding reaction to the binding reaction. The folding and unfolding rate constants at 5 and 6 m urea were determined by fitting the dependences of the observed rate constant on ligand concentration to Equation 3. The folding rate constants were found to fall on the extrapolated folding arm of the chevron (Fig. 6b). Moreover, the unfolding rate constants at these urea concentrations also matched the unfolding rate constants measured by urea-induced unfolding experiments (Fig. 6b). At 3.5 and 2.8 m urea, the folding and unfolding rate constants were determined by fitting the dependence of the observed rate constant on ligand concentration to Equation 4. The unfolding rate constants under renaturing conditions were found to fall on the extrapolated unfolding arm of the chevron (Fig. 6b). Additionally, the fraction of unfolded protein at every urea concentration, as calculated from the folding and unfolding rate constants, was found to be in agreement with the fraction of unfolded protein obtained from an equilibrium unfolding experiment (Fig. 6a).
Discussion
The mechanism of structure formation upon ligand binding has remained an open question for many years now. There has been a large body of work done on many systems to elucidate the mechanism of folding induced by binding (10–12, 35–42). In the current study, the kinetics of ligand-induced folding of the unfolded PI3K SH3 domain has been studied. The ligand-induced folding process appears to occur through both the CS and IF mechanisms. The reaction conditions dictate the mechanism that is operative. At high urea concentrations, folding occurs via the CS mechanism, whereas at low urea concentrations, a ligand concentration–dependent switch in the mechanism, from CS to IF, is observed. This kind of a switch in the mechanism is expected to occur on theoretical considerations (7) but has been observed only for RNase P from Bacillus subtilis (43).
Pro-rich peptide causes unfolded PI3K SH3 to fold
The Pro-rich peptide used as the ligand in this study is a polyproline-II helix (30). These secondary structural motifs, in which intramolecular H-bonds are absent, have been found in IDPs (44). Under native conditions, the value of KD of the PI3K SH3 domain for the Pro-rich peptide matches the previously reported value (Fig. 2b) (30). The observation that the PI3K SH3 domain gains stability upon ligand binding (Fig. 2c) suggested that the fraction of protein in the folded state at any urea concentration would be greater in the presence of the ligand than in its absence. This indicated that the addition of ligand would cause the protein to fold at high urea concentrations. This strategy was used to study the mechanism of ligand-induced folding of the unfolded protein.
Urea reduces the binding affinity of PI3K SH3 domain for the ligand
The population of N present at each urea concentration was found to bind to the ligand within the dead time of manual mixing in the burst phase (Fig. 4, a–d). The values of KDN, calculated from the burst phase amplitude indicated that urea has a significant effect on the binding affinity (Figs. 5 and S1a and Table 1). Urea is known to disrupt hydrophobic interactions in proteins (45, 46), and hydrophobic contacts are known to be important in the interaction between the SH3 domain and the ligand (30). A linear dependence of the logarithm of KDN on the concentration of urea (Fig. S1a) is expected when the interactions between the SH3 domain and the ligand, which are broken in the presence of urea, are hydrophobic in nature.
Binding-induced folding of the PI3K SH3 domain occurs by the CS mechanism under strongly denaturing conditions
In the case of the PI3K SH3 domain, because NL is more stable than U, the selective binding of N to the ligand causes U to convert to N by the law of mass action (Fig. 1), according to the CS mechanism, under strongly denaturing conditions. The ligand binds to the already existing N, thereby pulling the U ↔ N equilibrium toward N. Hence, overall, folding is followed by binding. Under strongly denaturing conditions, the PI3K SH3 domain is completely unstructured and hence will have very low affinity for the ligand. Finally, the rate constant of U binding to ligand and the rate constant of the formation of N from U determine the preferred pathway to be taken for the protein to fold (Fig. 1). Hence, the ligand binds to U slowly, and CS is the dominant pathway.
It has been surprising that binding-induced folding of IDPs invariably follows an IF mechanism (10–13). The current study of binding-induced folding of the PI3K SH3 domain suggests that the principal reason for a CS mechanism not being observed in the case of IDPs is that the binding affinity of the ligand for U is not low enough. Although the PI3K SH3 domain is obviously not an IDP, it could be artificially tuned to be unfolded by the addition of urea. The ability to modulate binding affinity by changing the urea concentration made it possible to study the mechanism of binding-induced folding over a very wide range of ligand concentrations, which is usually not possible with an IDP.
The interactions between native PI3K SH3 domain and its peptide ligand are primarily hydrophobic in nature (34) as are the interactions between IDPs and their ligands (47). Nevertheless, there is a major difference in the mode of binding. For almost all IDPs, the binding site is composed of a continuous stretch of amino acid residues (10–13, 48), which increases the probability of productive binding interactions with the ligand in their unfolded form. In the case of the PI3K SH3 domain, different segments of the protein together constitute the binding site (30), and hence, ligand binding is possible only after the tertiary structure is attained and the binding site is formed.
Ligand concentration–mediated switch in the mechanism of folding from CS to IF
The folding mechanism of a protein induced by ligand binding can be described by one of the two extreme possible mechanisms, CS and IF. In a pure CS mechanism, all the molecules fold by shifting the population toward N by selective binding of the ligand to the pre-existing N state. In contrast, if a pure IF mechanism is followed, structural induction occurs only after binding of the ligand to the U state. However, the energy landscape of ligand-induced folding could have contributions from both intramolecular (CS mechanism) and intermolecular (IF mechanism) interactions, and depending on the conditions, either intramolecular or intermolecular interactions could drive the process of folding. In an earlier study, a ligand concentration–mediated switch in the mechanism of coupled folding and binding was seen (43) wherein the rate constants of every step were obtained by fitting the kinetics of folding, induced by binding, to a complex model. The obtained rate constants were then used to calculate the flux along each pathway. The calculated flux indicated a ligand concentration–mediated switch in the mechanism. In contrast, in this study, direct evidence of a shift in the mechanism from CS to IF is observed. The observed rate constant of folding, induced by ligand binding, is seen to decrease at low ligand concentrations and increase at high ligand concentrations (Fig. 7, c and d), which are defining features of the CS and IF mechanisms, respectively. The calculation of relative flux along the CS and IF pathways also led to the same inference (Fig. 8, a and b).
In the current study, the mechanism is seen to switch from CS to IF at the urea concentrations of 3.5 and 2.8 m, and this switch in the mode of binding is brought about by the concentration of ligand. At urea concentrations of 3.5 and 2.8 m, binding-competent N constitutes 70 and 85%, respectively, of the population of protein molecules; nevertheless, the IF mechanism is followed at high ligand concentrations. Thus, this study indicates that the mere presence of a binding-competent form does not necessarily make the reaction follow a pure CS model. This is an important result because, with the advent of high-resolution NMR techniques, conformations structurally similar to the bound state have been shown to be populated to minor extents in equilibrium with the disordered state, and this observation of a minor binding-competent species in equilibrium with the major state was inferred to mean that the CS mechanism was operative (49–52).
Because two pathways are available for U to fold to NL (Fig. 1), the rate constants defining the pathways determine the dominant pathway. The apparently first-order rate constant of ligand binding to U is directly proportional to the concentration of ligand. Consequently, at a high concentration of ligand, the binding of ligand to U, albeit with low affinity (Table 1), appears to be more favorable energetically than the folding of U to N (Fig. 1), making IF the preferred pathway for folding. Finally, it is the fractional reaction flux along each pathway that determines the preferred pathway for a reaction. The switch in the mechanism occurs (Fig. 8, a and b) when the fractional flux along the IF pathway overrides that along the CS pathway at high ligand concentration. It is also important to note that the switch to the IF mechanism occurs at a higher concentration of ligand at 3.5 m than at 2.8 m urea because the equilibrium constant of dissociation of the ligand from the unfolded state (KDU) increases with an increase in urea concentration (Fig. S1b and Table 1).
It is important to understand the mode of interactions involved in the binding of U to the ligand because the binding site is not formed in the U state ensemble. The interactions between the native protein and the peptide ligand are primarily hydrophobic in nature. Leu-3 and Pro-4 in the peptide ligand interact with Trp-53, Pro-68, and Tyr-71 in the protein. Electrostatic interactions of Arg-1 and Arg-6 of the peptide with Asp-19 and Glu-49 of the protein are also critical for binding (30). Moreover, the protein is negatively charged, and the peptide is positively charged at pH 7.2. It has been shown that the U state ensemble in low concentrations of urea is more compact than in high concentrations of urea (53). It is conceivable that the ligand-binding interface might be partially formed in the compact unfolded state at low urea concentrations and that the charge density at the binding interface of the collapsed unfolded protein could be high. The partially formed, negatively charged binding interface in U would enable its binding to the ligand at high ligand concentrations, causing the folding reaction to proceed via the IF mechanism. In this context, it is important to note that proteins fold and unfold many times during their lifetime in a cell, and it is possible that a ligand whose binding induces folding may be present at a high enough local concentration in the cell such that the IF pathway is operative. It should be noted that the values of KDN and KDU are ∼7 and ∼90 μm in zero denaturant, and the local concentration of a structure-inducing ligand could conceivably reach these values.
Folding under unfolding conditions
The folding of the PI3K SH3 domain under strongly denaturing conditions was achieved by coupling the folding reaction to the binding reaction. Because binding-induced folding of the PI3K SH3 domain follows a CS mechanism under such conditions, folding and unfolding rate constants could be obtained from the dependence of the observed rate constants on ligand concentration. The observation that the folding rate constants determined from the ligand-induced folding experiments fall on the extrapolated folding arm of the chevron indicates that linear extrapolation of the chevron arms to higher denaturant concentrations (Fig. 6b) for determining the folding rate constant under denaturing conditions is valid for this protein. Previously, folding under unfolding condition had been studied in the case of barnase when its folding was induced by the binding of barstar (6) and in the case of a PDZ domain when its folding was induced by the binding of a specific peptide ligand (5).
Conclusion
Ligand binding experiments carried out under unfolding conditions reveal that the PI3K SH3 domain undergoes ligand-induced folding. Under strongly denaturing conditions, folding follows the CS mechanism. Thus, the rate constants of folding under denaturing conditions, which are otherwise difficult to determine, could be measured directly. In contrast, under mildly denaturing conditions, the reaction flux proceeds via both the CS and IF pathways. As a result, a ligand concentration–mediated switch in the mechanism is seen to occur, from CS at low ligand concentrations to IF at high ligand concentrations. This work experimentally shows that the mechanism of ligand-induced folding should never be categorized as being either CS or IF. For deducing the mechanism of a binding-induced folding process, the flux of molecules along both the pathways should be calculated from knowledge of the concentrations of all the species populated and the rate constants of all the steps over a wide range of ligand concentration. The ligand concentration–mediated switch in the mechanism of folding might have a role in regulating the functions of certain IDPs in the cell.
Materials and methods
Buffers and reagents
All the reagents used in this study were of the highest purity grade from Sigma. Urea of ultrahigh purity was obtained from United States Biochemical Corp. 20 mm sodium phosphate buffer (pH 7.2) was used as the native buffer, and unfolding buffer contained different concentrations of urea in the native buffer. All the experiments were carried out at 25 °C. The Pro-rich peptide (RPLPPRPSK) was obtained from Genscript. Its purity was found by electrospray ionization MS to be >95%.
Protein expression and purification
Protein expression and purification were carried out as described previously (54). The purity of the protein was checked by electrospray ionization MS and found to be >95%. The protein had the expected mass of 9276.8 Da. The protein concentration was determined by measuring the absorbance at 280 nm and using a molar extinction coefficient of 17,900 m−1 cm−1 (55).
Determination of peptide concentration
The concentration of a solution of the peptide was determined by comparing the methyl group resonance intensities of the peptide with that of a known concentration of Ala in a 1H NMR spectrum. A standard curve of absorbance at 205 nm versus peptide concentration was generated using this known concentration of peptide. A molar extinction coefficient of 29,613 m−1 cm−1 was obtained at 205 nm.
Establishing the monomeric nature of the peptide ligand
The DLS experiments were carried out on a DynaPro-99 unit (Protein Solutions Ltd.). The DLS profile of 8 mm peptide in 3.5 m urea was acquired, and the peptide was found to be monomeric at this concentration, which was the highest used in the binding experiments (Fig. S5b). Hence, the peptide ligand was monomeric at all the concentrations used.
Fluorescence emission spectra
Fluorescence emission spectra were acquired using a Fluoromax 3 (Horiba) spectrofluorimeter with the excitation wavelength set at 268 nm. The excitation slit width was 1 nm, and the fluorescence emission signal was collected from 280 to 430 nm with an emission slit width of 10 nm.
Equilibrium binding experiments
Equilibrium binding experiments were carried out using the MOS 450 optical system from Biologic and a cuvette of 1-cm path length. An excitation wavelength of 268 nm with a slit width of 4 nm was used. The fluorescence signal was collected at 320 nm using a bandpass filter (Asahi Spectra) with a bandwidth of 10 nm. The concentration of the protein used in the experiments was in the range of 1–5 μm. The wavelength of 320 nm was chosen for measuring fluorescence to monitor binding because the difference in fluorescence between the unbound and bound states was maximum at this wavelength as a consequence of ligand binding causing a blue shift in the fluorescence emission spectrum.
Stopped-flow refolding and unfolding experiments
All kinetic refolding and unfolding experiments in the millisecond time regime were carried out using a stopped-flow module (SFM 4) from Biologic. Unfolded protein in 6 m urea or native protein was diluted rapidly into solutions containing different concentrations of urea to achieve a final urea concentration in the range of 0.6–3.8 m and of 4–8 m for refolding and unfolding experiments, respectively. The excitation wavelength used was 268 nm for the specific excitation of Tyr, and the emitted fluorescence was collected using a 300-nm bandpass optical filter. The dead time was 11 ms when a cuvette of 0.2-cm path length and a flow rate of 5 ml/s were used.
Kinetic ligand-induced folding experiments
The kinetics of ligand-induced folding were monitored by fluorescence measurement using the MOS 450 optical system. Protein in different concentrations of urea was diluted into solutions containing different concentrations of ligand but the same concentration of urea to achieve the desired increase in the concentration of the ligand without affecting the urea concentration. The excitation wavelength used was 268 nm, and the emitted fluorescence was collected using a 320-nm bandpass optical filter. A dead time of 8–10 s was obtained by mixing the two solutions manually.
Equilibrium unfolding experiments
Equilibrium unfolding experiments were carried out using the MOS 450 optical system and a cuvette of 1-cm path length. The specific excitation of Tyr was achieved by using an excitation wavelength of 268 nm with a slit width of 4 nm. Fluorescence signals in the absence and presence of the ligand were collected using bandpass filters (Asahi Spectra) at 300 ± 10 and 320 ± 10 nm, respectively. The concentration of the protein used was in the range of 10–15 μm. The wavelength of 300 nm was chosen for measuring the change in fluorescence to monitor unfolding in the absence of ligand because the change in fluorescence upon unfolding was maximum at this wavelength.
Data analysis
Equilibrium unfolding experiments
The stability (ΔGNU) and its dependence on urea concentration (mNU value) were obtained by fitting each equilibrium unfolding curve to a two-state N ↔ U model (56).
Equilibrium binding curve
The equilibrium binding curve obtained in the absence of any ligand was fit to Equation 1.
| (Eq. 1) |
Here, F, F0, and ΔF are the measured fluorescence signal, the fluorescence signal in the absence of the ligand, and the change in the fluorescence signal upon complete ligand binding, respectively. L is the ligand concentration, and KDN is the equilibrium constant of dissociation from N.
The equilibrium ligand binding curve obtained at each urea concentration was fit to Equation 2.
| (Eq. 2) |
The fraction of protein bound to the peptide ligand (fb) includes both N (NL) and U (UL) in their bound form. KDU is the equilibrium constant of dissociation from U. KU is the equilibrium constant of unfolding of the unbound state. Equations 1 and 2 are valid when the concentration of protein is low (<KDN or KDU) so that, even when all the protein molecules have ligand bound to them, the free ligand concentration can be taken to be equal to the total ligand concentration.
Dependence of the observed rate constant of folding on ligand concentration
If folding follows only the CS mechanism
and when the folding step is the rate-limiting step of the overall process, then when the ligand concentration is much greater than the protein concentration, the observed rate constant of folding, kobs, is given by Equation 3.
| (Eq. 3) |
where kf and ku are the folding and unfolding rate constants, respectively, in the absence of the ligand.
If folding follows both the CS mechanism (Mechanism CS) and IF mechanism simultaneously (see Fig. 1)
and when the folding steps are the rate-limiting steps, then when the ligand concentration is much greater than the protein concentration, kobs is given by Equation 4.
| (Eq. 4) |
Where kfL and kuL are the folding and unfolding rate constants, respectively, for the ligand-bound states.
Calculation of fractional reaction flux
The flux along each pathway was calculated from the values of forward rate constants of each step and the concentration of each species (7). The flux along the CS pathway (FCS) is given by Equation 5.
| (Eq. 5) |
Here, KbN is the bimolecular rate constant of binding of L to N. If PT is the total protein concentration, then the fraction of protein that is unfolded is given by Equation 6.
| (Eq. 6) |
Combining Equations 6 and 5 and when kf ≪ KbN·[L], Equation 5 reduces to Equation 7.
| (Eq. 7) |
The flux along the IF pathway is given by Equation 8.
| (Eq. 8) |
Here, KbU is the bimolecular rate constant of binding of L to U.
| (Eq. 9) |
Combining Equations 9 and 8 and when kfL ≪ KbU·[L], Equation 9 reduces to Equation 10.
| (Eq. 10) |
The fractional flux of molecules along the CS pathway and along the IF pathway was calculated using Equations 7 and 10.
Kinetic refolding and unfolding experiments
The dependence on urea concentration of the observed rate constants of folding and unfolding in the absence of any ligand, obtained from urea-jump experiments, was fitted, using a two-state model, to Equation 11.
| (Eq. 11) |
Here, kobs is the observed rate constant; KfH2O and KuH2O are the rate constants of folding and unfolding in water; mf and mu are representative of the exposure of surface area that occurs due to the transition from U to the transition state (TS) and N to the TS, respectively; and [D] denotes the denaturant (urea) concentration.
Author contributions
S. S. and J. B. U. conceptualization; S. S. data curation; S. S. formal analysis; S. S. methodology; S. S. writing-original draft; J. B. U. supervision; J. B. U. funding acquisition; J. B. U. investigation; J. B. U. writing-review and editing.
Supplementary Material
Acknowledgments
We thank members of our laboratory for discussion. We thank Harish Kumar for help with DLS experiment.
This work was supported by the Tata Institute of Fundamental Research, the Indian Institute of Science Education and Research, and the Department of Science and Technology, Government of India. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- IF
- induced fit
- PI3K
- phosphatidylinositol 3-kinase
- SH3
- Src homology 3
- CS
- conformational selection
- IDP
- intrinsically disordered protein
- L
- ligand
- DLS
- dynamic light scattering
- TS
- transition state
- N
- native
- U
- unfolded.
References
- 1. Wright P. E., and Dyson H. J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 10.1006/jmbi.1999.3110 [DOI] [PubMed] [Google Scholar]
- 2. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., and Jones D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 10.1016/j.jmb.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 3. Koshland D. E. Jr., Némethy G., and Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 10.1021/bi00865a047 [DOI] [PubMed] [Google Scholar]
- 4. Monod J., Wyman J., and Changeux J.-P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 5. Ivarsson Y., Travaglini-Allocatelli C., Jemth P., Malatesta F., Brunori M., and Gianni S. (2007) An on-pathway intermediate in the folding of a PDZ domain. J. Biol. Chem. 282, 8568–8572 10.1074/jbc.M611026200 [DOI] [PubMed] [Google Scholar]
- 6. Sanz J. M., and Fersht A. R. (1994) Measurement of barnase refolding rate constants under denaturing conditions. FEBS Lett. 344, 216–220 10.1016/0014-5793(94)00384-X [DOI] [PubMed] [Google Scholar]
- 7. Hammes G. G., Chang Y.-C., and Oas T. G. (2009) Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 13737–13741 10.1073/pnas.0907195106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greives N., and Zhou H.-X. (2014) Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc. Natl. Acad. Sci. U.S.A. 111, 10197–10202 10.1073/pnas.1407545111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galburt E. A., and Rammohan J. (2016) A kinetic signature for parallel pathways: conformational selection and induced fit. Links and disconnects between observed relaxation rates and fractional equilibrium flux under pseudo-first-order conditions. Biochemistry 55, 7014–7022 10.1021/acs.biochem.6b00914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugase K., Dyson H. J., and Wright P. E. (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 10.1038/nature05858 [DOI] [PubMed] [Google Scholar]
- 11. Bachmann A., Wildemann D., Praetorius F., Fischer G., and Kiefhaber T. (2011) Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc. Natl. Acad. Sci. U.S.A. 108, 3952–3957 10.1073/pnas.1012668108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dogan J., Mu X., Engström Å., Jemth P. (2013) The transition state structure for coupled binding and folding of disordered protein domains. Sci. Rep. 3, 2076 10.1038/srep02076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rogers J. M., Oleinikovas V., Shammas S. L., Wong C. T., De Sancho D., Baker C. M., and Clarke J. (2014) Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. U.S.A. 111, 15420–15425 10.1073/pnas.1409122111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kloks C. P., Tessari M., Vuister G. W., and Hilbers C. W. (2004) Cold shock domain of the human Y-box protein YB-1. Backbone dynamics and equilibrium between the native state and a partially unfolded state. Biochemistry 43, 10237–10246 10.1021/bi049524s [DOI] [PubMed] [Google Scholar]
- 15. Moulick R., Das R., and Udgaonkar J. B. (2015) Partially unfolded forms of the prion protein populated under misfolding-promoting conditions. Characterization by hydrogen exchange mass spectrometry and NMR. J. Biol. Chem. 290, 25227–25240 10.1074/jbc.M115.677575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lou S.-C., Wetzel S., Zhang H., Crone E. W., Lee Y.-T., Jackson S. E., and Hsu S. D. (2016) The knotted protein UCH-L1 exhibits partially unfolded forms under native conditions that share common structural features with its kinetic folding intermediates. J. Mol. Biol. 428, 2507–2520 10.1016/j.jmb.2016.04.002 [DOI] [PubMed] [Google Scholar]
- 17. Baliga C., Selmke B., Worobiew I., Borbat P., Sarma S. P., Trommer W. E., Varadarajan R., and Aghera N. (2019) CcdB at pH 4 forms a partially unfolded state with a dry core. Biophys. J. 116, 807–817 10.1016/j.bpj.2019.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mok Y.-K., Kay C. M., Kay L. E., and Forman-Kay J. (1999) NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions11Edited by P. E. Wright. J. Mol. Biol. 289, 619–638 10.1006/jmbi.1999.2769 [DOI] [PubMed] [Google Scholar]
- 19. Pashley C. L., Morgan G. J., Kalverda A. P., Thompson G. S., Kleanthous C., and Radford S. E. (2012) Conformational properties of the unfolded state of Im7 in nondenaturing conditions. J. Mol. Biol. 416, 300–318 10.1016/j.jmb.2011.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gianni S., Dogan J., and Jemth P. (2014) Distinguishing induced fit from conformational selection. Biophys. Chem. 189, 33–39 10.1016/j.bpc.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 21. Paul F., and Weikl T. R. (2016) How to distinguish conformational selection and induced fit based on chemical relaxation rates. PLoS Comput. Biol. 12, e1005067 10.1371/journal.pcbi.1005067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma B., and Nussinov R. (2012) Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J. Mol. Biol. 421, 172–184 10.1016/j.jmb.2011.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Csermely P., Palotai R., and Nussinov R. (2010) Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546 10.1016/j.tibs.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uversky V. N., Oldfield C. J., and Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 10.1146/annurev.biophys.37.032807.125924 [DOI] [PubMed] [Google Scholar]
- 25. Uversky V. N. (2009) Intrinsic disorder in proteins associated with neurodegenerative diseases, in Protein Folding and Misfolding: Neurodegenerative Diseases, pp. 21–75, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 26. Hsieh M.-C., Liang C., Mehta A. K., Lynn D. G., and Grover M. A. (2017) Multistep conformation selection in amyloid assembly. J. Am. Chem. Soc. 139, 17007–17010 10.1021/jacs.7b09362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koch C. A., Anderson D., Moran M. F., Ellis C., and Pawson T. (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252, 668–674 10.1126/science.1708916 [DOI] [PubMed] [Google Scholar]
- 28. Pawson T., and Gish G. D. (1992) SH2 and SH3 domains: from structure to function. Cell. 71, 359–362 10.1016/0092-8674(92)90504-6 [DOI] [PubMed] [Google Scholar]
- 29. Pawson T., and Schlessingert J. (1993) SH2 and SH3 domains. Curr. Biol. 3, 434–442 10.1016/0960-9822(93)90350-W [DOI] [PubMed] [Google Scholar]
- 30. Yu H., Chen J. K., Feng S., Dalgarno D. C., Brauer A. W., and Schreiber S. L. (1994) Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76, 933–945 10.1016/0092-8674(94)90367-0 [DOI] [PubMed] [Google Scholar]
- 31. Batra-Safferling R., Granzin J., Mödder S., Hoffmann S., and Willbold D. (2010) Structural studies of the phosphatidylinositol 3-kinase (PI3K) SH3 domain in complex with a peptide ligand: role of the anchor residue in ligand binding. Biol. Chem. 391, 33–42 10.1515/BC.2010.003 [DOI] [PubMed] [Google Scholar]
- 32. Eftink M. R. (1991) Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 35, 127–205 [DOI] [PubMed] [Google Scholar]
- 33. Dasgupta A., and Udgaonkar J. B. (2012) Transient non-native burial of a Trp residue occurs initially during the unfolding of a SH3 domain. Biochemistry 51, 8226–8234 10.1021/bi3008627 [DOI] [PubMed] [Google Scholar]
- 34. Booker G. W., Gout I., Downing K. A., Driscoll P. C., Boyd J., Waterfield M. D., and Campbell I. D. (1993) Solution structure and ligand-binding site of the SH3 domain of the p85α subunit of phosphatidylinositol 3-kinase. Cell. 73, 813–822 10.1016/0092-8674(93)90259-S [DOI] [PubMed] [Google Scholar]
- 35. Dahal L., Kwan T. O. C., Hollins J. J., and Clarke J. (2018) Promiscuous and selective: how intrinsically disordered BH3 proteins interact with their pro-survival partner MCL-1. J. Mol. Biol. 430, 2468–2477 10.1016/j.jmb.2018.04.004 [DOI] [PubMed] [Google Scholar]
- 36. Haq S. R., Chi C. N., Bach A., Dogan J., Engström Å., Hultqvist G., Karlsson O. A., Lundström P., Montemiglio L. C., Strømgaard K., Gianni S., and Jemth P. (2012) Side-chain interactions form late and cooperatively in the binding reaction between disordered peptides and PDZ domains. J. Am. Chem. Soc. 134, 599–605 10.1021/ja209341w [DOI] [PubMed] [Google Scholar]
- 37. Giri R., Morrone A., Toto A., Brunori M., and Gianni S. (2013) Structure of the transition state for the binding of c-Myb and KIX highlights an unexpected order for a disordered system. Proc. Natl. Acad. Sci. U.S.A. 110, 14942–14947 10.1073/pnas.1307337110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hill S. A., Kwa L. G., Shammas S. L., Lee J. C., and Clarke J. (2014) Mechanism of assembly of the non-covalent spectrin tetramerization domain from intrinsically disordered partners. J. Mol. Biol. 426, 21–35 10.1016/j.jmb.2013.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shammas S. L., Travis A. J., and Clarke J. (2013) Remarkably fast coupled folding and binding of the intrinsically disordered transactivation domain of cMyb to CBP KIX. J. Phys. Chem. B 117, 13346–13356 10.1021/jp404267e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shammas S. L., Travis A. J., and Clarke J. (2014) Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc. Natl. Acad. Sci. U.S.A. 111, 12055–12060 10.1073/pnas.1405815111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dahal L., Kwan T. O. C., Shammas S. L., and Clarke J. (2017) pKID binds to KIX via an unstructured transition state with nonnative interactions. Biophys. J. 113, 2713–2722 10.1016/j.bpj.2017.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crabtree M. D., Mendonça C. A. T. F., Bubb Q. R., and Clarke J. (2018) Folding and binding pathways of BH3-only proteins are encoded within their intrinsically disordered sequence, not templated by partner proteins. J. Biol. Chem. 293, 9718–9723 10.1074/jbc.RA118.002791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daniels K. G., Tonthat N. K., McClure D. R., Chang Y.-C., Liu X., Schumacher M. A., Fierke C. A., Schmidler S. C., and Oas T. G. (2014) Ligand concentration regulates the pathways of coupled protein folding and binding. J. Am. Chem. Soc. 136, 822–825 10.1021/ja4086726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adzhubei A. A., Sternberg M. J., and Makarov A. A. (2013) Polyproline-II helix in proteins: structure and function. J. Mol. Biol. 425, 2100–2132 10.1016/j.jmb.2013.03.018 [DOI] [PubMed] [Google Scholar]
- 45. Herskovits T. T., and Jaillet H. (1969) Structural stability and solvent denaturation of myoglobin. Science 163, 282–285 10.1126/science.163.3864.282 [DOI] [PubMed] [Google Scholar]
- 46. Herskovits T. T., and Harrington J. P. (1975) Solution studies on heme proteins. Subunit structure, dissociation, and unfolding of Lumbricus terrestris hemoglobin by the ureas. Biochemistry 14, 4964–4971 10.1021/bi00693a027 [DOI] [PubMed] [Google Scholar]
- 47. Arai M., Ferreon J. C., and Wright P. E. (2012) Quantitative analysis of multisite protein–ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 134, 3792–3803 10.1021/ja209936u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narayanan R., Ganesh O. K., Edison A. S., and Hagen S. J. (2008) Kinetics of folding and binding of an intrinsically disordered protein: the inhibitor of yeast aspartic proteinase YPrA. J. Am. Chem. Soc. 130, 11477–11485 10.1021/ja803221c [DOI] [PubMed] [Google Scholar]
- 49. Song J., Guo L.-W., Muradov H., Artemyev N. O., Ruoho A. E., and Markley J. L. (2008) Intrinsically disordered γ-subunit of cGMP phosphodiesterase encodes functionally relevant transient secondary and tertiary structure. Proc. Natl. Acad. Sci. U.S.A. 105, 1505–1510 10.1073/pnas.0709558105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lange O. F., Lakomek N.-A., Farès C., Schröder G. F., Walter K. F., Becker S., Meiler J., Grubmüller H., Griesinger C., and de Groot B. L. (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 10.1126/science.1157092 [DOI] [PubMed] [Google Scholar]
- 51. Boehr D. D., Nussinov R., and Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 10.1038/nchembio.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kjaergaard M., Teilum K., and Poulsen F. M. (2010) Conformational selection in the molten globule state of the nuclear coactivator binding domain of CBP. Proc. Natl. Acad. Sci. U.S.A. 107, 12535–12540 10.1073/pnas.1001693107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dasgupta A., and Udgaonkar J. B. (2010) Evidence for initial non-specific polypeptide chain collapse during the refolding of the SH3 domain of PI3 kinase. J. Mol. Biol. 403, 430–445 10.1016/j.jmb.2010.08.046 [DOI] [PubMed] [Google Scholar]
- 54. Wani A. H., and Udgaonkar J. B. (2009) Revealing a concealed intermediate that forms after the rate-limiting step of refolding of the SH3 domain of PI3 kinase. J. Mol. Biol. 387, 348–362 10.1016/j.jmb.2009.01.060 [DOI] [PubMed] [Google Scholar]
- 55. Bader R., Bamford R., Zurdo J., Luisi B. F., and Dobson C. M. (2006) Probing the mechanism of amyloidogenesis through a tandem repeat of the PI3-SH3 domain suggests a generic model for protein aggregation and fibril formation. J. Mol. Biol. 356, 189–208 10.1016/j.jmb.2005.11.034 [DOI] [PubMed] [Google Scholar]
- 56. Agashe V. R., and Udgaonkar J. B. (1995) Thermodynamics of denaturation of barstar: evidence for cold denaturation and evaluation of the interaction with guanidine hydrochloride. Biochemistry 34, 3286–3299 10.1021/bi00010a019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.