

Abstract
The role of RNA methylation on the sixth N atom of adenylate (m6A) in acute kidney injury (AKI) is unknown. FTO (fat mass and obesity-associated protein) reverses the m6A modification in cisplatin-induced AKI. Here, we aimed to determine FTO's role in AKI. We induced AKI in c57BL/6 mice by intraperitoneal cisplatin injection and treated the animal with vehicle or an FTO inhibitor meclofenamic acid (MA) for 3 days. Moreover, as an in vitro model, human kidney proximal tubular cells (HK2 cells) were treated with cisplatin. We found that the cisplatin treatment reduces FTO expression and increases m6A levels in vivo and in vitro. MA aggravated renal damage and increased apoptosis in cisplatin-treated kidneys, phenotypes that were correlated with reduced FTO expression and increased m6A levels. Moreover, MA promoted apoptosis in cisplatin-treated HK2 cells, which was correlated with the reduced FTO expression and increased m6A in HK2 cells. FTO protein overexpression reduced m6A levels and inhibited apoptosis in cisplatin-treated HK2 cells and also blocked the MA-induced increase in m6A levels and apoptosis rates. In agreement, overexpression of the m6A-generating methyltransferase-like 3 and 14 (METTL3 and METTL14) or siRNA-mediated FTO knockdown promoted apoptosis and enhanced m6A levels in cisplatin-treated HK2 cells. MA increased p53 mRNA and protein levels in AKI both in vitro and in vivo, and FTO overexpression reduced p53 expression and reversed the MA-induced p53 increase in AKI. In conclusion, reduced renal FTO expression in cisplatin-induced AKI increases RNA m6A levels and aggravates renal damages.
Keywords: RNA modification; apoptosis; RNA methylation; kidney; p53; cell signaling; caspase; acute kidney injury, AKI; cisplatin; Fat mass and obesity-associated protein, FTO; N6-methyladenosine, m6A
Introduction
Cisplatin is a potent chemotherapy agent and is widely used to treat malignant solid tumors. Because 90% cisplatin is excreted through kidney, ∼30% of patients are suffered from renal damages after one high dose of cisplatin treatment, which hampered its clinical application (1). However, the mechanism of cisplatin-induced acute kidney injury (AKI)3 remains largely elusive.
Increasing evidence indicates that apoptosis plays a vital role in the progress of AKI (2). The apoptosis of renal tubular epithelial cells occurs several hours after the injection of cisplatin and reaches its highest level during 48–72 h in cisplatin-induced AKI. The p53 protein is an important regulator of apoptosis by regulating the homeostasis of the Caspase and Bcl family. In vitro and in vivo studies showed that the expression of p53 is significantly increased in cisplatin-induced AKI (3, 4). Inhibition of p53 attenuated cisplatin-induced AKIs (5).
m6A, defined as the RNA methylation on the sixth N atom of adenylate, is the most common internal modification of RNA (6). Recent studies showed that the levels of m6A determine the stability and translation efficiency of mRNA, and thus m6A modification is involved in a large number of biological processes including apoptosis (7). m6A is a reversible modification, which can be catalyzed by several enzymes, such as methyltransferase-like 3 and 14 (METTL3 and METTL14), and can also be erased by enzymes such as FTO (fat mass and obesity-associated protein) (8). Meclfemomic acid (MA) is a pharmaceutical compound that has been reported to modify cellular m6A levels by selectively targeting FTO (9).
The role of m6A modification in kidney disease is rarely studied in the mouse model of unilateral ureteral obstruction. The expression of FTO is increased, and ablation of the FTO gene attenuates fibrogenic responses in this model, implying that m6A modification may play a role in renal fibrosis (10). In this study, we aimed to explore the role of m6A modification in cisplatin-induced AKI and its downstream working mechanism.
Results
Cisplatin reduced FTO expression and enhanced m6A levels in mouse kidneys and HK2 cells
A mouse model of AKI was established by intraperitoneal injection of 17 mg/kg cisplatin, and kidneys were collected at day 3. As shown in Fig. 1A, cisplatin treatment significantly decreased FTO expression by 79% in mouse kidneys. The m6A levels were further analyzed in mouse kidneys, and dot-blot analysis showed that the m6A levels were significantly elevated by 14.7-fold in cisplatin-treated mouse kidneys as compared with that in vehicle-treated control kidneys (Fig. 1B).
Figure 1.

The expression of FTO was reduced, and the m6A levels were increased in cisplatin-induced kidney injury. Kidney samples were collected 3 days after cisplatin administration. A, the expression of FTO in kidneys was assessed by Western blotting. B, dot blot was performed to assess the level of total RNA m6A in kidneys. C, Western blotting was performed to measure FTO expression in human proximal tubular epithelial cells (HK2 cells). Protein samples were extracted from HK2 cells 24 h after cisplatin treatment. D, the m6A level in HK2 cells was assessed by dot blot. The data represent means ± S.D., and the results are representative of three independent experiments. Veh, vehicle; Cis, cisplatin; Con, control; MB, methylene blue.
Human proximal tubular epithelial line (HK2 cells) were exposed to 10 μmol/liter cisplatin for 24 h, and Western blotting analysis showed that FTO was significantly down-regulated by 67% in cisplatin-treated cells as compared with that in control cells (Fig. 1C). Fig. 1D showed that the m6A levels were increased by 17.5-fold in cisplatin-treated cells.
MA inhibited FTO expression, enhanced m6A levels, and promoted apoptosis in cisplatin-treated kidneys
The effect of MA was tested in cisplatin-induced AKI. Treatment with cisplatin increased blood urea nitrogen (BUN) levels by 134% and serum creatinine levels by 57% in mice, which were further elevated by 49 and 25%, respectively, after administration of MA (Fig. 2A). Pathological analysis by hematoxylin–eosin (HE) staining revealed a mild histology injury, such as loss of brush border membranes, tubular casts, and cast formation in cisplatin-treated kidneys, and these changes were much more prominent in MA-treated mice (Fig. 2B). Further quantification showed that the kidney injury score was significantly increased in cisplatin-treated kidneys, which was further aggravated by 44% by MA treatment (Fig. 2B). We further examined the effect of MA on apoptosis in cisplatin-treated kidneys. As shown in Fig. 2 (C and D), the ratio of Bax/Bcl-2 and Cleaved Caspase3 expression were increased by 109 and 147%, respectively, in mouse kidneys during cisplatin injury, which were further enhanced by 82 and 42%, respectively, by MA treatment. Immunofluorescencent staining revealed that TUNEL-positive cells in the cisplatin-treated kidneys were three times more than that in control kidneys, and administration of MA further increased TUNEL-positive signals by 118% in injured kidneys.
Figure 2.

MA deteriorated renal function and enhanced apoptosis in cisplatin-treated kidneys. Blood and kidney samples were collected 3 days after cisplatin administration. A, BUN and serum creatinine (SCr) levels. B, hematoxylin–eosin staining was performed, and representative pathological images (original magnification, ×400; bar, 100 μm) of kidneys are shown. #, renal tubular cast; arrowhead, dilated renal tubule. Scale bar, 100 μm. Tubular injury was semiquantitatively scored. C, the expression of Bcl-2, Bax, and Cleaved Caspase-3 in kidneys was assessed by Western blotting and then quantitatively analyzed. D, TUNEL staining was performed to measure apoptosis in the kidney tissue sections (original magnification, ×400; bar, 100 μm). The data represent means ± S.D., and the results are representative of three independent experiments. Veh, vehicle; Cis, cisplatin; NS, normal saline.
The effect of MA on FTO expression and m6A levels was assessed. As shown in Fig. 3A, treatment with MA did not inhibit FTO expression in control kidneys but reduced its expression by 80% in cisplatin-treated kidneys. In addition, MA did not increase m6A levels in control kidneys but significantly increased m6A levels in cisplatin-treated kidneys by 72% (Fig. 3B).
Figure 3.

MA inhibited FTO expression and enhanced m6A levels. Kidney samples were collected 3 days after cisplatin administration. A, the expression of FTO in kidneys was assessed by Western blotting, which was further quantified. B, dot blot was performed to measure the total RNA m6A level in kidneys and then was quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and the results are representative of three independent experiments. Veh, vehicle; Cis, cisplatin; NS, normal saline; ns, not significant.
MA inhibited FTO expression, enhanced m6A levels, and promoted apoptosis in cisplatin-treated HK2 cells
As an in vitro model, HK2 cells, a human proximal tubular epithelial cell line, were stimulated with normal saline or 10 μmol/liter cisplatin for 24 h with or without the addition of 100 mm MA. As shown in Fig. 4A, cisplatin increased the ratio of Bax and Bcl-2 by 184% and the expression of Cleaved Caspase3 by 331% in HK2 cells, which were further increased by 287 and 52%, respectively, after the addition of 100 mm MA for 24 h. Next, TUNEL staining showed that there was no TUNEL-positive cells in control cells, and there was 4% TUNEL-positive cells upon cisplatin exposure, which were further increased by 64% after the treatment with MA (Fig. 4B). Western blotting showed that the expression of FTO was decreased by 47% in cisplatin-treated HK2 cells, and administration of MA further reduced FTO expression by 67% (Fig. 4 C). However, m6A levels were enhanced by 360% upon cisplatin exposure and were further increased by 82% by MA treatment (Fig. 4D).
Figure 4.

MA inhibited FTO expression, enhanced m6A levels, and promoted apoptosis in cisplatin-treated HK2 cells. HK2 cells, a human proximal tubular epithelial cell line, were stimulated with 10 μmol/liter cisplatin for 24 h in the presence or absence of 100 mm MA. A, the expression of Bcl-2, Bax, and Cleaved Caspase-3 in HK2 cells was assessed by Western blotting and quantitatively analyzed. B, TUNEL staining was performed to detect cell apoptosis (original magnification, ×400; bar, 200 μm). C, Western blotting was performed to measure FTO expression in HK2 cells, which was further quantified. D, dot blot was used to assess the m6A levels in HK2 cell's total RNA and then was quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and the results are representative of three independent experiments. Con, control; Cis, cisplatin; NS, normal saline.
Overexpression of FTO blocked the pro-apoptotic effect of MA in vitro
FTO plasmids were transfected 24 h prior to the stimulation of 10 μmol/liter cisplatin and the treatment of 100 mm MA. As shown in Fig. 5A, transfection of the FTO plasmids reduced the ratio of Bax/Bcl-2 by 46% and the expression of Cleaved Caspase-3 by 40% in cisplatin-treated HK2. MA increased the ratio of Bax/Bcl-2 by 401% and the expression of Cleaved Caspase-3 by 344% in cisplatin-treated HK2 when transfected with the empty vector, but it did not increase the ratio of Bax/Bcl-2 and the expression of Cleaved Caspase-3 in cisplatin-treated HK2 when transfected with the FTO plasmid (Fig. 5A). Moreover, TUNEL staining showed that overexpression of FTO significantly reduced apoptosis by 82% in cisplatin-treated HK2 cells. MA increased TUNEL-positive cells by 87% in cisplatin-treated HK2 when transfected with the empty vector, but it did not increase TUNEL-positive cells in cisplatin-treated HK2 when transfected with the FTO plasmid (Fig. 5B). Western blotting analysis showed that the expression of FTO was increased in cisplatin-treated HK2 cells by 166-fold without the addition of MA or by 152-fold with the addition of MA when transfected with the FTO plasmid (Fig. 5C). Transfection of the FTO plasmid reduced m6A levels by 95% in cisplatin-treated HK2 (Fig. 5D). MA increased m6A levels by 135% in cisplatin-treated HK2 when transfected with the empty vector, but it did not increase m6A levels in cisplatin-treated HK2 when transfected with the FTO plasmid (Fig. 5D).
Figure 5.

Overexpression of FTO blocked the pro-apoptotic effect of MA in vitro. HK2 cells were transfected with empty vector (pCN) or the FTO (pFTO) plasmid. 24 h after the transfection, HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h in the presence or absence of 100 mm MA. A, the expression of Bcl-2, Bax, and Cleaved Caspase-3 in HK2 cells was assessed by Western blotting and then were quantified. B, TUNEL staining was performed to measure cell apoptosis (original magnification, × 400; bar, 200 μm) and then was quantified. C, Western blotting was performed to measure FTO expression in HK2 cells and then was quantified. D, dot blot was used to assess the m6A level in HK2 cells, which was further quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and the results are representative of three independent experiments. Cis, cisplatin; NS, normal saline; ns, not significant.
Knockdown of FTO promoted apoptosis in cisplatin-treated HK2 cells and enhanced m6A levels
FTO siRNA (Si-FTO) were transfected 24 h prior to the stimulation of 10 μmol/liter cisplatin, which treated HK2 cells for another 24 h. As shown in Fig. 6A, the expression of FTO protein in cisplatin-treated HK2 cells was significantly reduced by 66% after Si-FTO transfection, and knockdown of FTO expression by siRNA increased the ratio of Bax/Bcl-2 by 167% and the expression of Cleaved Caspase-3 by 85% in cisplatin-treated HK2. In addition, TUNEL staining indicated that knockdown of FTO expression significantly promoted apoptosis by 27% in cisplatin-treated HK2 cells. Moreover, transfection of the Si-FTO enhanced m6A levels by 34% in cisplatin-treated HK2 (Fig. 6C).
Figure 6.

Knockdown of FTO promoted apoptosis in cisplatin-treated HK2 cells and enhanced m6A levels. HK2 cells were transfected with nonsense siRNA (Si-NC) or the Si-FTO. 24 h after the transfection, HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h. A, the expression of FTO, Bcl-2, Bax, and Cleaved Caspase-3 in HK2 cells was assessed by Western blotting and then were quantified. B, TUNEL staining was performed to measure cell apoptosis (original magnification, ×400; bar, 200 μm) and then was quantified. C, dot blot was used to assess the m6A level in HK2 cells, which was further quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and results are representative of three independent experiments. Cis, cisplatin; NS, normal saline.
METTL3 promoted apoptosis and enhanced m6A levels in cisplatin-treated HK2 cells
As shown in Fig. 7A, cisplatin treatment significantly increased METTL3 expression by 4.7-fold in mouse kidneys. METTL3 plasmid (pMETTL3) were transfected 24 h before the stimulation of 10 μmol/liter cisplatin, and cells were harvested at 24 h after cisplatin treatment. As shown in Fig. 7B, cisplatin treatment increased the expression of METTL3 protein in HK2 by 4.1-fold, and transfection of METTL3 plasmids increased METTL3 expression by 25.1-fold in cisplatin-treated cells. Furthermore, overexpression of METTL3 significantly increased the ratio of Bax/Bcl-2 by 82.7-fold and the expression of Cleaved Caspase-3 by 87.9-fold in cisplatin-treated HK2 cells. Moreover, overexpression of METTL3 significantly increased TUNEL-positive cells by 64% in cisplatin-treated HK2 cells (Fig. 7C). In addition, transfection of METTL3 plasmids enhanced m6A levels by 138% in cisplatin-treated HK2 cells (Fig. 7D).
Figure 7.

Overexpression of METTL3 promoted apoptosis and enhanced m6A levels in cisplatin-treated HK2 cells. A, kidney samples were collected 3 days after cisplatin administration. The expression of METTL3 in kidneys was assessed by Western blotting. B, HK2 cells were transfected with empty vectors (pCN) or METTL3 (pMETTL3) plasmids, 24 h after the transfection; HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h. The expression of METTL3, Bcl-2, Bax, and Cleaved Caspase-3 in HK2 cells was assessed by Western blotting and then were quantified. C, TUNEL staining was performed to measure cell apoptosis (original magnification, ×400; bar, 200 μm) and then was quantified. D, dot blot was used to assess the m6A level in HK2 cells, which was further quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and the results are representative of three independent experiments. Cis, cisplatin; NS, normal saline; Veh, vehicle.
METTL14 promoted apoptosis and enhanced m6A levels in vitro
As shown in Fig. 8A, cisplatin treatment significantly increased METTL14 expression by 5.5-fold in mouse kidneys. METTL14 plasmids (pMETTL14) were transfected in HK2 cells for 24 h and then stimulated with 10 μmol/liter cisplatin for another 24 h. As shown in Fig. 8B, cisplatin treatment increased the expression of METTL14 protein in HK2 by 14.8-fold, and transfection of pMETTL14 increased METTL14 expression by 58.9-fold in cisplatin-treated cells. Overexpression of METTL14 significantly increased the ratio of Bax/Bcl-2 by 299% and the expression of Cleaved Caspase-3 by 477% in HK2 cells with cisplatin treatment. Moreover, TUNEL staining revealed that transfection of METTL14 plasmids significantly promoted apoptosis by 81% in cisplatin-treated HK2 cells (Fig. 8C). In addition, the m6A levels were increased by 9-fold after transfection of METTL14 plasmids in cisplatin-treated HK2 (Fig. 8D).
Figure 8.

Overexpression of METTL14 promoted apoptosis and enhanced m6A levels in vitro. A, kidney samples were collected 3 days after cisplatin administration. The expression of METTL14 in kidneys was assessed by Western blotting. B, HK2 cells were transfected with empty vectors (pCN) or METTL14 (pMETTL14) plasmids, 24 h after the transfection, HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h. The expression of METTL14, Bcl-2, Bax, and Cleaved Caspase-3 in HK2 cells was assessed by Western blotting and then were quantified. C, TUNEL staining was performed to measure cell apoptosis (original magnification, ×400; bar, 200 μm) and then was quantified. D, dot blot was used to assess the m6A level in HK2 cells, which was further quantified. Methylene blue (MB) staining was used to measure the quantity of RNA in membrane blots. The data represent means ± S.D., and the results are representative of three independent experiments. Cis, cisplatin; NS, normal saline; Veh, vehicle.
FTO inhibited p53 expression
The mRNA expression of p53 was measured. As shown in Fig. 9A, the expression of p53 mRNA increased by 67% in HK2 cells after cisplatin treatment for 12 h, which were further enhanced by 50% by MA treatment. Transfection of FTO plasmids reduced p53 mRNA by 36% at 12 h. FTO knockdown by siRNA promoted p53 mRNA by 20.4-fold at 24 h after cisplatin administration in HK2 cells (Fig. 9A). In kidney tissues, the expression of p53 mRNA was increased by 90% after cisplatin administration, which were further enhanced by 80% by MA treatment (Fig. 9B).
Figure 9.

FTO inhibited p53 mRNA and protein expression in vitro and in vivo. A, left panel, HK2 cells were treated with 10 μmol/liter cisplatin for 12 h in the presence or absence of 100 mm MA, and p53 mRNA was measured. Middle panel, HK2 cells were transfected with the FTO plasmid at 24 h prior to the stimulation of 10 μmol/liter cisplatin. The expression of p53 mRNA was measured 12 h after cisplatin stimulation. Right panel, HK2 cells were transfected with the Si-FTO 24 h prior to the stimulation of 10 μmol/liter cisplatin. The expression of p53 mRNA was measured 24 h after cisplatin stimulation. B, the expression of p53 mRNA levels in cisplatin-induced AKI kidneys treated with normal saline or MA. C, upper panel, HK2 cells were transfected with empty vector or the FTO (pFTO) plasmid. 24 h after the transfection, HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h in the presence or absence of 100 mm MA. Western blotting was performed to measure the expression of p53 in HK2 cells. Lower panel, HK2 cells were transfected with nonsense siRNA or the FTO siRNA 24 h after the transfection. HK2 cells were stimulated with 10 μmol/liter cisplatin for another 24 h. Western blotting was performed to measure the expression of p53 in HK2 cells. D, Western blotting was performed to measure p53 expression in in cisplatin-induced AKI kidneys treated with normal saline or MA. The data represent means ± S.D., and the results are representative of three independent experiments. Veh, vehicle; Con, control; Cis, cisplatin; NS, normal saline.
We further analysis the expression of p53 in protein levels. As shown in Fig. 9C, transfection of FTO plasmids reduced p53 expression by 74% in cisplatin-treated HK2 cells. Treatment with MA increased the expression of p53 by 58% in cisplatin-treated HK2 cells when transfected with the empty vector, but it did not increase the expression of p53 in cisplatin-treated HK2 cells when transfected with the FTO plasmid (Fig. 9C). Transfection of Si-FTO increased the p53 protein expression by 44% in HK2 cells treated with cisplatin (Fig. 9C). In kidney tissues, the expression of p53 protein increased by 192% after cisplatin administration, which were further enhanced by 60% by MA treatment (Fig. 9D).
Discussion
The role of m6A modification in AKI is unknown. In the current study, we showed that FTO is down-regulated in cisplatin-induced AKI in vivo and in vitro, and METTL3 and METTL14 are up-regulated in cisplatin-induced AKI in vivo and in vitro, which correlates with increased m6A modification. Moreover, inhibition of FTO by MA or FTO siRNA further increased m6A levels and promoted cisplatin-induced AKI. Furthermore, METTL3 and METTL14 overexpression increased m6A levels and promoted cisplatin-induced AKI. Our data suggest that increased m6A modification is detrimental in cisplatin-induced AKI.
AKI is characterized by increased apoptosis, and excessive apoptosis in AKI causes the deterioration of renal function (11). The effect of m6A modification on apoptosis has been studied (12, 13). Overexpression of FTO promoted cell proliferation and inhibited apoptosis in acute myeloid leukemia cell lines, which was associated with reduced global mRNA m6A levels (12). In contrast, knockdown of FTO increased m6A levels and promoted apoptosis of acute myeloid leukemia cells (12). In an animal model of lipopolysaccharide-induced liver injury, dietary curcumin increased the abundance of m6A, which was correlated with increased expression of pro-apoptotic genes (13). Thus, we hypothesized that m6A demethylation by FTO in cisplatin-induced kidney injury inhibits apoptosis. Indeed, we showed that increased m6A modification by the FTO inhibitor MA was paralleled with enhanced apoptosis in cisplatin-treated kidneys, which was further confirmed in vitro. We also showed that overexpression of FTO in cisplatin-treated HK2 cells reduced m6A levels and inhibited apoptosis. Moreover, overexpression of FTO reversed the enhanced m6A modification by MA and blocked the pro-apoptotic effect of MA. Furthermore, we confirmed the pro-apoptotic effect of m6A in cisplatin-induced AKI by knockdown of FTO or transfection of two methyltransferase METTL3 or METTL14.
p53, a central player in several types of programmed cell deaths, influences transcription of genes that are related to classical mitochondria apoptosis and nonclassical apoptosis (14). Thus, we postulated that FTO inhibited apoptosis through p53. We showed that overexpression of FTO inhibited the expression and methylation of p53 mRNA in cisplatin-treated HK2 cells. The FTO inhibitor MA and FTO siRNA increased the expression of p53 in mRNA and protein levels in cisplatin-treated cells and kidneys. Moreover, overexpression of FTO reversed the increased p53 expression by MA treatment in cisplatin-treated cells.
In summary, treatment with cisplatin reduced FTO expression and increased the m6A levels in kidneys, which leads to the up-regulation of p53. MA aggravated the renal damages in cisplatin-treated kidneys through p53-mediated apoptosis pathways (the Bax/Bcl-2 and Caspase3 pathways) (Fig. 10). We conclude that FTO is protective in cisplatin-induced AKI through inhibiting m6A RNA methylation.
Figure 10.
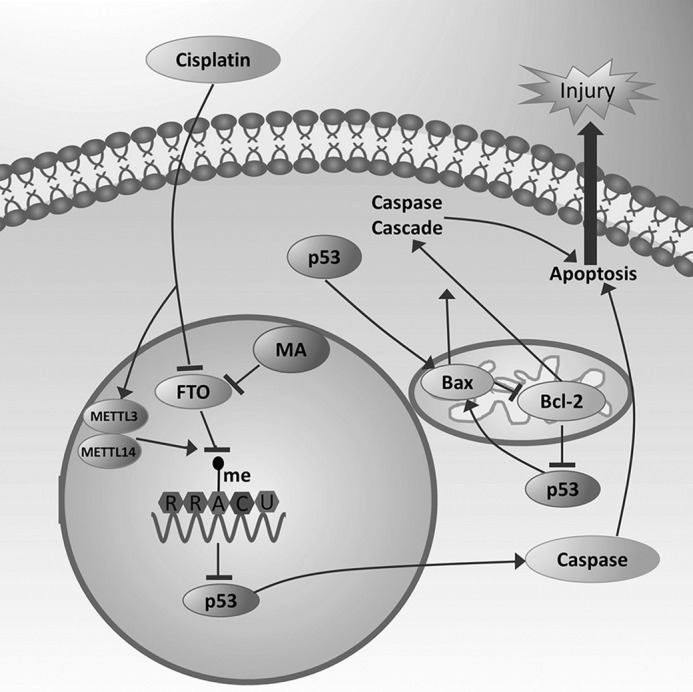
MA promotes cisplatin-induced acute kidney injury through inhibiting FTO-mediated m6A modification. Treatment with cisplatin reduced FTO expression and increased the m6A levels in kidneys, which leads to the up-regulation of p53. MA aggravated the renal damages in cisplatin-treated kidneys through p53-mediated apoptosis pathways (the Bax/Bcl-2 and Caspase3 pathways).
Experimental procedures
Animal
Male c57bl/6 mice were housed in the animal facilities of the Shanghai Jiao Tong University and fed a standard diet. The mice were randomly divided into four groups: vehicle + normal saline group, the vehicle + MA group, the cisplatin + normal saline group, and the cisplatin + MA group (n = 8/group). The mouse model of AKI was established by intraperitoneal injection of 17 mg/kg cisplatin (P4394; Sigma). MA (S5517; Selleck) was given intraperitoneally three times: 1 h prior to cisplatin injection and 24 and 48 h after cisplatin injection at the dose of 10 mg/kg. At day 3 after the cisplatin injection, all mice were sacrificed to collect blood and kidney samples. BUN and serum creatinine were assessed by automatic analyzer. The animal protocols were performed in accordance with the guide of the Care and Use of Laboratory Animals and were approved by Ethics Review Committee for Animal Experimentation of Shanghai Ninth People's Hospital.
Cell culture and treatment
Human proximal tubular epithelial cells (HK2) were bought from the cell bank of the Chinese Academy of Sciences, which were cultured in DMEM/F-12 medium (C11330500BT; Gibco, Thermo Fisher Scientific) with 10% fetal bovine serum (10270-106; Gibco, Thermo Fisher Scientific). To establish in vitro model of AKI, HK2 cells were stimulated with normal saline or 10 μmol/liter cisplatin for 24 h. In some experiments, HK2 cells were treated with normal saline or 100 mm MA. FTO plasmids were transfected by Lipofectamine 2000 (11668-027; Invitrogen) in HK2 cells according to the manufacturer's instruction. The FTO plasmids were constructed by subcloning the human FTO coding sequence into mammalian vector GV358. The empty GV358 vector was used as control. The human FTO siRNA sequences were as follows: forward, 5′-GGACAAUGAUGAUGUCUCU-3′; and reverse, 5′-AGAGACAUCAUCAUUGUCC-3′. The METTL3 plasmids were constructed by subcloning the human METTL3 coding sequence into mammalian vector pCMV3. The empty pCMV3 vector was used as control. The METTL14 plasmids were constructed by subcloning the human METTL14 coding sequence into mammalian vector GTP-C-3FLAG. The empty GTP-C-3FLAG vector was used as control.
HE staining and renal pathology
Kidney tissues were fixed in 4% formaldehyde solution and then embedded in paraffin. Sliced tissue sections were stained with hematoxylin and eosin. Tubular injuries were semiquantitatively scored as described previously (15): normal (score = 0), below 10% (score = 1), 10–25% (score = 2), 25–75% (score = 3), and over 75% (score = 4) of damaged areas.
Western blotting
Western blotting was performed as described previously (16). In brief, total proteins were extracted from kidney tissues or HK2 cells using radioimmune precipitation assay lysis buffer (P0013B; Beyotime, Shanghai, China). Protein samples were separated by SDS-PAGE electrophoresis and transferred to PVDF membrane. The membrane was incubated with primary antibodies overnight, which was followed by the incubation of the secondary antibody (A0216 or A0208; Beyotime). Primary antibodies for FTO (45980) and Cleaved Caspase-3 (9661) were purchased from Cell Signaling Technology (Beverly, MA). The antibodies for Bax (Ab32503) and Bcl-2 (Ab182858) were obtained from Abcam (Cambridge, UK), and the β-actin (60008-1-Ig) antibody was bought from Proteintech.
RNA extraction and PCR
Total RNA was extracted using TRIzol (15596-026; Sigma) according to the manufacturer's instruction, which was reverse transcribed to cDNA by TaKaRa PrimeScript RT reagent kit (RR0036A; Kyoto, Japan). The primer sequences listed in Table 1 were used for quantitative PCR (RR820A).
Table 1.
Specific primer sequences for PCR
| Gene name | Primer sequence | |
|---|---|---|
| Mouse p53 | Forward | 5′-CCCCTGTCATCTTTTGTCCCT-3′ |
| Reverse | 5′-AGCTGGCAGAATAGCTTATTGAG-3′ | |
| Mouse β-actin | Forward | 5′-GGCTGTATTCCCCTCCATCG-3′ |
| Reverse | 5′-CCAGTTGGTAACAATGCCATGT-3′ | |
| Human p53 | Forward | 5′-CAGCACATGACGGAGGTTGT-3′ |
| Reverse | 5′-TCATCCAAATACTCCACACGC-3′ | |
| Human β-actin | Forward | 5′-AAGGAGCCCCACGAGAAAAAT-3′ |
| Reverse | 5′-ACCGAACTTGCATTGATTCCAG-3′ | |
Dot blot
Total RNA was preheated to 95 °C for 3 min and then was spotted on positively charged nylon membrane (RPN303B; Amersham Biosciences). After blocking with nonfat milk, the m6A was probed with a specific antibody for m6A (ABE572; Millipore), followed by the second antibody (A0208; Beyotime). The signals were detected using ECL detection reagents (P36599; Millipore). The same membrane was further stained with methylene blue (319112; Sigma) to measure the quantity of RNA in membrane blots. ImageJ (National Institutes of Health) was used to quantify the signals of dot blot.
TUNEL staining
TUNEL kit (12156792910; Roche) and Hoechst (C1011; Beyotime) reagent were used to identify apoptotic cells in kidney sections and cultured cells according the manufacturer's instructions. In brief, deparaffinized tissue sections were hydrated and then digested with proteinase K (P6556; Sigma) for 30 min at room temperature. HK2 cells were fixed with 4% paraformaldehyde for 1 h at room temperature. Subsequently, kidney tissue sections or cells were washed with PBS and then incubated with TUNEL reagent for 1 h at 37 °C. A fluorescence microscope was used to measure TUNEL-positive cells.
Statistical analysis
All data were analyzed using GraphPad Prism version 5.0 software. The unpaired t test or one-way analysis of variance was used for statistical analysis. p < 0.05 was considered as statistical significance.
Author contributions
P. Z. and Q. X. data curation; P. Z. and M. W. formal analysis; P. Z. and M. W. methodology; P. Z. writing-original draft; P. Z. writing-review and editing; M. W. and L. W. conceptualization; M. W. and L. W. supervision; C. Y. and L. W. project administration; L. W. funding acquisition.
This work was supported by grants from the Science and Technology Commission of Shanghai Municipality (No. 16ZR1419400). The authors declare that they have no conflicts of interest with the contents of this article.
- AKI
- acute kidney injury
- MA
- meclofenamic acid
- m6A
- RNA modification N6-methyladenosine
- BUN
- blood urea nitrogen
- HE
- hematoxylin–eosin
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- Si-FTO
- FTO siRNA.
References
- 1. Bellomo R., Kellum J. A., and Ronco C. (2012) Acute kidney injury. Lancet 380, 756–766 10.1016/S0140-6736(11)61454-2 [DOI] [PubMed] [Google Scholar]
- 2. Miller R. P., Tadagavadi R. K., Ramesh G., and Reeves W. B. (2010) Mechanisms of cisplatin nephrotoxicity. Toxins 2, 2490–2518 10.3390/toxins2112490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hafner A., Bulyk M. L., Jambhekar A., and Lahav G. (2019) The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 10.1038/s41580-019-0110-x [DOI] [PubMed] [Google Scholar]
- 4. Jiang M., Wang C. Y., Huang S., Yang T., and Dong Z. (2009) Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am. J. Physiol. Renal Physiol. 296, F983–F993 10.1152/ajprenal.90579.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Molitoris B. A., Dagher P. C., Sandoval R. M., Campos S. B., Ashush H., Fridman E., Brafman A., Faerman A., Atkinson S. J., Thompson J. D., Kalinski H., Skaliter R., Erlich S., and Feinstein E. (2009) siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 20, 1754–1764 10.1681/ASN.2008111204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maity A., and Das B. (2016) N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. 283, 1607–1630 10.1111/febs.13614 [DOI] [PubMed] [Google Scholar]
- 7. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., and Rechavi G. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- 8. Shi H., Wei J., and He C. (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 10.1016/j.molcel.2019.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang Y., Yan J., Li Q., Li J., Gong S., Zhou H., Gan J., Jiang H., Jia G. F., Luo C., and Yang C. G. (2015) Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384 10.1093/nar/gku1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang C. Y., Shie S. S., Tsai M. L., Yang C. H., Hung K. C., Wang C. C., Hsieh I. C., and Wen M. S. (2016) FTO modulates fibrogenic responses in obstructive nephropathy. Sci. Rep. 6, 18874 10.1038/srep18874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Havasi A., and Borkan S. C. (2011) Apoptosis and acute kidney injury. Kidney Int. 80, 29–40 10.1038/ki.2011.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Z., Weng H., Su R., Weng X., Zuo Z., Li C., Huang H., Nachtergaele S., Dong L., Hu C., Qin X., Tang L., Wang Y., Hong G. M., Huang H., et al. (2017) FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 10.1016/j.ccell.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu N., Li X., Yu J., Li Y., Wang C., Zhang L., Wang T., and Zhong X. (2018) Curcumin attenuates lipopolysaccharide-induced hepatic lipid metabolism disorder by modification of m6A RNA methylation in piglets. Lipids 53, 53–63 10.1002/lipd.12023 [DOI] [PubMed] [Google Scholar]
- 14. Yamada K., and Yoshida K. (2019) Mechanical insights into the regulation of programmed cell death by p53 via mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 1866, 839–848 10.1016/j.bbamcr.2019.02.009 [DOI] [PubMed] [Google Scholar]
- 15. Oh D. J., Dursun B., He Z., Lu L., Hoke T. S., Ljubanovic D., Faubel S., and Edelstein C. L. (2008) Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am. J. Physiol. Renal Physiol. 294, F264–F271 10.1152/ajprenal.00204.2007 [DOI] [PubMed] [Google Scholar]
- 16. Tian H., Wu M., Zhou P., Huang C., Ye C., and Wang L. (2018) The long non-coding RNA MALAT1 is increased in renal ischemia–reperfusion injury and inhibits hypoxia-induced inflammation. Ren. Fail. 40, 527–533 10.1080/0886022X.2018.1487863 [DOI] [PMC free article] [PubMed] [Google Scholar]