

Abstract
Toxoplasma gondii is an important neurotropic pathogen that establishes latent infections in humans that can cause toxoplasmosis in immunocompromised individuals. It replicates inside host cells and has developed several strategies to manipulate host immune responses. However, the cytoplasmic pathogen-sensing pathway that detects T. gondii is not well-characterized. Here, we found that cyclic GMP-AMP synthase (cGAS), a sensor of foreign dsDNA, is required for activation of anti-T. gondii immune signaling in a mouse model. We also found that mice deficient in STING (Stinggt/gt mice) are much more susceptible to T. gondii infection than WT mice. Of note, the induction of inflammatory cytokines, type I IFNs, and interferon-stimulated genes in the spleen from Stinggt/gt mice was significantly impaired. Stinggt/gt mice exhibited more severe symptoms than cGAS-deficient mice after T. gondii infection. Interestingly, we found that the dense granule protein GRA15 from T. gondii is secreted into the host cell cytoplasm and then localizes to the endoplasmic reticulum, mediated by the second transmembrane motif in GRA15, which is essential for activating STING and innate immune responses. Mechanistically, GRA15 promoted STING polyubiquitination at Lys-337 and STING oligomerization in a TRAF protein-dependent manner. Accordingly, GRA15-deficient T. gondii failed to elicit robust innate immune responses compared with WT T. gondii. Consequently, GRA15−/− T. gondii was more virulent and caused higher mortality of WT mice but not Stinggt/gt mice upon infection. Together, T. gondii infection triggers cGAS/STING signaling, which is enhanced by GRA15 in a STING- and TRAF-dependent manner.
Keywords: Toxoplasma gondii, TNF receptor-associated factor (TRAF), interferon, innate immunity, signal transduction, cyclic GMP-AMP synthase (cGAS), protozoan parasite, stimulator of interferon genes protein (STING), type I IFNs
Introduction
The protozoan parasite Toxoplasma gondii can infect nearly all warm-blooded animals (1, 2). As for humans, nearly 30% of the world's population is infected with T. gondii (3). In healthy adults, T. gondii is controlled by the immune system and remains dormant in the brain. However, in immunocompromised individuals, a defect of the immune system leads to the reactivation of the T. gondii parasites and the development of toxoplasmosis. Reactivated parasite replication causes life-threatening brain damage with brain abscesses and necrotic areas (4). Thus, HIV/AIDS patients, cancer patients, and organ transplant recipients are highly susceptible to T. gondii infection.
The infection of T. gondii parasites is recognized by pattern recognition receptors (PRRs).4 Previous studies showed that TLR11 is the PRR of T. gondii in murine cells. TLR11 is able to detect the actin-binding protein Profilin, which is required for entry of T. gondii during infection. TLR11 and TLR12 form a heterodimer in murine dendritic cells (DC) after sensing Profilin and activate adaptor protein MyD88 to initiate downstream signaling for defense against T. gondii (5). Moreover, TLR7 and TLR9 are able to compensate for the loss of TLR11 by activating MyD88 in TLR11-deficient mice (6). Interestingly, TLR7 is activated by T. gondii RNA and triggers innate immune signaling only in TLR11-deficient cells (7). In addition, TLR7 expression is undetectable in CD9α + DCs, which is believed to be the primary DC subset for surveillance of the infection of T. gondii (8). Therefore, the importance of TLR7 in defense against T. gondii is unclear. TLR9 recognizes the methylated DNA of T. gondii (9, 10). However, TLR9 expression is not always detected in the T. gondii–infected DC, whose expression needs priming by IFNγ (6). In addition to those receptors localized in endosomes, the cell plasma membrane receptors TLR2 and TLR4 are shown to recognize the glycosylphosphatidylinositol from the surface of T. gondii and T. gondii-derived heat-shock protein 70 (HSP70) (11, 12). However, some studies showed that T. gondii suppressed innate immune responses via TLR4. Leng and Denkers (13) found that chromatin remodeling in T. gondii–infected macrophages inhibited cytokine production via TLR4. Lee et al. (14) also reported that T. gondii suppressed the production of pro-inflammatory cytokines after TLR4–ligand interactions. The well-studied murine PRRs of T. gondii are TLR11 and TLR12. However, the human genome does not encode TLR11 or TLR12 proteins. Nearly all the known T. gondii PRRs are membrane-harbored on plasma membrane or endosome. Cytoplasmic PPR pathways are also crucial for sensing invading pathogens and initiating host defense. The cytoplasmic sensors for virus and bacterium have been well-characterized (15–18). Moreover, T. gondii is an intracellular pathogen. A previous study (19) showed that NLRP1 is an inflammasome sensor for T. gondii, which influences susceptibility to human congenital toxoplasmosis (20). However, T. gondii was not able to activate NLRP1 signaling without pretreatment of lipopolysaccharide. Moreover, the inflammasome mainly mediated maturation of proinflammatory cytokines and cell pyroptosis but not induction of immune genes. Therefore, the PRRs of T. gondii, particularly the cytoplasmic pathogen-sensing pathway modulating induction of immune effectors, wait to be uncovered.
The classic antiviral cytokine interferon (IFN) β can be induced during parasitic infection. It has been shown that IFNβ was able to inhibit replication of T. gondii (21, 22). A recent study showed that a small group of atypical strains are able to induce type I IFN production in bone marrow-derived macrophages (BMDM) in a RIG-I- and IRF3-dependent manner. The canonical type I, II, or III strains failed to trigger type I IFN induction (23). However, Robey and co-workers (24) found inflammatory monocytes produce IFNβ in response to type II T. gondii infection, suggesting different types of cells respond to T. gondii infection differently.
cGMP-AMP synthase (cGAS) is the cytoplasmic double-stranded DNA (dsDNA) sensor recognizing foreign dsDNA and abnormal host dsRNA, which is critical for defending against DNA viruses and regulating homeostasis of immune system (25, 26). cGAS synthesizes secondary messenger cGAMP, which in turn activates STING (stimulator of interferon genes protein). STING is a key signal molecule localized in ER. Activated STING recruits kinase TBK1 that phosphorylates IRF3/IRF7 to initiate transcription of type I IFNs. It is believed that post-translational modification of STING modulates its activity and protein stability. TBK1 phosphorylates STING at serine 366, which is critical for IRF3 recruitment and type I IFN induction (27). E3 ligase TRIM56 is able to attach Lys-63–linked polyubiquitin chain to STING and promotes dimerization and activation of STING (28). E3 protein complex autocrine motility factor receptor and insulin-induced gene 1 (INSIG1) promote Lys-27–linked polyubiquitination of STING and direct its translocation from the ER to the perinuclear vesicle (29). RNF5 ubiquitinates STING at lysine 150 resulting in its degradation by the proteasome, which could be antagonized by RNF26 that attaches Lys-11-linked polyubiquitin to the same lysine of STING (30, 31). However, the role of cGAS/STING in the host defense against T. gondii has not been well-characterized.
Accumulating studies have shown that T. gondii virulence is mediated by effector proteins secreted by parasites into the host cell. Dense granule proteins (GRAs) are produced by T. gondii, many of which are secreted into the parasitophorous vacuole (PV) and contribute to the maturation and structural integrity of the PV (32). GRA proteins also participate in the modulation of host defense during infection (33, 34). GRA proteins mainly suppress host immune responses and facilitate parasite replication (35). However, GRA15 activates NF-κB signaling in a TRAF6-dependent way, and in turn it up-regulates the production of IL12 (36). Moreover, GRA15 promotes the secretion of IL1β via inflammatory monocytes during infection (37).
In this study, we found that cGAS is required for innate immune response against T. gondii. cGAS-deficient mice are much more susceptible to lethal infection of T. gondii than WT mice. Loss of STING results in a more severe phenotype during T. gondii infection. Mechanistically, we demonstrated that secreted dense granule protein GRA15 promotes oligomerization and activation of STING. GRA15 promotes polyubiquitination of STING at lysine 337 via TRAF proteins, which is required for activation of STING. Together, GRA15 enhances cGAS/STING signaling by potentiating STING activity during T. gondii infection.
Results
cGAS/STING axis is critical for anti-T. gondii immunity in vivo and in vitro
The cGAS/STING axis recognizes cytoplasmic DNA and triggers innate immune responses. Here, we investigated the role of STING in host defense against the infection of T. gondii. The STING loss–of–function Tmem173gt/gt mice (also known as Golden ticket and here referred to Stinggt/gt) (38) and WT mice were infected with type II T. gondii (ME49 strain). Interestingly, Stinggt/gt mice were more susceptible to T. gondii infection than WT animals (Fig. 1A). Consistent with the survival results, the parasite burden (as assessed by T. gondii B1 DNA) was significantly higher in the spleen of Stinggt/gt mice than WT mice after T. gondii infection (Fig. 1B). Furthermore, we detected less type I IFNs, including IFNβ and IFNa4 in the spleen of Stinggt/gt mice than WT mice during infection (Fig. 1C and Fig. S1A). Consequently, induction of interferon-stimulated genes, including ISG15 and CXCL10, was significantly impaired in STING mutant mice. In addition to type I IFNs, STING is also required for the induction of inflammatory cytokine interleukin 12 (IL12), which is believed critical for controlling T. gondii infection (Fig. 1C). Consistently, the induction of IFNγ is modestly decreased in Stinggt/gt mice (Fig. 1C). Therefore, STING is required for host defense against infection with T. gondii in vivo. At the cellular level, we isolated peritoneal macrophages from WT and Stinggt/gt mice and infected these cells with T. gondii. In line with the animal experiments, macrophages with mutant STING failed to mount production of IFNβ, ISG15, CXCL10, and IL12 during T. gondii infection (Fig. 1D).
Figure 1.

Stinggt/gt mice are susceptible to T. gondii infection. A, C57BL/6 wildtype (WT, black lines, n = 6) and Stinggt/gt (red lines, n = 8) mice were infected with 105 T. gondii tachyzoites per mouse, and survival and body weight were monitored. B, mice (n = 4) were infected with T. gondii as in A, and spleens from live animals were harvested 5 days post-infection, followed by DNA isolation. Parasite growth was quantified by qRT-PCR of TgB1 primers and normalized against tubulin. C, RNA was isolated after spleens were harvested as in B. Ifng, Il12a, Ifnb1, and Isg15 mRNA was quantified by qRT-PCR. D, qRT-PCR analysis of Il12a, Ifnb1, Isg15, and Cxcl10 in WT or Stinggt/gt peritoneal macrophages after 12 h of T. gondii infection. E, parasite growth was qualified by qRT-PCR in WT or Sting−/− iBMDM cells after 24 h of T. gondii infection. F, qRT-PCR analysis of Il12a, Isg15, and Cxcl10 mRNA in WT or Sting−/− iBMDM cells after 24 h of T. gondii infection. G, C57BL/6 wildtype (WT, black lines, n = 6) and Cgas−/− (red lines, n = 7) mice were infected with 105 T. gondii tachyzoites per mouse, and survival was monitored. Statistical analysis was performed by log rank test for A and G and t test for B–F. Data were from three independent experiments. NS, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. Error bars, S.D.
To further confirm these findings, we infected immortalized Bone Marrow-Derived Macrophages (iBMDM) with T. gondii after deleting endogenous STING by using CRISPR/Cas9. The lacking of STING resulted in more parasite replication in iBMDM, which was measured by immunofluorescence and T. gondii B1 DNA qRT-PCR (Fig. 1E and Fig. S1B). The induction of ISGs and IL12 was also significantly decreased in STING-deficient iBMDM, although the induction of these cytokines in WT iBMDM was not as robust as that in WT primary peritoneal macrophages (Fig. 1F). Interestingly, we did not observe the induction of IFNβ by T. gondii infection in this cell type (Fig. S1C). This could be due to the specificity of cell type–dependent pathways or immune evasion strategies utilized by T. gondii.
As cGAS is a cytoplasmic DNA sensor that functions upstream of STING (39), we reasoned that cGAS might also be involved in anti-T. gondii responses by sensing the cytoplasmic DNA. We then evaluated the involvement of cGAS in anti-T. gondii immune responses. The mouse survival experiment showed that cGAS was required for anti-T. gondii responses (Fig. 1G). In addition to DNA, RNA from the invading pathogen can also trigger innate immunity of the host. RLRs recognize foreign RNA in the cytoplasm and activate adaptor protein MAVS. To this end, we infected MAVS-deficient mice, whose cytoplasmic RNA-sensing pathway was blocked, or WT mice with T. gondii. The survival and body weight results showed that MAVS was dispensable for anti-T. gondii immune responses (Fig. S1D). Together, cGAS and STING are crucial for innate immunity against T. gondii.
GRA15 is required for mounting innate immune response during T. gondii infection
During T. gondii infection in cGAS-deficient mice, interestingly, we noticed that loss of cGAS resulted in less severe symptoms and delayed death than STING deficiency, indicating an additional mechanism contributing to STING-mediated defense exists. We next focused on revealing this additional mechanism by which T. gondii activates STING signaling. In addition to nucleic acids, proteins from pathogens are also able to regulate host immune responses (35, 40). It is believed that the effector proteins secreted by T. gondii are able to modulate host immune responses (41). Dense granule proteins (GRAs) have emerged as important determinants of T. gondii pathogenesis. To determine whether the T. gondii proteins affect innate immune activation, we constructed plasmids encoding various dense granule proteins of T. gondii and transfected these plasmids together with cGAS, STING, and IFNβ luciferase reporter plasmids. We found that only GRA15, which was reported to regulate NF-κB activation (36), was able to significantly enhance cGAS/STING-mediated induction of IFNβ (Fig. 2A). We then investigated the effect of GRA15 on parasite virulence. We generated GRA15-deficient T. gondii by using CRISPR/Cas9 (Fig. 2B). In contrast to the previous finding (36), C57BL/6 mice were more susceptible to GRA15−/− T. gondii than WT T. gondii (Fig. 2C). Consistent with the survival data, mice infected with GRA15−/− T. gondii showed significantly higher parasite burden in the spleen than mice infected with WT T. gondii (Fig. 2D). Moreover, replication of GRA15−/− T. gondii significantly increased in MEFs compared with WT parasites (Fig. S2A). Furthermore, GRA15−/− T. gondii induced less IFNβ, ISG15, CXCL10, IFNγ, and IL12 than WT T. gondii did in the spleen (Fig. 2E), although the decrease of IFNβ induction was not as significant as other cytokines. Consequently, GRA15−/− parasite infection caused more severe mortality of mice. These results suggested that GRA15 is required for competent induction of innate immune responses. To further confirm this, we infected iBMDM with WT or GRA15−/− T. gondii. qRT-PCR assay showed that the production of cytokines was severely impaired during GRA15−/− T. gondii infection compared with WT parasite infection (Fig. 2F). To examine whether GRA15 is involved in STING signaling in vivo, we infected Stinggt/gt mice with WT or GRA15−/− parasite. The infection of WT or GRA15−/− parasite resulted in similar phenotypes in Stinggt/gt mice (Fig. 2G). Similarly, the GRA15−/− parasite failed to cause more severe toxoplasmosis in Cgas−/− mice (Fig. S2B), suggesting that GRA15 might enhance innate immunity in a cGAS/STING-dependent manner.
Figure 2.

GRA15 is vital for T. gondii-stimulated immune response. A, in HEK293 cells overexpressing cGAS and STING, luciferase activity of IFNβ-luc was measured after transfection of plasmids encoding different genes of T. gondii. B, GRA15−/− T. gondii was constructed by CRISPR/Cas9. Mutation was checked by DNA sequencing of GRA15. Total infected cell proteins at 48 h post-infection were analyzed by immunoblotting to detect T. gondii GRA15 and SAG1 proteins. C, WT mice were infected with 105 WT (black lines, n = 6) or GRA15−/− (red lines, n = 6) T. gondii tachyzoites per mouse, and survival and body weight were monitored. D, mice (n = 3) were infected with T. gondii as in C, and spleens from live animals were harvested 5 days post-infection, followed by DNA isolation. Parasite growth was quantified by qPCR of TgB1 primers and normalized against tubulin. E, RNA was isolated after spleens were harvested as in D. Il12a, Ifng, Ifnb1, Isg15, and Cxcl10 mRNA was quantified by qRT-PCR. F, qRT-PCR analysis of Il12a, Isg15, and Cxcl10 mRNA in WT iBMDM cells after 24 h of WT or GRA15−/− T. gondii infection. G, C57BL/6 Stinggt/gt mice were infected with 105 WT (black lines, n = 8) or GRA15−/− (red lines, n = 8) T. gondii tachyzoites, and survival and body weight were monitored. Statistical analysis was performed by log rank test for C and G and t test for A and D–F. Data were from three independent experiments. NS, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. Error bars, S.D.
GRA15 enhances innate immunity via STING
To strengthen the point that GRA15 facilitates immune responses in a cGAS/STING-dependent way, we cotransfected GRA15 and cGAS/STING into cells and performed IFNβ reporter assay. GRA15 significantly enhanced the induction of IFNβ by STING but not by MyD88 (Fig. S3A). Strikingly, a robust induction of IFNβ was observed in the presence of cGAS/STING and GRA15, which was confirmed by qRT-PCR (Fig. S3A). It is known that GRA15 is able to activate NF-κB, which also contributes to transcriptional activation of IFNβ. To this end, we knocked out RELA (also termed as p65), a subunit of NF-κB (Fig. S3B), from HEK293 cells and investigated the activation of IFNβ. The activation of the NF-κB reporter was abrogated in RELA-deficient cells (Fig. S3C). However, GRA15 was still able to synergize IFNβ induction by cGAS/STING (Fig. 3A). Consistently, infection with WT but not GRA15−/− T. gondii enhanced activation of IFNβ and IL12 (Fig. 3B). Accordingly, IFNβ enhancement was also observed in RELA-deficient cells (Fig. S3D). These results suggested that GRA15 also activates NF-κB–independent innate immune signaling, which contributes to type I IFN enhancement. To further determine the detailed mechanism by which GRA15 enhances innate immunity, we delivered GRA15 into iBMDM with lentivirus and performed genome-wide RNA-seq analysis. GRA15 induced expression of a group of ISGs, including Ddx58, Ifih1, Ifit1, Ifit2, Ifit3, Irf1, Irf7, and Isg15 (Fig. 3, C and D). Ddx58 and Ifih1 encode RIG-1 and MDA5, respectively, which are important PRRs recognizing RNA viruses in cytoplasm. IRF1 and IRF7 are key transcription factors that direct transcription of type I IFNs. The rest of these ISGs are also critical for host defense against different kinds of pathogens. Consistent with the above data that GRA15 enhances cGAS/STING signaling, the RNA-seq analysis showed that GRA15 induced the expression of the genes involved in innate immune responses, defense response to virus, and herpes simplex infection (Fig. 3E). Moreover, induction of these ISGs by GRA15, including GBP3, was significantly attenuated in STING-deficient macrophages (Fig. 3F). Some ISG activation was impaired in cGAS-deficient macrophages (Fig. S3E), suggesting that cGAS is also important for GRA15-mediated signaling.
Figure 3.

GRA15 enhances STING-mediated anti-T. gondii immunity. A, luciferase activity of IFNβ-luc in p65−/− HEK293 cells 24 h after transfection with indicated reporter plasmids and empty vector (Vec) or GRA15. B, luciferase activity of IFNβ-luc and IL12-luc was measured 24 h after WT or GRA15−/− T. gondii infection in HEK293 cells overexpressing cGAS and STING for 24 h. C, selected activated genes in iBMDM cells overexpressing GRA15. D, RT-PCR experiments showing the expression of some ISGs, IRG, and GBP were increased in iBMDM cells overexpressing GRA15. E, histogram of the combined score (https://david.ncifcrf.gov/tools.jsp) (58, 59) for biological processes (up) and pathway (down) for the activated genes in iBMDM cells overexpressing GRA15. F, qRT-PCR experiments showing some genes were activated in WT iBMDM cells overexpressing GRA15 but not in Sting−/− iBMDM cells. G, luciferase activity of IFNβ-luc was measured in HEK293 cells overexpressing full-length (FL-GRA15) and different deletions of GRA15 with STING. H, luciferase assay of IFNβ-luc of HEK293 cells transfected with plasmids encoding empty vector (Vec), GRA15 full-length (FL-GRA15), GRA15 without transmembrane domain 1 (GRA15 Δmem1), or GRA15 without transmembrane domain 2 (GRA15 Δmem2) with cGAS and STING. Statistical analysis was performed by t test for A, B, and F–H. Data were from three independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001. Error bars, S.D.
To gain further insight into the mechanism by which GRA15 facilitates innate immunity, we constructed a series of GRA15 truncations and examined their function. Reporter assay showed that the first 100 amino acids were essential for the induction of both IFNβ and NF-κB (Fig. 3G and Fig. S3F). We then analyzed these amino acids and found two transmembrane motifs within the first 100 amino acids (Fig. S3G). However, the role of these transmembrane motifs has not been characterized (42). We deleted these two transmembrane motifs and examined their function in activating IFNβ. We found that the first transmembrane motif was dispensable for the enhancement of IFNβ. However, the lack of the second transmembrane motif almost completely blocked the synergistic effect of GRA15 on IFNβ induction (Fig. 3H). The second transmembrane motif is also required for activation of NF-κB by GRA15 (Fig. S3H), although we did not observe a synergistic effect of GRA15 and STING on NF-κB activation (Fig. S3F). Taken together, GRA15 enhances the innate immune responses by cGAS/STING, which relies on the second transmembrane motif.
GRA15 anchors to the ER of host cells and activates STING
It is believed that GRA15 is secreted into host cells during T. gondii infection (36). We found that the second transmembrane motif is critical for the function of GRA15, which prompted us to investigate the distribution of GRA15 in the host cell. Surprisingly, we observed that GRA15 is mainly located on ER, as assessed by immunofluorescence (Fig. 4A and Fig. S4A). To confirm this, we performed subcellular fractionation to isolate the ER, and we detected the localization of GRA15. Consistent with the immunofluorescence results, GRA15 was exclusively presented in the fraction of the ER. However, most of the GRA15 translocated into cytosol when the second transmembrane motif was deleted, suggesting this transmembrane motif is responsible for anchoring GRA15 to the ER (Fig. 4B). It is known that STING localizes on the ER, which prompted us to examine the possibility that GRA15 might interact with STING. To this end, we transfected STING and GRA15 together into cells and performed coimmunoprecipitation. The result confirmed that GRA15 could interact with STING, but not cGAS (Fig. 4C). Moreover, ER localization was critical for the interaction between STING and GRA15 (Fig. 4D). More interestingly, GRA15 promoted the oligomerization of STING, which is believed to be the active form of STING (Fig. 4E). To strengthen this point, we stained STING in HeLa cells transfected with GRA15 or T. gondii actin. The immunofluorescence results further confirmed that GRA15 promoted the oligomerization of STING (Fig. 4F). Furthermore, the translocation of STING to the Golgi apparatus and increased recruitment of TBK1 induced by GRA15 implied that STING was further activated by GRA15 (Fig. S4, B and C). Therefore, GRA15 interacts with STING and promotes its oligomerization, translocation, and interaction with TBK1 to activate immune response.
Figure 4.

GRA15 located on ER to activate immune response. A, immortalized MEF cells were infected by T. gondii and at 24 h post-infection stained for nucleus (DAPI, blue), T. gondii (green), GRA15 (red), and endoplasmic reticulum (purple) and visualized by confocal microscopy. The scale bar length is 10 μm. B, HEK293 cells were transfected with plasmids encoding GRA15 FL or GRA15 Δmem2, following immunoblot analysis of WCL, isolated ER, and cytosol. C, immunoblot analysis of HEK293 cells transfected plasmids encoding His-GRA15, Flag-STING, or Flag-cGAS, lysed, and immunoprecipitated (IP) with anti-Flag. WCLs were immunoblotted with antibodies to the indicated proteins (* indicates nonspecific band). D, immunoblot analysis of HEK293 cells transfected with plasmids encoding His-GRA15 ΔMEM2, Flag-STING, lysed, and immunoprecipitated with anti-Flag. WCLs were immunoblotted with antibodies to the indicated proteins. E, SDD-AGE and SDS-PAGE of HEK293 cells expressing Flag-STING and His-GRA15. F, 2fTGH cells were transfected with plasmids encoding Flag-GRA15 or Flag-TgActin and at 24 h stained for STING (green) and Flag (red) and visualized by confocal microscopy. The scale bar length is 10 μm.
GRA15 promotes ubiquitination and activation of STING
The previous report showed that STING could be targeted by TRIM56 for lysine 63–linked ubiquitination, which induced STING dimerization. This dimerization is a prerequisite for the recruitment of antiviral kinase TBK1 and subsequent induction of IFNβ (28). Therefore, to determine whether the post-translational modification (PTM) of STING is affected by GRA15, we overexpressed GRA15 in HEK293 cells transfected with plasmids encoding STING and purified Flag-STING by immunoprecipitation (Fig. S5, A and B). PTM analysis of STING by MS identified lysine 337 of STING was ubiquitinated in 293T cells expressing GRA15 (Fig. 5A and Fig. S5C). Next, we constructed a Flag-STING mutant in which lysine 337 was substituted with an alanine residue (K337A). Notably, STING K337A was not able to potentiate IFNβ-luc activation effectively. Moreover, the synergistic effect between GRA15 and STING was dampened when lysine 337 of STING is replaced with alanine (Fig. 5B). We then mutated this alanine back to lysine (K337A re), which completely rescued the function of STING (Fig. 5B). Accordingly, GRA15 promotes ubiquitination of WT STING but not the K337A mutant (Fig. 5C). As noted above, GRA15 promoted the oligomerization of STING. We then determined whether this ubiquitination is related to the oligomerization of STING. The SDD-AGE assay showed lysine 337 is required for oligomerization of STING (Fig. 5D). It is well-established that STING functions via IRF3 and NF-κB (26). We examined the effect of this mutation on transcription activation. The K337A mutation was able to induce NF-κB activation, which is comparable with WT STING. However, luciferase activity induction of P561, which is believed to be an IRF3-specific activating element (43), was significantly impaired by this mutant (Fig. 5E). Therefore, GRA15 can promote lysine 337 ubiquitination of STING, leading to oligomerization and activation. The activated STING stimulates downstream signals via IRF3.
Figure 5.
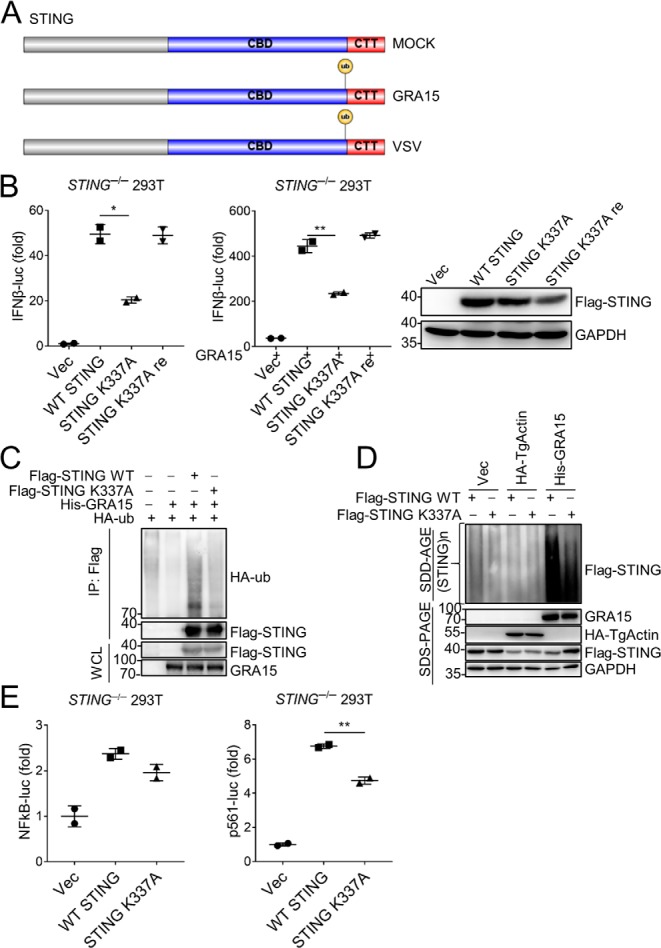
GRA15 promotes activation of STING. A, diagram detailing STING domains (CBD and CTT) and ubiquitinated site. B, left: luciferase assay of IFNβ-luc in STING−/− HEK293 cells 24 h after transfection with plasmids encoding WT, mutant STING (K337A), or STING recover (K337A re). Middle: luciferase assay as left in STING−/− HEK293 cells overexpressing GRA15. Right: immunoblot analysis of HEK293 cells transfected with plasmids encoding empty vector (Vec), WT, mutant STING (K337A), or STING recover (K337A re). C, immunoblot analysis of HEK293 cells transfected with plasmids encoding HA-ub, His-GRA15, Flag-STING WT, or Flag-STING mutant (K337A), lysed, and immunoprecipitated (IP) with anti-Flag. WCLs were immunoblotted with antibodies to indicated proteins. D, SDD-AGE and SDS-PAGE of STING−/− HEK293 cells expressing WT STING, STING K337A, Flag-GRA15, or Flag-TgActin. E, luciferase assay of NF-κB-luc and P561-luc in STING−/− HEK293 cells 24 h after transfection with plasmids encoding WT or mutant STING (K337A). Statistical analysis was performed by t test for B and E. Data were from three independent experiments. *, p < 0.05; **, p < 0.01. Error bars, S.D.
GRA15 relies on TRAFs to activate STING
To more clearly understand GRA15-related immunity and to identify GRA15-interacting factors, we overexpressed Flag-GRA15 in HeLa cells and purified Flag-GRA15 by immunoprecipitation. Mass spectrometry analysis identified TRAF2 and TRAF6 as GRA15-interacting proteins (Fig. S6A). The interactions were confirmed by coimmunoprecipitation (Fig. S6B). To further evaluate the importance of TRAF2 and TRAF6, we knocked out TRAF2/TRAF6 separately or together in HEK293 cells by using CRISPR/Cas9. NF-κB activation was abrogated in TRAF6-deficient cells and decreased in TRAF2-deficient cells (Fig. S6C). However, we observed that GRA15 was still able to synergize with cGAS and STING in TRAF6−/−, TRAF2−/−, or TRAF2/6−/− cells (Fig. 6A and Fig. S6D). This result suggests again there is an NF-κB–independent way that mediated GRA15/cGAS/STING immune response. It is known that TRAF proteins form heterodimers and function redundantly (44). We thus did not observe complete abolishment of IFNβ enhancement by GRA15 in TRAF2- and TRAF6-deficient cells. To this end, we performed this experiment with TRAF2, TRAF3, TRAF5, and TRAF6 four gene-deficient cells (TRAFs−/−). The results showed that GRA15 was not able to enhance IFNβ induction by cGAS/STING without TRAF proteins (Fig. 6, B and C). Accordingly, the oligomerization of STING induced by GRA15 was abolished in TRAFs−/− 293T cells (Fig. 6D). In line with this, GRA15 was not able to interact with STING in TRAFs-deficient cells (Fig. 6E), suggesting that TRAFs bridged the interaction between GRA15 and STING. To this end, we performed yeast two-hybridization to confirm the interaction among these proteins. GRA15 directly binds to TRAF2 or TRAF6 but not STING (Fig. S6E). In line with that, the coimmunoprecipitation results indicated that TRAF2, -5, and -6 could bind to STING significantly, whereas the interaction between TRAF3 and STING was much weaker (Fig. 6F). Accordingly, TRAF2 and TRAF6 can be recruited to ER by GRA15 (Fig. S6F). Taken together, TRAF proteins play a vital role in cGAS/STING/GRA15-mediated immune response by bridging the interaction between GRA15 and STING.
Figure 6.

GRA15 depends on TRAFs to activate STING. A, qRT-PCR analysis of IFNB1 mRNA in WT, TRAF6−/−, and TRAF2−/− HEK293 cells overexpressing GRA15, cGAS, and STING. B, qRT-PCR analysis of IFNB1 mRNA in TRAFs−/− HEK293 cells overexpressing GRA15, cGAS, and STING. C, enhancement of IFNβ-luc activity in TRAF2−/−, TRAF6−/−, TRAF2/6−/−, TRAF3−/−, TRAF5−/−, or TRAFs−/− HEK293 cells overexpressing GRA15, cGAS, and STING. D, SDD-AGE and SDS-PAGE of WT or TRAFs−/− HEK293 cells overexpressing Flag-STING and His-GRA15. E, immunoblot analysis of WT and TRAFs−/− HEK293 cells transfected with indicated plasmids, lysed, and immunoprecipitated with anti-Flag antibody. WCLs were immunoblotted with antibodies to the indicated proteins. F, immunoblot analysis of HEK293 cells transfected with plasmids encoding HA-STING, Flag-TRAF2, Flag-TRAF3, Flag-TRAF5, or Flag-TRAF6, lysed, and immunoprecipitated with anti-Flag antibody. WCLs were immunoblotted with antibodies to the indicated proteins. Statistical analysis was performed by t test for A–C. Data were from three independent experiments. NS, not significant; *, p < 0.05; **, p < 0.01; and ***, p < 0.001. Error bars, S.D.
Discussion
T. gondii is an important intracellular pathogen that usually establishes latent infection in the nervous system. As an intracellular pathogen, its cytoplasmic PRR has not been well-characterized as yet. We identified here that cGAS participates in antiparasite immune responses during T. gondii infection. The source of the dsDNA-activating cGAS, T. gondii-derived or host-derived, remains to be elucidated. ME49 used in this study is a type II strain of T. gondii, one of the canonical strains. Different from the atypical T. gondii strains (23), canonical strains could not activate the RLR-signaling pathway to induce IFNβ production. MAVS-deficient mice showed similar symptoms as WT mice after challenge with T. gondii ME49 (Fig. S1D). In addition to cGAS, other cytoplasmic dsDNA sensors, including DDX41, IFI16, DAI, IFI204, and AIM2 (45–48), may compensate for loss of cGAS or also function in activating the immune response against T. gondii, which could explain the fact that the delayed death of cGAS-deficient mice compared with STING-deficient mice after T. gondii infection. It will be interesting to investigate the function of these receptors in host defense against T. gondii.
After recognizing dsDNA, cGAS activates STING and downstream signaling. In addition to the signals from cGAS, T. gondii dense granule protein GRA15 promotes activation of STING in a TRAF protein–dependent way. We confirmed the direct interaction between TRAF2 or TRAF6 and GRA15 with coimmunoprecipitation and yeast two-hybridization. However, we still observed enhancement of type I interferon induction by GRA15 in TRAF2-deficient, TRAF6-deficient, or TRAF2 and TRAF6 double-knockout cells. This enhancement is completely abolished in TRAF2, TRAF3, TRAF5, and TRAF6 four gene knockout cells. TRAF2/TRAF5 and TRAF6 are believed to play important roles in NF-κB activation. It is known that GRA15 activates NF-κB via TRAF6. TRAF6 deficiency abrogates NF-κB activation but not IFNβ enhancement by GRA15. GRA15 activates IFNβ production, at least partially, in an NF-κB–independent way. Consistently, we still observed IFNβ up-regulation by GRA15 in p65-deficient cells. However, TRAF3 is believed to play a role in activation of TBK1 and IRF3/IRF7. Deficiency of TRAF3 could not block the enhancement of IFNβ induction by GRA15 either. Each TRAF protein seems to be required for this enhancement of IFNβ induction by GRA15 (Fig. 6). Therefore, these data suggested that GRA15 activated both NF-κB and IRF pathways. Our results suggested that STING activated by GRA15 mainly depends on IRF3 to induce target gene expression (Fig. 5E).
Consistent with a previous study (23), we did not observe type I IFN induction in iBMDM during T. gondii ME49 infection (Fig. S1C). However, IFNβ was significantly induced in peritoneal macrophages by T. gondii ME49 infection (Fig. 1D). Moreover, the induction of IL12, ISG15, and CXCL10 was also more significant in peritoneal macrophages than in iBMDM during T. gondii ME49 infection (Fig. 1, D and F). We also observed type I IFN induction in mouse spleen during T. gondii infection (Fig. 1C and Fig. 2E). These results suggested that different types of cells develop different strategies to counteract T. gondii evasion. Therefore, T. gondii induces type I IFN in a cell type–dependent manner. However, we found here that type I IFNs and ISGs are up-regulated via cGAS/STING in vitro and in vivo. The importance of type I IFNs in controlling T. gondii infection is still controversial (49, 50). Barik and co-workers (51) showed that induction of ISGs by IRF3 promotes replication of T. gondii when they infected mice with a tachyzoite of type I T. gondii (RH strain). Robey and co-workers (24) found type I IFN is required for defense against T. gondii when they challenged mice with cysts of type II T. gondii (Prugniaud strain). In this study, we used type II T. gondii (ME49 strain) and observed that the cGAS/STING/IRF3 axis is required for inhibiting the infection of this parasite.
We found here that GRA15 promoted ubiquitination of STING at lysine 337 in WT cells but not in TRAF-deficient cells. TRAF proteins are known as the Ring finger domain containing ubiquitin E3 ligase (52). Further studies should be done to clarify the mechanistic details of which TRAFs act with GRA15 to direct ubiquitination of STING, and which type of polyubiquitin has been attached to STING. Our yeast two-hybridization assay showed that GRA15 directly interacted with TRAF2 and TRAF6 but not STING. Moreover, there was significant interaction between TRAF proteins and STING (Fig. 6). These results suggested that TRAF proteins function as an adaptor to bridge the association between GAR15 and STING. In line with this, we observed coimmunoprecipitation of GRA15 and STING in WT cells but not in TRAF-deficient cells. However, the mechanism by which GRA15 activates TRAF proteins is unknown.
GRA15 is a secreted dense granule protein of T. gondii. Interestingly, we here found that the second transmembrane motif of GRA15 directed its localization on ER. The ER localization is essential for its activity to potentiate innate immune responses, including NF-κB activation and type I IFN induction. Our immunofluorescence results suggested that GRA15 could recruit TRAF2 and TRAF6 to ER (Fig. S6F), allowing their engagement with STING. However, it is puzzling why ER localization is critical for GRA15 to activate NF-κB by TRAF6.
In conclusion, we demonstrated that T. gondii-derived protein GRA15 promotes STING polyubiquitination at Lys-337 to activate STING via TRAF proteins, which in turn enhances the DNA–cGAS/STING pathway for host defense against T. gondii infection (Fig. S7). The ER-localized GRA15 enhances activation of innate immune signaling to suppress the propagation of T. gondii, which might be needed for moderate replication of the parasite and establishment of latent infection in host cells because excessive T. gondii replication would kill hosts before cysts formation. This study found a cytoplasmic innate immune signaling modulator derived from invading microbes and shed new insights on how a microbial protein modulates host immune activation.
Experimental procedures
Viruses and growth of T. gondii
Vesicular stomatitis virus (Indiana strain) was a gift from J. Rose (Yale University).
All experiments in this paper used the T. gondii ME49 strain, a type II strain provided by Dr. Yang Zhao (Yale University). T. gondii stain was grown in HFF cells.
Mice and T. gondii infection
The care and use of all animals adhered to the Guide for the Care and Use of Laboratory Animals of the Chinese Association for Laboratory Animal Science. All procedures of animal handling were approved by the Animal Care and Use Committee of Peking University Health Science Center (permit number LA 2016240). Stinggt/gt mice and Mavs−/− mice have been described previously (53). WT B6 mice were purchased from the Department of Laboratory Animal Science of Peking University Health Science Center, Beijing, China. Mice were kept and bred in pathogen-free conditions.
Age- and sex-matched C57BL/6 littermates were produced and used in all the in vivo experiments. T. gondii was purified by filtration through 5-μm syringe filters, suspended in PBS, and counted in a hemocytometer under a microscope. Six-week-old mice were infected with 105 WT T. gondii or GRA15−/− T. gondii per mouse by intraperitoneal injection. For cytokine studies and T. gondii genome copy number measurements, mice were sacrificed at day 5 post-infection, and spleens were harvested.
Cells
iBMDM cells were a gift from Dr. Feng Shao (National Institute of Biological Sciences, Beijing, China). HFF cells were given by Dr. Guang Yang (Jinan University). HEK293 cells were purchased from American Type Culture Collection (ATCC) and cultured in DMEM supplemented with 10% FBS, 100 units/ml penicillin/streptomycin.
Luciferase reporter analysis
HEK293 cells seeded on 24-well plates were transiently transfected with 50 ng of the luciferase reporter plasmid together with a total of equal amounts of various expression plasmids or empty control plasmids. Then, 24 h later, reporter gene activity was analyzed using the Dual-Luciferase Reporter 1000 assay system (Promega) and measured with a TD-20/20 luminometer (Turner Designs) according to the manufacturers' instructions.
Regular RT-PCR and qRT-PCR
Total RNA from cells and homogenized tissues was isolated using the RNA simple total RNA kit (TIANGEN). 1 μg of RNA was reverse-transcribed using a FastKing RT kit (TIANGEN). Levels of the indicated genes were analyzed by RT-PCR for 28–35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 20 s or analyzed by qRT-PCR amplified using SYBR Green (Transgene). Data shown are the relative abundance of the indicated mRNA normalized to GAPDH or tubulin. The primers are listed in Table S1.
Coimmunoprecipitation
HEK293 cells seeded on 10-cm2 dishes (1 × 107 cells/dish) were transfected with a total of 10 μg of empty plasmid or various expression plasmids. At 24–36 h after transfection, cells were lysed in lysis buffer (0.5% Triton X-100, 20 mm HEPES (pH 7.4), 150 mm NaCl, 12.5 mm β-glycerophosphate, 1.5 mm MgCl2, 2 mm EGTA, 10 mm NaF, 1 mm Na3VO4, 2 mm DTT) containing protease inhibitors. Lysates were centrifuged and incubated with anti-Flag antibodies at 4 °C overnight. The next day, prewashed protein A/G beads (Pierce) were added and incubated at 4 °C for 4 h. The beads were washed with cold PBS four times and eluted with DTT-containing SDS sample buffer by boiling for 10 min for Western blotting.
Immunofluorescent microscopy
HeLa and MEF cells on coverslips were washed once with pre-warmed phosphate-buffered saline (PBS) and fixed in 4% (w/v) paraformaldehyde for 15 min. After three washes in PBS, cells were permeabilized with 0.2% (v/v) Triton X-100 for 5 min. After three washes in PBS, cells were blocked in PBS containing 5% (w/v) bovine serum albumin (BSA) for 30 min and incubated with the indicated antibodies in PBS containing 3% (w/v) BSA for 2 h at 37 °C. After three washes, cells were incubated with Alexa Fluor 488-conjugated secondary antibodies or Alexa Fluor 555-conjugated secondary antibodies for 1 h at 37 °C, and then with 4′,6-diamidino-2-phenylindole (DAPI, Roche Applied Science) for 15 min. The coverslips were washed extensively and mounted onto slides. Imaging of the cells was carried out using N-STORM5.0 (Nikon) microscope under a ×100 oil objective.
CRISPR/Cas9 system
Sting−/−, Cgas−/− iBMDM, and STING−/−, TRAF2−/−, TRAF6−/−, TRAF3−/−, TRAF5−/−, TRAF2/6−/−, and p65−/− HEK293 cells were constructed by CRISPR/Cas9 system. Specific guide RNA was ligated into the BsmBI restriction site of the inducible lentiviral vector (lentiGuide-Puro). Lentivirus particles were produced by cotransfected HEK293 cells with guide RNA plasmids (2 μg), packaging plasmids pCMV–VSV-G (800 ng, AddGene 8454), and psPAX2 (800 ng, AddGene 12260). The medium was changed to fresh DMEM containing 10% FBS at 12 h post-transfection, and the viral supernatant was collected at 48–72 h. Then, a total of 1 × 105 iBMDMs or HEK293 cells were infected with 2 ml of viral supernatant supplemented with 8 μg/ml Polybrene and incubated for 48 h. Possible knockout cells were screened by puromycin (1 μg), and each monoclone was confirmed by sequencing. Cells were negative for mycoplasma.
HEK293 cells were transfected by standard calcium phosphate precipitation method. HeLa cells were transfected by Lipofectamine 2000 (Invitrogen) according to procedures recommended.
GRA15−/− T. gondii were constructed as described previously (54). The high-efficiency gRNA was designed on http://grna.ctegd.uga.edu/5 (55). The forward primer is GAAGCGACTTCTAAACACGTGGTTTTAGAGCTAGAAATAGC and the reverse primer is AACTTGACATCCCCATTTAC. Then, the GRA15 gRNA replaced the target gRNA of parental plasmid pDHFR-SAG1::Cas9-U6::sgUPRT using the Q5® site-directed mutagenesis kit (New England Biolabs, E0552S). Next, for transfection and selection of the mutated parasites, T. gondii was nucleofected with 10 μg of circular pDHFR-SAG1::Cas9-U6::sgGRA15 plasmid using the Basic Parasite Nucleofector® kit 1 (Lonza, Basel, Switzerland) according to the manufacturer's instructions. Briefly, 106 to 107 T. gondii cells were suspended in 100 μl of nucleofection buffer (82 μl of Basic Parasite Nucleofector® solution 1 supplemented with 18 μl of Supplement P1). The 100 μl of the parasite suspension was mixed with DNA solution and transferred to a cuvette (provided as a component of the kit). T. gondii cells were nucleofected using program U-033 and were immediately inoculated into confluent HFF cells. After 2 days post-nucleofection, T. gondii cells were selected by 1 μm pyrimethamine in the 10% FBS/DMEM. The guiding RNAs are listed in Table S2.
Antibodies
The following antibodies were used: anti-HA (H3663); anti-tubulin (AF7011); anti-Flag (mouse, M183-3L); anti-Flag (rabbit, PM020); and anti-Lamin B1 (66095) from Proteintech. The antibodies were diluted 1000 times for immunoblots and 200 times in immunofluorescence. The mouse anti-GRA15 polyclonal antibodies were raised against bacterial pET-28a (+)–GRA15 (full-length) protein.
Mass spectrometry
To identify proteins that interact with Flag-GRA15 and post-translational modification of Flag-STING, samples pulled down with Flag antibody were sent for MS.
Plasmid constructs
For the plasmids coding human STING, MAVS have been described previously (53). Genes of T. gondii, including GRA15, were amplified by PCR and were cloned into pCMV7.1 plasmids or pcDNA3.1 plasmids to generate Flag-tagged or His-tagged expression constructs.
RNA sequencing
We harvested iBMDM-overexpressing GRA15 and control cells and purified whole RNA by using the RNeasy mini kit (Qiagen no. 74104). The transcriptome library for sequencing was generated using VAHTSTM mRNA-seq version 2 Library Prep kit for Illumina® (Vazyme Biotech Co., Ltd., Nanjing, China) following the manufacturer's recommendations. After clustering, the libraries were sequenced on IlluminaHiseq × Ten platform using (2 × 150 bp) paired-end module. The raw images were transformed into raw reads by base calling using CASAVA (Illumina). Then, raw reads in a fastq format were first processed using in-house perl scripts. Clean reads were obtained by removing reads with adaptors, reads in which unknown bases were more than 5%, and low-quality reads (the percentage of low-quality bases was over 50% in a read, and we defined the low-quality base to be the base whose sequencing quality was no more than 10). At the same time, Q20 and Q30 GC content of the clean data were calculated (Vazyme Biotech Co., Ltd., Nanjing, China). The original data of the RNA-seq was uploaded to the GEO DataSets.
SDD-AGE
Cells from a six-well plate were lysed with 100 μl of pre-cold lysis buffer (0.5% Triton X-100, 50 mm Tris, 150 mm NaCl, 10% glycerol, proteinase mixture, and phosphatase mixture) on ice for 40 min. Supernatants were collected by centrifugation at 13,000 rpm for 10 min at 4 °C. Protein loading buffer was added into the supernatants and loaded onto the gel. We performed vertical agarose electrophoresis for 35 min with a constant voltage of 100 V on ice, followed by standard Western blotting procedure.
Statistical analysis
RNA-seq data were analyzed using Enrichr (http://amp.pharm.mssm.edu/Enrichr)5 (56, 57). For all the scatter plots, data were expressed as mean ± S.D. Prism 7 software (graphic software) was used for survival curves, charts, and statistical analyses. Survival curves were analyzed using a log-rank (mantel-cox) test. For the in vitro results and animal results, a standard two-tailed unpaired Student's t test was used. The results with p value <0.05 were considered significant. The sample sizes (biological repeats), specific statistical tests used, and the main effects of our statistical analyses for each experiment are detailed in each figure legend. All attempts at replication were successful. The investigators were blinded to group allocation during data collection and analyses.
Author contributions
Peiyan Wang and F. Y. conceptualization; Peiyan Wang and S. Li data curation; Peiyan Wang software; Peiyan Wang formal analysis; Peiyan Wang, S. Li, Y. Z., B. Z., Y. L., and S. Liu validation; Peiyan Wang, S. Li, Y. Z., B. Z., Y. L., S. Liu, L. C., M. O., X. Y., P. L., and X. G. investigation; Peiyan Wang and S. Li visualization; Peiyan Wang and S. Li methodology; Peiyan Wang, H. D., and F. Y. writing-original draft; X. G., Penghua Wang, C. J., F. S., G. Y., and F. Y. supervision; F. Y. funding acquisition; F. Y. project administration; F. Y. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Zhengfan Jiang (Peking University, Beijing, China) for providing TRAF-deficient cell- and STING-deficient cells.
This work was supported by National Natural Science Foundation of China Grants 31570891 and 31872736 and National Key Research and Development Program of China Grant 2016YFA0500302. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Tables S1 and S2.
This article was selected as one of our Editors' Picks.
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE122906.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PRR
- pattern recognition receptor
- qRT-PCR
- quantitative RT-PCR
- WCL
- whole-cell lysate
- ER
- endoplasmic reticulum
- SDD-AGE
- semidenaturing detergent–agarose gel electrophoresis
- DC
- dendritic cell
- PV
- parasitophorous vacuole
- GRA
- granule
- IFN
- interferon
- TRAF
- TNF receptor-associated factor
- DAPI
- 4′,6-diamidino-2-phenylindole
- ISG
- interferon-stimulated gene
- cGAS
- cyclic GMP-AMP synthase
- iBMDM
- immortalized bone marrow-derived macrophage
- PTM
- post-translational modification
- HFF
- human foreskin fibroblast
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- RNA-seq
- RNA-sequencing
- MEF
- mouse embryo fibroblast
- RLR
- RIG-I-like (retinoic acid-inducible gene I-like) receptors
- MAVS
- mitochondrial antiviral-signaling protein.
References
- 1. Robert-Gangneux F., and Dardé M. L. (2012) Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 25, 264–296 10.1128/CMR.05013-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Q., Wang Z. D., Huang S. Y., and Zhu X. Q. (2015) Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit. Vectors 8, 292 10.1186/s13071-015-0902-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montoya J. G., and Liesenfeld O. (2004) Toxoplasmosis. Lancet 363, 1965–1976 10.1016/S0140-6736(04)16412-X [DOI] [PubMed] [Google Scholar]
- 4. Yarovinsky F. (2014) Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 14, 109–121 10.1038/nri3598 [DOI] [PubMed] [Google Scholar]
- 5. Koblansky A. A., Jankovic D., Oh H., Hieny S., Sungnak W., Mathur R., Hayden M. S., Akira S., Sher A., and Ghosh S. (2013) Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38, 119–130 10.1016/j.immuni.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrade W. A., Souza Mdo C., Ramos-Martinez E., Nagpal K., Dutra M. S., Melo M. B., Bartholomeu D. C., Ghosh S., Golenbock D. T., and Gazzinelli R. T. (2013) Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13, 42–53 10.1016/j.chom.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pisitkun P., Deane J. A., Difilippantonio M. J., Tarasenko T., Satterthwaite A. B., and Bolland S. (2006) Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672 10.1126/science.1124978 [DOI] [PubMed] [Google Scholar]
- 8. Edwards A. D., Diebold S. S., Slack E. M., Tomizawa H., Hemmi H., Kaisho T., Akira S., and Reis e Sousa C. (2003) Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 10.1002/eji.200323797 [DOI] [PubMed] [Google Scholar]
- 9. Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., and Akira S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- 10. Gissot M., Choi S. W., Thompson R. F., Greally J. M., and Kim K. (2008) Toxoplasma gondii and Cryptosporidium parvum lack detectable DNA cytosine methylation. Eukaryot. Cell 7, 537–540 10.1128/EC.00448-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Debierre-Grockiego F., Campos M. A., Azzouz N., Schmidt J., Bieker U., Resende M. G., Mansur D. S., Weingart R., Schmidt R. R., Golenbock D. T., Gazzinelli R. T., and Schwarz R. T. (2007) Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J. Immunol. 179, 1129–1137 10.4049/jimmunol.179.2.1129 [DOI] [PubMed] [Google Scholar]
- 12. Mun H. S., Aosai F., Norose K., Piao L. X., Fang H., Akira S., and Yano A. (2005) Toll-like receptor 4 mediates tolerance in macrophages stimulated with Toxoplasma gondii-derived heat shock protein 70. Infect. Immun. 73, 4634–4642 10.1128/IAI.73.8.4634-4642.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leng J., and Denkers E. Y. (2009) Toxoplasma gondii inhibits covalent modification of histone H3 at the IL-10 promoter in infected macrophages. PLoS One 4, e7589 10.1371/journal.pone.0007589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee E. J., Heo Y. M., Choi J. H., Song H. O., Ryu J. S., and Ahn M. H. (2008) Suppressed production of pro-inflammatory cytokines by LPS-activated macrophages after treatment with Toxoplasma gondii lysate. Korean J. Parasitol. 46, 145–151 10.3347/kjp.2008.46.3.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delbridge L. M., and O'Riordan M. X. (2007) Innate recognition of intracellular bacteria. Curr. Opin. Immunol. 19, 10–16 10.1016/j.coi.2006.11.005 [DOI] [PubMed] [Google Scholar]
- 16. Akira S., Uematsu S., and Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 17. Neyen C., and Lemaitre B. (2016) Sensing Gram-negative bacteria: a phylogenetic perspective. Curr. Opin. Immunol. 38, 8–17 10.1016/j.coi.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 18. Wilkins C., and Gale M. Jr. (2010) Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22, 41–47 10.1016/j.coi.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ewald S. E., Chavarria-Smith J., and Boothroyd J. C. (2014) NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 82, 460–468 10.1128/IAI.01170-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Witola W. H., Mui E., Hargrave A., Liu S., Hypolite M., Montpetit A., Cavailles P., Bisanz C., Cesbron-Delauw M. F., Fournié G. J., and McLeod R. (2011) NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect. Immun. 79, 756–766 10.1128/IAI.00898-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitz J. L., Carlin J. M., Borden E. C., and Byrne G. I. (1989) β-Interferon inhibits Toxoplasma gondii growth in human monocyte-derived macrophages. Infect. Immun. 57, 3254–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Orellana M. A., Suzuki Y., Araujo F., and Remington J. S. (1991) Role of β-interferon in resistance to Toxoplasma gondii infection. Infect. Immun. 59, 3287–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Melo M. B., Nguyen Q. P., Cordeiro C., Hassan M. A., Yang N., McKell R., Rosowski E. E., Julien L., Butty V., Dardé M. L., Ajzenberg D., Fitzgerald K., Young L. H., and Saeij J. P. (2013) Transcriptional analysis of murine macrophages infected with different Toxoplasma strains identifies novel regulation of host signaling pathways. PLoS Pathog. 9, e1003779 10.1371/journal.ppat.1003779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han S. J., Melichar H. J., Coombes J. L., Chan S. W., Koshy A. A., Boothroyd J. C., Barton G. M., and Robey E. A. (2014) Internalization and TLR-dependent type I interferon production by monocytes in response to Toxoplasma gondii. Immunol. Cell Biol. 92, 872–881 10.1038/icb.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai X., Chiu Y. H., and Chen Z. J. (2014) The cGAS–cGAMP–STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 10.1016/j.molcel.2014.03.040 [DOI] [PubMed] [Google Scholar]
- 26. Chen Q., Sun L., and Chen Z. J. (2016) Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 10.1038/ni.3558 [DOI] [PubMed] [Google Scholar]
- 27. Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y. T., Grishin N. V., and Chen Z. J. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 10.1126/science.aaa2630 [DOI] [PubMed] [Google Scholar]
- 28. Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., and Akira S. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 10.1016/j.immuni.2010.10.013 [DOI] [PubMed] [Google Scholar]
- 29. Wang Q., Liu X., Cui Y., Tang Y., Chen W., Li S., Yu H., Pan Y., and Wang C. (2014) The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 10.1016/j.immuni.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 30. Qin Y., Zhou M. T., Hu M. M., Hu Y. H., Zhang J., Guo L., Zhong B., and Shu H. B. (2014) RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 10, e1004358 10.1371/journal.ppat.1004358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhong B., Zhang L., Lei C., Li Y., Mao A. P., Yang Y., Wang Y. Y., Zhang X. L., and Shu H. B. (2009) The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 10.1016/j.immuni.2009.01.008 [DOI] [PubMed] [Google Scholar]
- 32. Rommereim L. M., Bellini V., Fox B. A., Pètre G., Rak C., Touquet B., Aldebert D., Dubremetz J. F., Cesbron-Delauw M. F., Mercier C., and Bzik D. J. (2016) Phenotypes associated with knockouts of eight dense granule gene loci (GRA2-9) in virulent Toxoplasma gondii. PLoS One 11, e0159306 10.1371/journal.pone.0159306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hiszczyńska-Sawicka E., Olędzka G., Holec-Gąsior L., Li H., Xu J. B., Sedcole R., Kur J., Bickerstaffe R., and Stankiewicz M. (2011) Evaluation of immune responses in sheep induced by DNA immunization with genes encoding GRA1, GRA4, GRA6, and GRA7 antigens of Toxoplasma gondii. Vet. Parasitol. 177, 281–289 10.1016/j.vetpar.2010.11.047 [DOI] [PubMed] [Google Scholar]
- 34. Nam H. W. (2009) GRA proteins of Toxoplasma gondii: maintenance of host–parasite interactions across the parasitophorous vacuolar membrane. Korean J. Parasitol. 47, S29–S37 10.3347/kjp.2009.47.S.S29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunter C. A., and Sibley L. D. (2012) Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778 10.1038/nrmicro2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosowski E. E., Lu D., Julien L., Rodda L., Gaiser R. A., Jensen K. D., and Saeij J. P. (2011) Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208, 195–212 10.1084/jem.20100717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gov L., Karimzadeh A., Ueno N., and Lodoen M. B. (2013) Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio 4, e00255–13 10.1128/mBio.00255-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sauer J. D., Sotelo-Troha K., von Moltke J., Monroe K. M., Rae C. S., Brubaker S. W., Hyodo M., Hayakawa Y., Woodward J. J., Portnoy D. A., and Vance R. E. (2011) The N-ethyl-N-nitrosourea–induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79, 688–694 10.1128/IAI.00999-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun L., Wu J., Du F., Chen X., and Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melo M. B., Jensen K. D., and Saeij J. P. (2011) Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends Parasitol. 27, 487–495 10.1016/j.pt.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hakimi M. A., and Bougdour A. (2015) Toxoplasma's ways of manipulating the host transcriptome via secreted effectors. Curr. Opin. Microbiol. 26, 24–31 10.1016/j.mib.2015.04.003 [DOI] [PubMed] [Google Scholar]
- 42. Hsiao C. H., Luisa Hiller N., Haldar K., and Knoll L. J. (2013) A HT/PEXEL motif in Toxoplasma dense granule proteins is a signal for protein cleavage but not export into the host cell. Traffic 14, 519–531 10.1111/tra.12049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiang Z., Mak T. W., Sen G., and Li X. (2004) Toll-like receptor 3–mediated activation of NF-κB and IRF3 diverges at Toll–IL-1 receptor domain-containing adapter inducing IFN-β. Proc. Natl. Acad. Sci. U.S.A. 101, 3533–3538 10.1073/pnas.0308496101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu S., Chen J., Cai X., Wu J., Chen X., Wu Y. T., Sun L., and Chen Z. J. (2013) MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2, e00785 10.7554/eLife.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Z., Yuan B., Bao M., Lu N., Kim T., and Liu Y. J. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 10.1038/ni.2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., and Bowie A. G. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 10.1038/ni.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takaoka A., Wang Z., Choi M. K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., Ohba Y., and Taniguchi T. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 10.1038/nature06013 [DOI] [PubMed] [Google Scholar]
- 48. Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., and Fitzgerald K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1–activating inflammasome with ASC. Nature 458, 514–518 10.1038/nature07725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Silva-Barrios S., and Stäger S. (2017) Protozoan parasites and type I IFNs. Front. Immunol. 8, 14 10.3389/fimmu.2017.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beiting D. P. (2014) Protozoan parasites and type I interferons: a cold case reopened. Trends Parasitol. 30, 491–498 10.1016/j.pt.2014.07.007 [DOI] [PubMed] [Google Scholar]
- 51. Majumdar T., Chattopadhyay S., Ozhegov E., Dhar J., Goswami R., Sen G. C., and Barik S. (2015) Induction of interferon-stimulated genes by IRF3 promotes replication of Toxoplasma gondii. PLoS Pathog. 11, e1004779 10.1371/journal.ppat.1004779 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Bradley J. R., and Pober J. S. (2001) Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20, 6482–6491 10.1038/sj.onc.1204788 [DOI] [PubMed] [Google Scholar]
- 53. You F., Wang P., Yang L., Yang G., Zhao Y. O., Qian F., Walker W., Sutton R., Montgomery R., Lin R., Iwasaki A., and Fikrig E. (2013) ELF4 is critical for induction of type I interferon and the host antiviral response. Nat. Immunol. 14, 1237–1246 10.1038/ni.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sugi T., Kato K., and Weiss L. M. (2016) An improved method for introducing site-directed point mutation into the Toxoplasma gondii genome using CRISPR/Cas9. Parasitol. Int. 65, 558–562 10.1016/j.parint.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peng D., and Tarleton R. (2015) EuPaGDT: A web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb. Genom. 1, e000033 10.1099/mgen.0.000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., and Ma'ayan A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., and Ma'ayan A. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang D. W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 59. Huang D. W., Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.