

Abstract
Historically, methods to remove the 4-methoxybenzyl (Mob) protecting group from selenocysteine (Sec) in peptides have used harsh and toxic reagents. The use of 2,2’- dithiobis-5-nitropyridine (DTNP) is an improvement over these methods, however many wash steps are required to remove the by-product contaminant 5-nitro-2-thiopyridine. Even with many washes, excess DTNP adheres to the peptide. The final product needs excess purification to remove these contaminants. It was recently discovered by our group that hindered hydrosilanes could be used to reduce Cys(Mob). We sought to apply a similar methodology to reduce Sec(Mob), which we expected to be even more labile. Here we present a gentle and facile method for deprotection of Sec(Mob) using triethylsilane (TES), phenol, and a variety of other scavengers often used in deprotection cocktails. The different cocktails were all incubated at 40 °C for 4 hours. The combination of TFA/TES/thioanisole (96:2:2) appeared to be the most efficient of the cocktails tested, providing complete deprotection and yielded peptide that was mainly in the diselenide form. This cocktail also showed no evidence of side reactions or significant contaminants in the HPLC and MS analyses. We envision that our new method will allow for a simple and gentle “one pot” deprotection of Sec(Mob) following solid-phase peptide synthesis and will minimize the need for extensive purification steps.
Graphical Abstract
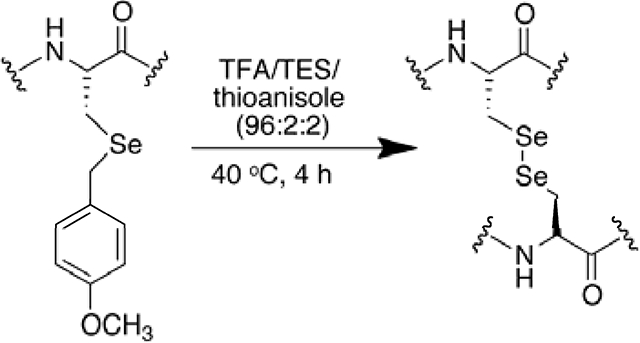
Here in we demonstrate the very facile removal of the 4-methoxybenzl protecting group from selenocysteine after solid phase peptide synthesis. The most effective condition is a deprotection cocktail consisting of TFA/TES/thioanisole at 40 °C for 4 hours. This method is a great improvement over other methods that use harsh reagents for deprotection of the 4-methoxybenzyl group and results in a purer peptide after minimal purification washes and precipitation into ether.
INTRODUCTION
Historically, the 4-methoxybenzyl (Mob) group has been the protecting group of choice for protecting the selenol side chain of selenocysteine (Sec) in solid-phase peptide synthesis (SPPS).1, 2 The C–Se bond of Sec(Mob) is more stable in comparison to the acid labile C–Se bond of Sec(Trt), but is less prone to racemization than Sec(Bzl) and other similar Sec-protecting groups.3–7 Though we note that the xanthyl group has been used successfully to protect Sec.8 A significant challenge when working with either Mob protected cysteine (Cys) or Sec(Mob) is the difficulty in removing the Mob group after SPPS. Some of the conditions used in the past have included hydrofluoric acid, silyl-Lewis acids, or heavy metals.6, 9–12 Not only are these reagents toxic but they tend to be difficult to work with often requiring special apparatus. These deprotection conditions can also result in unwanted side reactions and often leave an impure or oily peptide that is difficult to work with after precipitation and lyophilization.13 The discovery of 2,2′-dithiobis(5-nitropyridine) (DTNP) as a deprotection reagent for Sec(Mob) and Cys(Mob) was a major improvement over these harsh conditions.13 However, the use of DTNP has its own limitations.
We set out to conduct this study because deprotection of a Sec(Mob)-containing peptide with DTNP results in the formation of a 2-thio(5-nitropyridyl) (5-Npys) adduct. In order to get to the free selenol form of the peptide, it must be treated with either a large excess of free thiol or with ascorbate.13, 14 These methods require many washing steps for initial purification as the free 5-Npys group tends to adhere to the peptide and is difficult to remove even after many ether washes. This leaves the final lyophilized peptide with an off-white to yellowish color and the 5-Npys group is a contaminant when the peptide is redissolved in buffer for subsequent applications.
A previous study by our group demonstrated the ability of hindered hydrosilanes, such as triisopropylsilane (TIS) and triethylsilane (TES), to reduce the C–S bond of a protected Cys peptide, yielding a reduced protecting group and either a Cys-SH or a disulfide containing peptide upon deprotection.15 The Cys(Mob) group was shown to be particularly labile to reduction by TIS and TES as the C–Se bond is weaker than the C–S bond. In light of this study, we reasoned that Sec(Mob) should be even more labile to deprotection by TIS or TES. The aim of this present study was to develop the most gentle and facile deprotection of Sec(Mob) residues reported to date using the ability of hydrosilanes to reduce the C–Se bond of a Sec(Mob) residue in a peptide.
Here we show that TES in combination with water, thioanisole, or phenol will deprotect Sec(Mob) when incubated at 40 °C for 4 hours. This deprotection method can be used to conduct peptide cleavage and Sec(Mob) deprotection all in one pot. Our method is gentle, facile, and results in a purer peptide after ether precipitation than other reported methods.
RESULTS AND DISCUSSION
A previous study by us showed that Cys(Mob) can be reduced with TIS and a variety of other scavengers.15 Here, we chose TES instead of TIS because TES resulted in higher conversion of Cys(Mob) into Cys-SH.15
Here we report that trifluoroacetic acid (TFA)/TES/thioanisole (96:2:2), TFA/TES/H2O (96:2:2), and TFA/phenol/H2O (96:2:2) afforded the highest degree of Mob removal when incubated with the peptide at 40 °C for 4 hours. The same peptide incubated in TFA at 40 °C for 4 hours showed ~30% conversion (control). The extent of deprotection was determined by analytical HPLC and the results of our deprotection assays are illustrated in Figure 1. We have consistently found that when incubating Sec(Mob)-containing peptides in neat TFA at room temperature, a small amount (in the range of 5%−10%) of deprotection occurs (data not shown). It is therefore not surprising that a significant amount of deprotection occurs at 40 °C in neat TFA. We note that our “neat TFA” is not dry and certainly contains a small amount of water due to the hydroscopic nature of TFA.
Figure 1: Extent of deprotection of Sec(Mob) using different deprotection cocktails at 40 °C for 4 hours as measured by analytical HPLC.

A) ~30% deprotection of Sec(Mob) was observed after incubation in neat TFA as seen by the appearance of the deprotected peptide peak at 10 min in the chromatogram. B) Nearly 100% deprotection of Sec(Mob) was observed after incubation in TFA/TES/H2O (96:2:2). C) ~90% deprotection of Sec(Mob) was observed after incubation in TFA/TES/phenol (96:2:2). An unidentified peak appears at 21 minutes. D) 100% deprotection of Sec(Mob) was observed after incubation in TFA/TES/thioanisole (96:2:2). E) ~98% deprotection of Sec(Mob) was observed after incubation in TFA/phenol/H2O (96:2:2). F) The diselenide peak near 13 minutes decreases substantially when the sample shown in E) was treated with 5-fold excess TCEP for 12 h. The extent of deprotection was determined by peak areas in all cases.
We found that the deprotected peptide exists mostly in the diselenide form. The presence of the peptide diselenide was empirically determined by reducing the diselenide with tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl). After incubation in TFA/TES/H2O (96:2:2) for 4 hours at 40 °C, the peptide sample was precipitated in ether and dried with a stream of argon. The dried peptide was then dissolved in water/acetonitrile (5:1) and lyophilized. Following lyophilization, the peptide sample was redissolved in water/acetonitrile (5:1) and divided into two aliquots. One aliquot was injected into the analytical HPLC, while the other sample was incubated at room temperature for 12 hours with a 5-fold excess TCEP-HCl and then analyzed by HPLC. The sample treated with TCEP-HCl showed a significant reduction in intensity of the peptide diselenide peak and an increase in the intensity of the selenol peak (compare Figure 1B with 1F). We note that TCEP has been found to promote deselenization of Sec-containing peptides,16 but was not observed here. This may be due to the low pH of the reaction.
The mass spectral analyses (Figure 2) confirmed the observations from the HPLC analyses shown in Figure 1. Some peptide samples that were stored in water/acetonitrile (5:1) for 24 h prior to lyophilization and mass spectral analysis were observed to have oxidized to the diselenide. Therefore, the diselenide form appears to be more abundant in the mass chromatograms (especially the control) in comparison to the corresponding HPLC trace.
Figure 2: Mass chromatograms of the deprotection assays corresponding to the HPLC analyses in Figure 1.

The arrow points to the observed m/z while the number inside the parentheses is the theoretical m/z value. All species are singly charged unless otherwise indicated. A) The mass spectrogram of the control shows mostly the protected peptide is present. The inset shows the characteristic isotope pattern of selenium for the two peaks indicated. B) – E) Mass spectrograms corresponding to the HPLC traces shown in Figure 1B – 1E, respectively. As is evident by the data in Figures 1D and 2D, deprotection is the most effective with TFA/TES/thioanisole (96:2:2). We note that we do not show the mass chromatogram of the sample reduced with TCEP.
We note that there is an unidentified peak near 21 minutes in the HPLC chromatogram when using deprotection cocktails containing phenol (C and E in Figures 1 and 2). Mass spectral analyses of reaction C show the presence of a mass ion at 873.29 m/z. We suspect that this is either a phenol adduct to the peptide, or the result of a reaction between phenol and Mob molecules, but we cannot make this identification based on the mass. We also note that the large unlabeled peak that appears in many of the samples at 530.29 m/z is a truncated peptide with the sequence H-PTVTGG-OH•H2O.
Of the conditions tested, TFA/TES/thioanisole (96:2:2) incubated for 4 hours at 40 °C proved to be the most efficient and complete method for reduction of Sec(Mob) (Figure 1 and Figure 2), and results in the peptide being almost exclusively in the diselenide form. The lability of the Mob protecting group on Sec in this deprotection cocktail at 40 °C is very consistent with our earlier report on the lability of Cys(Mob) in a cocktail of TFA/TIS/H2O (96:2:2) at 37 °C.15 Cocktails containing TES and phenol were also effective at deprotecting Sec(Mob) at 40 °C, but contained more impurities that we were unable to identify. Cocktails containing either phenol or water did result in a significant amount of the peptide-selenol however.
This simple deprotection method for Sec(Mob) at slightly elevated temperature, requires minimal purification and it also allows for a “one pot” cleavage and side-chain deprotection following SPPS using reagents already commonly used in SPPS cleavage cocktails. To show that we could use our protocol in a “one pot” manner, we synthesized the peptide RGUN (corresponding to the active site of glutathione peroxidase) on a 0.1 mmol scale. The resin was dried and split evenly into two glass beakers. One portion of the resin was cleaved using a “standard” protocol for two hours at room temperature with a cleavage cocktail consisting of TFA/TES/H2O (96:2:2), while the other portion was cleaved with our modified protocol at 40 °C for four hours with the same cocktail. The reactions were then filtered, the TFA solution was reduced in volume, precipitated in ether, and then lyophilized. HPLC and mass spectral analyses clearly show that our modified protocol resulted in fully deprotected peptide, though mainly in the diselenide form (Figure S1 of the Supporting Information). However, the peptide treated with the standard conditions resulted in a more complex HPLC chromatogram and contained a substantial amount of Mob-protected peptide, while our modified protocol resulted in a much cleaner HPLC chromatogram.
In summary, the deprotection of Sec(Mob) has now evolved in our hands from the treatment with silyl Lewis acids, iodine, or mercuric acetate,9–12 to the use of DTNP in TFA,13 and finally to the use of TIS or TES in TFA at 40 °C.15 While we used an incubation time of four hours in this study, we recommend trying the cleavage between 2–4 hours. This protocol makes for an excellent addition to the peptide chemist’s toolbox when working with Cys and Sec containing peptides.
MATERIALS AND METHODS
Materials
Solvents for peptide synthesis were purchased from Fisher Scientific (Pittsburgh, PA). Triethylsilane (99%) and thioanisole (99%) were purchased from Acros Organics (Pittsburgh, PA). All other chemicals were purchased from either Sigma–Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA). The HPLC system is from Shimadzu with a Symmetry® C18–5 μm column from Waters Corp. (Milford, MA) (4.6 × 150 mm). Mass spectral analysis was performed on a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (ThermoFisher).
Peptide Synthesis
Fmoc-Sec(Mob)-OH was synthesized as had previously been reported by us.17,18 This Sec-derivative was used to synthesize peptides H-Pro-Thr-Val-Thr-Gly-Gly-Sec-Gly-OH (H-PTVTGGUG-OH) and H-Arg-Gly-Sec-Asn-OH (H-RGUN-OH). Both peptides were synthesized according to solid-phase methods on a 0.1 mmol scale using a glass vessel shaken with a model 75 Burrell wrist action shaker. We used 300 mg of 2-chlorotrityl chloride resin (100–200 mesh, Chem-Impex Int’l Inc.) for each peptide. This resin was swelled in dichloromethane (DCM) for 20 min. The first amino acid was directly coupled to the resin using N-methylmorpholine (NMM)/DCM (2:98), shaking for 1 h. The resin was then capped using DCM/methanol/NMM (8:1:1). Fmoc-Sec(Mob)-OH was coupled using 0.2 mmol of Fmoc-protected amino acid, 0.4 mmol of 1-hydroxy-7-azabenzotriazole (HOAt), and 0.607 mmol N, N’-diisopropylcarbodiimide (DIC) in dimethylformamide (DMF), shaking for 2 h. Subsequent amino acids were coupled using 0.2 mmol of Fmoc-protected amino acid, 0.4 mmol of HOAt, and 0.607 mmol DIC in DMF, shaking for 1 h. Between amino acid couplings, the Fmoc protecting group was removed by two 10 min washes with piperidine/DMF (2:8). The success of Fmoc removal and amino acid couplings were monitored qualitatively using a ninhydrin test.19 Peptides were cleaved from the resin via a 1.5-h reaction with a cleavage cocktail consisting of TFA/H2O (96:4). Following cleavage, the resin was washed with DCM, and the volume of the cleavage solution was reduced by evaporation with argon gas. Each peptide was precipitated with cold, anhydrous ether. Centrifugation at 3000 rpm on a clinical centrifuge (International Equipment Co., Boston, MA) for 5 min pelleted the peptide. Peptides were dried under argon gas, then dissolved in a minimal amount of water/HPLC-grade acetonitrile (5:1), lyophilized, and used without further purification.
Deprotection of Sec(Mob) by triethylsilane and phenol cocktails
Lyophilized peptide H-PTVTGGUG-OH cleaved from the solid support was dissolved in TFA and the solution was divided into five aliquots as follows: aliquot 1 consisted of peptide in neat TFA, aliquot 2 consisted of TFA/TES/H2O (96:2:2), aliquot 3 consisted of TFA/TES/phenol (96:2:2), aliquot 4 consisted of TFA/TES/thioanisole (96:2:2), and the fifth aliquot consisted of TFA/phenol/H2O (96:2:2). All five aliquots were incubated in a 40 °C water bath for 4 h. For all reactions, the final volume was 1.0 mL and contained 4.4 mM peptide in the TFA deprotection cocktail. After incubation, the reaction volume was reduced by evaporation with argon gas and peptides were precipitated with cold diethyl ether and pelleted via centrifugation. For purification, the ether was decanted and fresh ether was added to the peptide pellets and then vortexed. The peptide was then pelleted once again using centrifugation and dried under argon. Dried peptide pellets were dissolved in water/HPLC-grade acetonitrile (5:1), lyophilized, and analyzed without further purification.
High Pressure Liquid Chromatography
High pressure liquid chromatography analysis of all samples was performed using a Shimadzu with a Symmetry C18 5 μm column from Waters (4.6 × 150 mm). The aqueous and organic phases were 0.1% TFA in distilled, deionized water (buffer A) and 0.1% TFA in HPLC–grade acetonitrile (buffer B), respectively. Beginning with 100% buffer A, buffer B was increased by 1% up to 50% over 50 min with a 1.4 mL/min gradient elution. Buffer B was then increased from 50% to 100% over 5 min. This method was used for analysis of each sample. Peptide elution was monitored via absorbance at both 214 and 254 nm.
Reduction of peptide diselenide by TCEP
Peptide incubated in TFA/TES/H2O (96:2:2) for 4 h at 40 °C was precipitated and lyophilized as described above. The peptide was then dissolved in water/acetonitrile (5:1). To this solution, 5 mole equivalents of TCEP HCl was added and allowed to react for 12 h at room temperature. HPLC analysis was then performed without further purification.
Supplementary Material
Acknowledgments
These studies were supported by National Institutes of Health Grants GM094172 to RJH.
REFERENCES
- 1.Koide T, Itoh H, Otaka A, Yasui H, Kuroda M, Esaki N, Soda K, Fuji N. Synthetic study on selenocystine-containing peptides. Chem. Pharm. Bull 1993; 41: 5020–506. [DOI] [PubMed] [Google Scholar]
- 2.Koide T, Itoh H, Otaka A, Furuya M, Kitajaima Y, Fuji N. Synthesis and biological activities of selenium analogs of α-rat atrial natiruretic peptide. Chem. Pharm. Bull 1993; 41: 1596–1600. [DOI] [PubMed] [Google Scholar]
- 3.Han Y, Albericio F, Barany G. Occurrence and minimization of cysteine racemization during stepwise solid-phase peptide synthesis. J. Org. Chem 1997; 62: 4307–4312. [DOI] [PubMed] [Google Scholar]
- 4.Hibino H, Nishiuchi Y. 4-Methoxybenzyloxymethyl group, a racemization-resistant protecting group for cysteine in Fmoc solid phase peptide synthesis. Org. Lett 2012; 14: 1926–1929. [DOI] [PubMed] [Google Scholar]
- 5.Moroder L, Besse D, Musiol HJ, Rudolph-Bohner S, Siedler F. Oxidative folding of cystine-rich peptides vs regioselective cysteine pairing strategies. Biopolymers (Peptide Science) 1996; 40: 207–234. [DOI] [PubMed] [Google Scholar]
- 6.Isidro-Llobet A, Álvarez M, Albericio F. Amino acid-protecting groups. Chem. Rev 2009; 109: 2455–2504. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara Y, Akaji K, Kiso Y. Racemization-free synthesis of C-terminal cysteine-Peptide Using 2-Chlorotrityl Resin. Chem. Pharm. Bull 1994; 42: 724–726. [DOI] [PubMed] [Google Scholar]
- 8.Flemer S Jr. Fmoc-Sec(Xan)-OH: Synthesis and Utility of Fmoc Selenocysteine SPPS Derivatives with Acid-Labile Sidechain Protection. J. Pep. Sci 2015; 21: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuji N, Otaka A, Watanabe T, Okamachi A, Tamamura H, Yajima H, Inagaki Y, Nomizu M, and Asano K. Silver trifluoromethanesulphonate as an S-deprotecting reagent for the synthesis of cystine peptides. J. Chem. Soc. Chem. Commun 1989; 283–284. [Google Scholar]
- 10.Fujii N, Otaka A, Funakoshi S, Bessho K, Yajima H. New procedure for the synthesis of cysteine-peptides by oxidation of S-substituted cysteine-peptides with thallium(III) trifluoroacetate. J. Chem. Soc. Chem. Commun 1987; 3: 163–164. [DOI] [PubMed] [Google Scholar]
- 11.Osamu N, Kitada C, Fujino M. New method for removing the S-p-methoxybenzyl and S-t-butyl groups of cysteine residues with mercuric trifluoroacetate. Chem. Pharm. Bull 1978; 26: 1576–1585. [Google Scholar]
- 12.Besse D, Moroder L. Synthesis of selenocysteine peptides and their oxidation to diselenide-bridged compounds. J. Pep. Sci 1997; 3: 442–453. [DOI] [PubMed] [Google Scholar]
- 13.Harris KM, Flemer S Jr., Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J. Pep. Sci 2007; 13: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ste. Marie EJ, Ruggles EL, Hondal RJ. Removal of the 5-nitro-2-pyridine-sulfenyl protecting group from selenocysteine and cysteine by ascorbolysis. J. Pep. Sci 2016; 22: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ste. Marie EJ, Hondal RJ. Reduction of cysteine-S-protecting groups by triisopropylsilane. J. Pep. Sci 2018; 24: e3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dery S, Reddy PS, Dery L, Mousa R, Dardashti RN, Metanis N. Insights into the deselenization of selenocysteine into alanine and serine. Chem. Sci 2015; 6: 6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J. Am. Chem. Soc 2001; 123: 5140–5141. [DOI] [PubMed] [Google Scholar]
- 18.Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol 2002; 347: 70–83. [DOI] [PubMed] [Google Scholar]
- 19.Kaiser E, Colescott R, Bossinger C, Cook P. Color test for detection of free terminal amino groups in the solid–phase synthesis of peptides. Anal. Biochem 1970; 34:595–598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.