

Abstract
Several studies have shown that the airways of asthma patients contain higher diversity of bacteria and are enriched in pathogenic species. However, sampling the airways in children is challenging. Here we aimed to identify differences in the salivary bacterial composition between African Americans children with and without asthma. Saliva samples from 57 asthma cases and 57 healthy controls were analyzed by means of 16S rRNA amplicon profiling. Measurements of bacterial diversity and genus relative abundance were compared between cases and controls using the non-parametric Wilcoxon test and multivariate regression models. A total of five phyla and a mean of 56 genera were identified. Among them, 15 genera had a relative abundance greater than 1%, being Prevotella, Haemophilus, Streptococcus, and Veillonella the most abundant genera. Differences between cases and controls were found in terms of diversity, as well as in relative abundance for Streptococcus genus (13.0% in cases vs 18.3% in controls, p=0.003) and Veillonella genus (11.1% in cases vs 8.0% in controls, p=0.002). These differences remained significant after correction for multiple comparisons and when potential confounders were taken into account in logistic regression models. In conclusion, we identified changes in the salivary microbiota associated with asthma among African Americans.
Keywords: 16S rRNA sequencing, next-generation sequencing, respiratory disease, bacterial composition
1. Introduction
Asthma is a chronic heterogeneous disease that affects the lower respiratory tract. The most characteristic symptoms of this disease are dyspnea, chest tightness, wheezing, and cough1. Typical characteristics of this pathology are the inflammation and hyperresponsiveness of the airways as a response to different triggering agents, such as air pollution, tobacco smoke, airborne allergens, the practice of sports, stress, certain drugs, or viral and bacterial respiratory infections1.
All population groups, from children to elder people, are affected by asthma1. It has an estimated prevalence ranging from 1 to 16% of the population depending on the country2, resulting nowadays in 340 million people affected worldwide by this disease3. Additionally, within countries, there are also differences in asthma prevalence depending on the racial and/or ethnic group. For instance, in the United States of America, asthma prevalence among children is lowest in Mexican Americans (3.1%) and European Americans (8.2%) and highest in African Americans (14.6%) and Puerto Ricans (21.9%)4. These differences in prevalence have been partially attributed to the distinctive genetic makeup of these populations, since it has been shown that genetic African ancestry increases the risk for asthma5.
Recent microbiome studies based on culture-independent techniques, such as massive sequencing or microarrays, have shown that the lung is not sterile as previously thought6. Furthermore, it has been highlighted the role of bacteria residing in the human lung in the development of asthma and the severity of the disease7, and how bacteria interact with the airways in asthma susceptibility8. Additionally, it has been shown that there is a connection between the inflammation of the airways and early environmental exposures in life, being those children who have had contact with animals and other allergens during their first period of life the ones less prone to develop the disease9. In fact, a rich microbial exposure (i.e. being exposed to a wide range of microbes from pets, other children in daycare and animals in farm environment) has shown to reduce the risk of asthma10.
Furthermore, the knowledge generated until now suggests that the airways of asthma patients have higher bacterial diversity than the ones from healthy individuals11,12 and that a higher microbial burden is present in the upper airways compared to the lower airways13,14. In most of these studies, invasive sampling methods have been used to characterize the microbiota from the lower airways, such as induced sputum, lung or bronchus biopsies, protected brushings, or bronchoalveolar lavage15,16. However, Marri et al. have shown that the dysbiosis (i.e. an imbalance of the microbial communities) observed in the induced sputum and lower respiratory tract from asthmatic patients might reflect changes occurring in the whole respiratory airways, and not in any specific compartment17.
Since asthma is the most prevalent chronic disease during childhood1,3, the search for alternative non-invasive sample sources for microbiome biomarkers is needed. Previous studies have suggested that there is a continuum in the microbiota from the lower and the upper airways from healthy individuals13,14, being this particularly evident for the oral microbiome and the right upper lobe microbiome, due to anatomical reasons and repeated microaspirations of oropharyngeal secretions18. Additionally, it has been shown that an individual’s lung microbiome is more likely to be similar to its salivary microbiome than to the lung microbiome of another individual14. Therefore, saliva could be an accessible and non-invasive sample to study the microbiome, since it is in direct contact with one of the entry points of the airways, and it has been proposed as the source of microorganisms that colonize the lung niche13,16. Despite its potential, saliva samples have barely been analyzed in previous metagenomics studies of asthma, with only one study performed in European populations and focused on allergy development in early life19.
In this study, we aimed to assess whether there are differences in the bacterial microbiome composition of saliva samples from children and young adults with and without asthma, focusing for the first time on one of the populations that is most affected by asthma, African Americans. For that purpose, we analyzed a total of 114 samples from the Study of African American, Asthma, Genes & Environments (SAGE II)20 using 16S rRNA gene-targeted sequencing.
2. Materials and Methods
2.1. Sample description
The study subjects for the current analysis were drawn from the Study of African American, Asthma, Genes & Environments (SAGE II)20, an ongoing case-control study of childhood and youth asthma in African Americans initiated in December 2006. This study has been approved by the Human Research Protection Program Institutional Review Board of the University of California, San Francisco (San Francisco, United States) (ethics approval number: 210362), and all subjects/parents provided written assent/consent, respectively. Eligibility criteria were age between 8 to 21 years and having all four grandparents self-identified as African American. Exclusion criteria were: 10 or more pack-years of smoking; any smoking within one year of recruitment date; pregnancy in the third trimester; or history of one of the following conditions: sickle cell disease, cystic fibrosis, sarcoidosis, cerebral palsy, or history of heart or chest surgery. Cases had a physician-diagnosis of asthma, and they were required to show active symptoms (coughing, wheezing, or shortness of breath) in the two years preceding enrollment. Data was available for the atopic status, medication prescription, the severity of the disease, and the presence of asthma exacerbations in the past 12 months. More details can be found in the E-Methods of Online Supporting Information.
Controls were non-allergic, non-asthmatic individuals and they were ineligible for recruitment if they reported: a history of allergic conditions (asthma, eczema, hives, hay fever, and/or allergic rhinitis), the use of medication for allergies, or the presence of symptoms of wheezing or shortness of breath during their lifetime.
From the 1,705 subjects included in the SAGE II study, saliva samples were available for a subset of 245 individuals. From those, we prioritized 204 subjects that had passed genome-wide genotyping quality controls21, to ensure that individuals included had not familiar relationship and that genetic ancestry could be assessed. Then, we selected all the asthma patients available in that subset, and we chose a similar number of controls matching the recruitment areas where the cases were enrolled. Therefore, in the current study, a subset of 57 cases and 57 controls was analyzed.
Based on 1,000 simulations, we estimated that this sample size sequenced with at least 10,000 reads per individual would provide 95% power to detect differences in the microbiome composition between cases and controls with moderate effect sizes (Cramer’s ϕ>0.05), and at a significance level of α=0.0122.
One milliliter of spit saliva samples was collected using Oragene DNA Discover OGR-500 self-collection kits (DNA Genotek, Inc., Ontario, Canada) following the manufacturer’s protocol. The participants were required not to eat, drink, smoke or chew gum in 30 minutes preceding the sampling. Additionally, as part of the procedures of pulmonary function testing, they were instructed to withhold all bronchodilator medications prior to recruitment. Specifically, they withhold short-acting bronchodilators for 8 hours, intermediate-acting bronchodilators for 24 hours and long-acting bronchodilators for 48 hours.
2.2. Genetic ancestry estimation
Estimates of African and European ancestries were obtained using an unsupervised analysis in ADMIXTURE assuming two ancestral populations (African and European), as previously described21. Haplotypes from individuals from HapMap phase II (http://hapmap.ncbi.nlm.nih.gov) were used as a reference for the European and African components: Utah residents with Northern and Western European ancestry from the CEPH collection (CEU) and Yorubans from Ibadan, Nigeria (YRI).
2.3. Sequencing of the V4 16S rRNA region
The V4 16S rRNA hypervariable region was amplified by PCR using a HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany) and specific fusion primers23. PCRs included a negative control (water) and a positive control consisting of a mix of DNA from 20 different bacterial species from the Microbial Mock Community B (BEI Resources, Manassas, VA) in each 96-well plate. PCR products were purified using the AxyPrep™Mag FragmentSelect-I purification kit (Axygen Biosciences, Union City, CA) and the concentration of each library was normalized using the SequalPrep™ normalization Plate (96) Kit (Invitrogen, Frederick, MD). Libraries were pooled and quantified by means of a 7500 Fast Real-Time PCR System (Life Technologies, Carlsbad, CA) using the KAPA Library Quantification Kit Illumina® platforms (Kapa Biosystems, Wilmington, MA). The size distribution of amplicons was evaluated with the Agilent 2100 Bioanalyzer using a High Sensitivity DNA Analysis Kit (Agilent Technologies Inc., Palo Alto, CA). Sequencing of the libraries was performed at The College of Biological Sciences UCDNA Sequencing Facility at the University of California, Davis, using the MiSeq Reagent Kit v2 with 250 bp paired-end reads in a MiSeq sequencer (Illumina, San Diego, CA), including ~20% of the PhiX Control Library.
2.4. Microbial community analysis
Primary image analysis and base calling were performed on the MiSeq instrument (Illumina). The processing of the 16S rRNA sequencing data, from filtering of raw reads to creating operational taxonomic units (OTUs) and assigning of taxonomic rank, was performed using the open-source bioinformatics tool Quantitative Insights Into Microbial Ecology (QIIME) v1.824.
Quality control included the removal of reads with a Phred score lower than 30 and ensuring that all samples had at least 10,000 reads. Sequences were clustered based on similarity, using an open-reference OTU picking approach with UCLUST25. BLAST26 was used to identify chimeric sequences to be excluded from the analyses based on a 97% similarity. Taxonomic assignment was performed using the Greengenes database27 and a 97% sequence identity threshold. Contaminant taxa, mainly sequences belonging to Burkholderia genus, found in negative controls were removed from the OTU tables for further analyses.
2.5. Statistical analysis
R v3.3.228 was used to assess the differences in sequencing depth between cases and controls, in order to discard the effect of the number of reads as a confounder. R package vegan 2.5.129 was used to normalize the count reads into relative abundance. Shannon diversity index and the number of genera were calculated based on the number of reads of genera with a relative abundance greater than 1% and for all genera separately. Additionally, the Shannon diversity index was normalized by the natural logarithm of the number of genera also known as Pielou index. Differences in bacterial diversity and genera abundance between asthma cases and controls were assessed by means of a non-parametric Wilcoxon test, considering only those genera with more than 1% of relative abundance. Adjustment by multiple comparisons was carried out by means of Bonferroni correction, taking into account the number of genera analyzed. Additionally, for the genera significantly associated with asthma, multivariate logistic regression models were used to assess whether results could be confounded by differences in age, sex, or genetic ancestry between cases and controls. Moreover, we assessed whether associated genera had differences within asthma cases by means of logistic regression models adjusted by age, sex, and genetic ancestry, analyzing as outcomes: atopy (atopic versus non-atopic asthma), the use of asthma medication (users versus non-users for each drug), severity (comparing moderate/severe asthma cases against mild cases), and the presence of asthma exacerbations in the past year. All the regression models were run in R28.
3. Results
3.1. Sample characteristics and quality control of the sequencing data
Demographic and clinical characteristics of the subjects are presented in Table 1. Cases and controls had a similar gender and age distribution, ranging from 8.5 to 21.7. However, cases with asthma had higher proportions of African ancestry than controls (83.3% versus 75.6%, p=3.34×10−4).
Table 1.
Characteristics of the subjects included in the study
| Case (n=57) | Control (n=57) | p | |
|---|---|---|---|
| Sex (% female) | 28 (49.1) | 36 (63.2) | 0.186 |
| Age | 15.6±3.3 | 15.0±3.9 | 0.493 |
| African ancestry (%) | 83.3±7.6 | 75.6±14.0 | 3.34 × 10−4** |
| FEV1 (% predicted) | 94.8±13.1 | NA | NA |
| FVC (% predicted) | 102.0±14.5 | NA | NA |
| Medication (%) | |||
| Short acting beta agonists | 57 (100) | NA | NA |
| Inhaled corticosteroids1 | 34 (59.7) | NA | NA |
| Long acting beta agonists1 | 12 (21.1) | NA | NA |
| Leukotriene receptor antagonists | 5 (8.8) | NA | NA |
| Positive skin prick test (%)2 | |||
| Any allergen | 27 (58.7) | NA | NA |
| Dust mites | 5 (10.9) | NA | NA |
| Animals | 12 (26.1) | NA | NA |
| Pollens | 20 (43.5) | NA | NA |
| Molds | 8 (17.4) | NA | NA |
| Asthma severity (%) | |||
| Mild Intermittent | 23 (40.4) | NA | NA |
| Mild persistent | 19 (33.3) | NA | NA |
| Moderate persistent | 13 (22.8) | NA | NA |
| Severe persistent | 2 (3.5) | NA | NA |
| Any exacerbation in the past year (%)3 | 18 (34.0) | NA | NA |
Subjects taking combo medications are included in these categories.
Data available for 46 subjects.
Data available for 53 subjects.
For continuous variables, the mean ± the standard deviations are shown.
Abbreviations: FEV1: Forced Expiratory Volume in 1 second; FVC: Forced Vital Capacity; NA: Non-Applicable.
p < 0.01
After filtering out reads based on their quality (Phred score<30) and the quality of the pairing, approximately 7 million reads were retained. All the samples included in the study had more than 10,000 reads. The mean number of reads per sample was 57,647±24,164, being the lowest and the highest output 12,598 and 179,622 reads, respectively. Additionally, no differences were found in sequencing depth between asthma cases and controls (p=0.830).
3.2. Microbial composition of saliva samples
Both case and control samples were dominated by 5 major phyla (Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, and Fusobacteria) that accounted for more than 98% of the reads (Figure 1). Nevertheless, none of the phyla presented statistically significant differences between cases and controls (p> 0.05, E-Table 1). At this taxonomic level, reads that were not assigned to any taxonomic rank (i.e. the unknown category) represents 0.02%.
Figure 1:

Phyla distribution in the two study population groups. Rare category represents the reads belonging to phyla with a relative abundance lesser than 1%. Unknown category represents reads that could not be assigned to any taxonomic rank.
At the genus level, 5.2% of the reads were classified into higher taxonomic levels (i.e. family, order, etc), which were grouped under the unclassified category. Additionally, 1.9% of the reads were not assigned to any taxonomic rank (unknown category). On average, the salivary microbiome was composed of 56 genera, having 15 of them a relative abundance greater than 1% in the group of cases and/or controls (Figure 2). The most abundant genera were Prevotella, Haemophilus, Streptococcus, and Veillonella.
Figure 2:

Overview of the microbiome composition and relative abundance of bacterial genera in the two groups studied. Rare category represents the reads belonging to genera with a relative abundance lesser than 1%. Unknown category represents reads that could not be assigned to any taxonomic rank.
3.3. Differences in the salivary microbiome composition by asthma status
Shannon diversity index was significantly different between cases (2.12±0.23) and controls (2.01±0.24) (p=0.013) (Figure 3, panel A). Pielou index also showed significant differences between cases (0.81±0.04) and controls (0.79±0.05) (p=0.01) (Figure 3, panel B). Additionally, diversity differences between cases and controls were also found when calculated for all the genera detected (Shannon index: 2.44±0.23 versus 2.34±0.24 in cases and controls, respectively, p=0.027; and Pielou index: 0.61±0.06 versus 0.58±0.06 in cases and controls, respectively, p=0.013).
Figure 3:
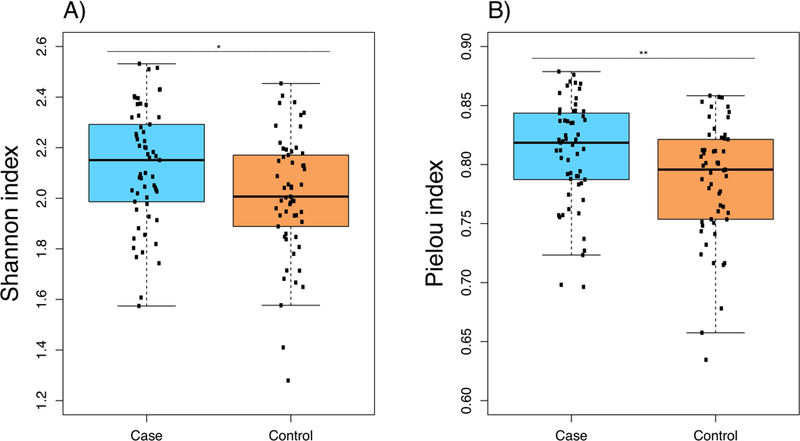
Boxplot of the diversity at genus level in cases and controls assessed for the genera with a relative abundance higher than 1% in at least one of the groups. (a) Diversity based on Shannon index (b) Diversity based on Pielou index.
Either analyzing the Shannon or Pielou diversity indices, the diversity was higher in cases than in controls. In order to confirm that the differences found were not a consequence of the effect of other confounding variables, multivariate logistic regression was performed adjusting by age, gender, and genetic ancestry. In these regression models, both diversity indexes remained associated with asthma status (p=0.005 for the Shannon index and p=0.01 for the Pielou index).
Since differences in diversity were noticed, changes in relative abundance differences were also sought. For that purpose, those genera with a relative abundance greater than 1% in at least one of the groups (cases/controls) were interrogated. Statistically significant differences in relative abundance were found between cases and controls for the Streptococcus genus (13.0% in cases and 18.3% in controls; p=0.003, Bonferroni p=0.039) and Veillonella genus (11.1% in cases and 8.0% in controls; p=0.002, Bonferroni p=0.035;) (Table 2).
Table 2.
Genera mean relative abundance (%) between asthma cases and controls for the main genera detected.
| Case (n=57) | Control (n=57) | p | Bonferroni adjusted p | |
|---|---|---|---|---|
| Actinomyces | 2.5 | 2.2 | 0.313 | 1 |
| Aggregatibacter | 2.3 | 1.6 | 9.8 × 10−3 ** | 0.147 |
| Atopobium | 1.2 | 0.9 | 0.048 * | 0.735 |
| Campylobacter | 1.2 | 1.4 | 0.713 | 1 |
| Fusobacterium | 3.6 | 3.8 | 0.991 | 1 |
| Haemophilus | 15.6 | 16.4 | 0.892 | 1 |
| Leptotrichia | 1.0 | 1.1 | 0.683 | 1 |
| Megasphaera | 1.1 | 0.8 | 0.051 | 0.735 |
| Neisseria | 5.8 | 3.7 | 0.073 | 1 |
| Oribacterium | 1.5 | 1.2 | 0.371 | 1 |
| Porphyromonas | 2.0 | 1.7 | 0.212 | 1 |
| Prevotella | 19.7 | 20.6 | 0.671 | 1 |
| Rothia | 4.7 | 4.3 | 0.383 | 1 |
| Streptococcus | 13.0 | 18.3 | 2.6 × 10−3 ** | 0.039 * |
| Veillonella | 11.1 | 8.0 | 2.3 × 10−3 ** | 0.035 * |
Statistically significant p-values after Bonferroni correction are shown in bold.
p < 0.05,
p < 0.01
The differences observed for the genera Streptococcus and Veillonella by asthma status remained statistically significant when testing a multivariate regression model adjusted by age, gender and genetic ancestry (Streptococcus, OR: 0.92, 95% CI: 0.87–0.97, p=4.4×10−3; Veillonella, OR=1.12, 95% CI=1.02–1.22, p=0.015), suggesting that these factors were not confounding the results. Additionally, no statistically significant differences were found for these genera when comparing different asthma subgroups based on atopic status, medication use, severity, or presence of exacerbations in the past year (E-Table 2).
4. Discussion
In this study, we aimed to assess the differences in the salivary microbial composition between asthma cases and controls among a high-risk population for asthma. To the best of our knowledge, this is the largest study of the salivary microbiome in relation to this disease and the only one performed in African Americans. Our results show the existence of differences in diversity between asthma cases and controls, as well as distinctive relative abundance of two bacterial genera, Streptococcus and Veillonella.
Regarding the composition of the samples, all the phyla, as well as the main genera detected in the saliva samples analyzed in our study, are the ones typically found in the oral cavity30,31. Additionally, when we compared the group of asthma patients with controls, our results were in agreement with what has been seen in other studies analyzing the airways regarding alpha diversity (mainly Shannon index H’ and species richness)12,17. Furthermore, although those studies only reached the family resolution and ours was able to get to the genus level, the association we observed for the relative abundances are also concordant with what has been previously described in induced sputum samples of young adults with and without asthma (mean age =26.3±0.5)17. Specifically, the family Veillonellaceae has been found to be more abundant in asthmatics, while Streptococcaceae has been found to be present in a higher proportion in control subjects17. Conversely, we did not find any correlation with Moraxella, Neisseria or Haemophilus genera that have been extendedly associated with asthma in bronchial brushings, bronchoalveolar lavages, and induced sputum samples32. However, the phylum in which these genera are included, Proteobacteria, showed a trend to be more abundant in cases (27.7%) than in controls (25.4%), being concordant with what has been reported in previous studies32. The lack of statistical significance could be due to the analysis of a different sample type. Despite this, our results indicate that the differences in the airways microbiota composition are partially captured sampling saliva. Therefore, our results have important implications in the use of saliva as a non-invasive and more accessible alternative sample type than lung biopsies, protected brushings, or induced sputum to study the microbiome in asthma.
The fact that Streptococcus was found to be more prevalent in healthy controls in this study could be explained by a higher abundance of commensal species among non-asthmatic individuals, such as Streptococcus salivarius and Streptococcus oralis, which would maintain the stability of the community against pathogenic species. In fact, previous studies have shown that the administration of certain strains of both species via nasal spray reduced the occurrence of upper respiratory tract infections33,34, significantly diminishing the occurrence of the episodes, as well as the number of school and work absences35. Therefore, our results suggest that future studies are needed to understand whether topical administration of probiotics may effectively change the oral or lung microbiome and, consequently, become a new strategy in asthma management and/or prevention. In contrast to the results obtained for Streptococcus, the genus Veillonella was found to be associated with higher risk for asthma, which is consistent with the differences observed in a previous study in the gut microbiome composition, focused on children at 1-year old that later developed asthma at the age of 5 years36. Since the oral cavity represents the sole entry point to the gastrointestinal tract, the concordance found between the salivary and gut microbiomes for Veillonella is plausible.
Several studies have assessed the role of the airways and fecal microbiome in asthma37. However, despite the oral cavity possesses the second most diverse microbial community in the body38, it has only been analyzed in a previous study of asthma among Europeans19. As part of a longitudinal study comparing 15 children with asthma and 32 controls that did not develop the disease, Dzidic et al.19 described different results from the ones presented in the current study. In terms of diversity, asthma patients at the age of 7 years old had lower diversity than controls, as opposed to our results and what has been described in previous studies focused on the airways11,12,17. Additionally, distinct genera were associated with asthma in the previous study19. These differences could be due to the fact the previous study was performed as part of a randomized double-blind trial evaluating the effect of the probiotic Lactobacillus reuteri in the prevention of allergy development19. Additionally, the current study is focused on African Americans and the previous study in Europeans, and ethnicity has been shown to influence the bacterial composition of the oral microbiome39. For that reason, our study took into account the effect of genetic ancestry in the regression models, allowing confirming that our results were not confounded by this variable. Another distinctive aspect of the current study is that we analyzed older individuals than the previous study since the mean age in our study is 15 versus 7 years. Additionally, the previous study required a preamplification of the V1-V5 16S hypervariable regions, due to the low input of sample, which could have affected the results.
Although bacteria from the oral cavity have been proposed as the main contributors to the bacterial composition in the lung due to microaspirations, in children these also involve secretions from the nasopharynx due to increased production of nasal secretions and the peculiar anatomy of the upper airways40. Additionally, recent studies have shown that in children the mouth and nasal microbiome have a different composition than the bronchial microbiome, in terms of predominant phyla, diversity, and burden of bacteria41,42. Therefore, to study causal changes of the bronchial microbiota associated with childhood asthma, sampling the lower airway is needed.
The current study has some limitations. First, the use of saliva samples reduced our ability to detect bacteria that could be causal of the disease, rather than markers. Second, the analysis of just one of the hypervariable regions of the most commonly used marker in microbiome studies, the V4 region of the 16S rRNA gene, only allowed us to achieve the bacterial genus resolution. Differences in the abundance of specific species or strains within each genus could be relevant to the disease and should be explored by future studies. Third, we did not take into account as confounder factors the use of antibiotics or diet since this information was not surveyed during recruitment. However, the salivary microbiota composition is very stable over time and has been found that is not easily altered by antibiotic use43. In fact, a previous study analyzing the salivary microbiome in relation to asthma showed that antibiotic treatment was not a confounder factor19. Similarly, the composition of the saliva microbiota has been found to be independent of the diet habits in a study performing a comprehensive food frequency questionnaire44, and presents stability over several years even involving changes in oral hygiene and diet45. Fourth, our study did not include data regarding the presence of oral cavities or periodontitis, which have been previously related to the saliva composition46. Therefore, oral health status is a potential confounder of the results of this study that remains unexplored. Fifth, we analyzed together both children and young adults. Although age was included as one of the covariates in the regression models, we acknowledge that asthma is not a static condition in different age groups.
The strengths of our study include studying African Americans, in contrast to most of the previous studies focused on European populations6,17,19,47. Minority populations are commonly underrepresented in biomedical studies, even in those focused on respiratory diseases48, despite their high burden of certain diseases such as asthma. The sample size analyzed is the largest studying the salivary microbiome19 and it is amongst the largest analyzing any type of sample in the context of this disease17, except for one study47. Additionally, the individuals included in the study had genome-wide genotyping data, which allowed us to ensure that only non-related individuals were included, and that genetic ancestry could be taken into account in the analyses.
In conclusion, our results show that the differences in bacterial diversity described in the airways among asthma patients are also found in the saliva. Additionally, while the relative abundance of the Streptococcus was higher in healthy controls, Veillonella was more prevalent in asthma cases. Therefore, saliva samples could be a non-invasive source for microbiome biomarkers for asthma.
Supplementary Material
Acknowledgments:
The authors acknowledge the SAGE II recruiters, participants, and study coordinator Sandra Salazar. The Microbial Mock Community B (BEI Resources, Manassas, VA) was obtained through BEI Resources, NIAID, NIH, as part of the Human Microbiome Project: Genomic DNA from Microbial Mock Community B (Even, Low Concentration), v5.1L, for 16S rRNA Gene Sequencing, HM-782D.
Funding: This research was funded by the Sandler Family Foundation, the American Asthma Foundation, the Robert Wood Johnson Clinical Harold Amos Medical Faculty Development Program, Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, National Institutes of Health (1R01HL117004, R01Hl128439, R01HL135156, and 1X01HL134589), National Institute of Environmental Health Sciences (R01ES015794, and R21ES24844), the National Institute on Minority Health and Health Disparities (1P60MD006902, RL5GM118984, and 1R01MD010443), and the Tobacco-Related Disease Research Program under Award Number 24RT-0025 to E.G.B. This work was also funded by Instituto de Salud Carlos III (ISCIII), co-financed by the European Regional Development Funds, “A way of making Europe” (PI14/00844) and by grant SAF2017-83417R from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO/AEI/FEDER, UE). N.H-P was funded by a fellowship from ISCIII and co-funded by the European Social Funds from the European Union (ESF) “ESF invests in your future” (FI16/00136). MP-Y was supported by the Ramón y Cajal Program by the Spanish Ministry of Economy, Industry and Competitiveness (RYC-2015-17205). SSO and CE were funded by the U.S. Department of Defense (PR141896).
Abbreviations
- CI
Confidence interval
- DNA
Deoxyribonucleic acid
- OR
Odds ratio
- OUT
Operational taxonomic unit
- PCR
Polymerase chain reaction
- SAGE II
Study of African Americans, Asthma, Genes and Environments
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.GINA. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention, 2019. http://www.ginasthma.org. 2019:1–28. [Google Scholar]
- 2.Masoli M, Fabian D, Holt S, Beasley R, Global Initiative for Asthma (GINA) Program. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59(5):469–78. [DOI] [PubMed] [Google Scholar]
- 3.The Global Asthma Report 2018. Auckland, New Zealand: Global Asthma Network, 2018. [Google Scholar]
- 4.Moorman JE, Zahran H, Truman BI, Molla MT, Centers for Disease Control and Prevention (CDC). Current asthma prevalence - United States, 2006–2008. MMWR Suppl. 2011;60(1):84–6. [PubMed] [Google Scholar]
- 5.Hernandez-Pacheco N, Flores C, Oh SS, Burchard EG, Pino-Yanes M. What Ancestry Can Tell Us About the Genetic Origins of Inter-Ethnic Differences in Asthma Expression. Curr Allergy Asthma Rep. 2016;16(8):53. [DOI] [PubMed] [Google Scholar]
- 6.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan A, Hunt E, MacSharry J, Murphy DM. The Microbiome and the Pathophysiology of Asthma. Respir Res. 2016;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Cicco M, Pistello M, Jacinto T, Ragazzo V, Piras M, Freer G, Pifferi M, Peroni D. Does lung microbiome play a causal or casual role in asthma? Pediatr Pulmonol. 2018;53(10):1340–1345. [DOI] [PubMed] [Google Scholar]
- 9.Ver Heul A, Planer J, Kau AL. The Human Microbiota and Asthma. Clinic Rev Allerg Immunol. 2018:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ege MJ, Mayer M, Normand A-C, Genuneit J, Cookson WOCM, Braun-Fahrländer C, Heederik D, Piarroux R, von Mutius E. Exposure to Environmental Microorganisms and Childhood Asthma. N Engl J Med. 2011;364(8):701–709. [DOI] [PubMed] [Google Scholar]
- 11.Fujimura KE, Lynch S V. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe. 2015;17(5):592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. Analysis of the Upper Respiratory Tract Microbiotas as the Source of the Lung and Gastric Microbiotas in Healthy Individuals. MBio. 2015;6(2):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rivas MN, Crother TR, Arditi M. The Microbiome in Asthma. Curr Opin Pediatr. 2016;28(6):764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, Curtis JL. Bacterial Topography of the Healthy Human Lower Respiratory Tract. MBio. 2017;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, Curtis JL. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann Am Thorac Soc. 2015;12(6):821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dzidic M, Abrahamsson TR, Artacho A, Collado MC, Mira A, Jenmalm MC. Oral microbiota maturation during the first 7 years of life in relation to allergy development. Allergy. 2018;73(10):2000–2011. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, Thyne S, Farber HJ, Serebrisky D, Kumar R, et al. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188(3):309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White MJ, Risse-Adams O, Goddard P, Contreras MG, Adams J, Hu D, Eng C, Oh SS, Davis A, Meade K, et al. Novel genetic risk factors for asthma in African American children: Precision Medicine and the SAGE II Study. Immunogenetics. 2016;68(6–7):391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes I, Harris K, Quince C. Dirichlet Multinomial Mixtures: Generative Models for Microbial Metagenomics Gilbert JA, editor. PLoS One. 2012;7(2):e30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. [DOI] [PubMed] [Google Scholar]
- 27.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria; R Foundation for Statistical Computing, Vienna, Austria. 2017. [Google Scholar]
- 29.Jari Oksanen F.Guillaume Blanchet, Friendly Michael, Kindt Roeland, Legendre Pierre, McGlinn Dan, Minchin Peter R., O’Hara RB, Simpson Gavin L., Solymos Peter, Stevens ES M. Henry H. and vegan HW. : Community Ecology Package. R package version 2.5–1. 2018. [Google Scholar]
- 30.Nasidze I, Li J, Quinque D, Tang K, Stoneking M. Global diversity in the human salivary microbiome. Genome Res. 2009;19(4):636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raju SC, Lagström S, Ellonen P, de Vos WM, Eriksson JG, Weiderpass E, Rounge TB. Gender-Specific Associations Between Saliva Microbiota and Body Size. Front Microbiol. 2019;10:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung KF. Airway microbial dysbiosis in asthmatic patients: A target for prevention and treatment? J Allergy Clin Immunol. 2017;139(4):1071–1081. [DOI] [PubMed] [Google Scholar]
- 33.Passali D, Passali GC, Vesperini E, Cocca S, Visconti IC, Ralli M, Bellussi LM. The efficacy and tolerability of Streptococcus salivarius 24SMB and Streptococcus oralis 89a administered as nasal spray in the treatment of recurrent upper respiratory tract infections in children. Eur Rev Med Pharmacol Sci. 2019;23(1 Suppl):67–72. [DOI] [PubMed] [Google Scholar]
- 34.Tarantino V, Savaia V, D’Agostino R, Silvestri M, Passali FM, Di Girolamo S, Ciprandi G. Bacteriotherapy in children with recurrent upper respiratory tract infections. Eur Rev Med Pharmacol Sci. 2019;23(1 Suppl):39–43. [DOI] [PubMed] [Google Scholar]
- 35.Ciprandi G, Savaia V, D’Agostino R, Silvestri M, Ciprandi G. Bacteriotherapy for preventing recurrent upper respiratory infections in children: a real-world experience. Otolaryngol Pol. 2018;72(3):30–35. [DOI] [PubMed] [Google Scholar]
- 36.Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, Schoos A-MM, Kunøe A, Fink NR, Chawes BL, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Meel ER, Jaddoe VW V., Bønnelykke K, de Jongste JC, Duijts L. The role of respiratory tract infections and the microbiome in the development of asthma: A narrative review. Pediatr Pulmonol. 2017;52(10):1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kilian M, Chapple ILC, Hannig M, Marsh PD, Meuric V, Pedersen AML, Tonetti MS, Wade WG, Zaura E. The oral microbiome – an update for oral healthcare professionals. Br Dent J. 2016;221(10):657–666. [DOI] [PubMed] [Google Scholar]
- 39.Mason MR, Nagaraja HN, Camerlengo T, Joshi V, Kumar PS. Deep Sequencing Identifies Ethnicity-Specific Bacterial Signatures in the Oral Microbiome. PLoS One. 2013;8(10):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsh RL, Kaestli M, Chang AB, Binks MJ, Pope CE, Hoffman LR, Smith-Vaughan HC. The microbiota in bronchoalveolar lavage from young children with chronic lung disease includes taxa present in both the oropharynx and nasopharynx. Microbiome 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An S, Warris A, Turner S. Microbiome characteristics of induced sputum compared to bronchial fluid and upper airway samples. Pediatr Pulmonol. 2018;53(7):921–928. [DOI] [PubMed] [Google Scholar]
- 42.Kloepfer KM, Deschamp AR, Ross SE, Peterson-Carmichael SL, Hemmerich CM, Rusch DB, Davis SD. In children, the microbiota of the nasopharynx and bronchoalveolar lavage fluid are both similar and different. Pediatr Pulmonol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaura E, ten Cate JM. Towards Understanding Oral Health. Caries Res. 2015;49(1):55–61. [DOI] [PubMed] [Google Scholar]
- 44.Belstrøm D, Holmstrup P, Nielsen CH, Kirkby N, Twetman S, Heitmann BL, Klepac-Ceraj V, Paster BJ, Fiehn N-E. Bacterial profiles of saliva in relation to diet, lifestyle factors, and socioeconomic status. J Oral Microbiol. 2014;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stahringer SS, Clemente JC, Corley RP, Hewitt J, Knights D, Walters WA, Knight R, Krauter KS. Nurture trumps nature in a longitudinal survey of salivary bacterial communities in twins from early adolescence to early adulthood. Genome Res. 2012;22(11):2146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherkasov S V, Popova LY, Vivtanenko TV, Demina RR, Khlopko YA, Balkin AS, Plotnikov AO. Oral microbiomes in children with asthma and dental caries. Oral Dis. 2019;25(3):898–910. [DOI] [PubMed] [Google Scholar]
- 47.Depner M, Ege MJ, Cox MJ, Dwyer S, Walker AW, Birzele LT, Genuneit J, Horak E, Braun-Fahrländer C, Danielewicz H, et al. Bacterial microbiota of the upper respiratory tract and childhood asthma. J Allergy Clin Immunol. 2017;139(3):826–834.e13. [DOI] [PubMed] [Google Scholar]
- 48.Burchard EG, Oh SS, Foreman MG, Celedón JC. Moving toward true inclusion of racial/ethnic minorities in federally funded studies. A key step for achieving respiratory health equality in the United States. Am J Respir Crit Care Med. 2015;191(5):514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.