

Graphical Abstract
Fibrillar fibronectin (fFn) networks are freely suspended across porous polymer structures without the use of cells fFn networks were engineered through the combination of the interfacial activity of fibronectin and fluid shear. fFn networks enable in vivo implantation or in vitro expansion of various cell types including patient breast cancer cells that otherwise fail to survive on tissue culture polystyrene.
Keywords: fibrillar fibronectin, extracellular matrix, tumor microenvironment, 3D cell culture, protein-polymer composite
Extracellular matrix (ECM) proteins, and most prominently, fibronectin (Fn), are routinely used in the form of adsorbed pre-coatings in an attempt to create a cell-supporting environment in both two- and three-dimensional cell culture systems. However, these protein coatings are typically deposited in a form which is structurally and functionally distinct from the ECM-constituting fibrillar protein networks naturally deposited by cells. Here, we report the cell-free and scalable synthesis of freely-suspended, and mechanically robust three-dimensional (3D) networks of fibrillar fibronectin (fFn) supported by tessellated polymer scaffolds. We discovered that hydrodynamically induced Fn fibrillogenesis at the three-phase contact line between air, a Fn solution, and a tessellated scaffold microstructure yields extended protein networks. Importantly, engineered fFn networks promoted cell invasion and proliferation, enabled in vitro expansion of primary cancer cells, and induced an epithelial-to-mesenchymal transition in cancer cells. Engineered fFn networks were seeded with cells from plural effusions of 14 cancer patients, where less than 5% of the initial population were cancer cells, and, in all cases, formed complex 3D multi-cellular structures with cancer cells constituting up to 36% of the total cell population after six days. With further work, engineered fFn networks could have a transformative impact on fundamental cell studies, precision medicine, pharmaceutical testing and preclinical diagnostics.
The extracellular matrix (ECM) constitutes the proteinaceous microenvironment of all multicellular tissues[1] and serves as a model for three-dimensional (3D) in vitro cell culture design.[2] Changes in ECM structure and composition drive biological function in both healthy and diseased states.[3] Fibronectin (Fn) is one of the most abundant ECM proteins in normal tissues,[4] and increased Fn expression is associated with multiple cancers.[5] During breast cancer progression, fibrillar Fn promotes tumorigenesis and metastasis, partly due to an epithelial-to-mesenchymal transition (EMT) in malignant cells.[6] Furthermore, upregulation of Fn has been identified to be a key step during pre-metastatic niche formation.[7]
A mechanosensitive protein, fibronectin exists in a compact state when solubilized such as in blood plasma, and in various stretched states such as fibrillar fibronectin (fFn) in ECM.[4, 8] The structure and conformation of fibronectin governs the exposure or sequestration of binding sites that impact its biological function and cell response[8b, 9]. Fn fibrillogenesis involves unfolding of the protein via mechanical translocation of integrins bound to cell surface receptors. Structurally, unfolding occurs largely at the mechanosensitive type III domain (Fn-3) exposing self-association sites for subsequent fibrillogenesis.[4, 8b, 10] Despite years of study, a comprehensive understanding of each region of the type I, II, and III Fn domains and how they interact with one another or cells to govern fibril formation is still a topic of ongoing investigation.[11]
We discovered that hydrodynamically induced fibrillogenesis at the three-phase contact line between air, a Fn solution, and a tessellated porous scaffold yields remarkably stable fibrillar Fn networks that promote cell invasion and proliferation, enable in vitro expansion of primary cancer cells, and induce EMT. These engineered fFn networks can serve as a platform for the design of in vitro 3D culture systems, including tumor models which would impact fundamental studies of tumorigenesis and advance our ability to directly expand primary patient cancer cells.
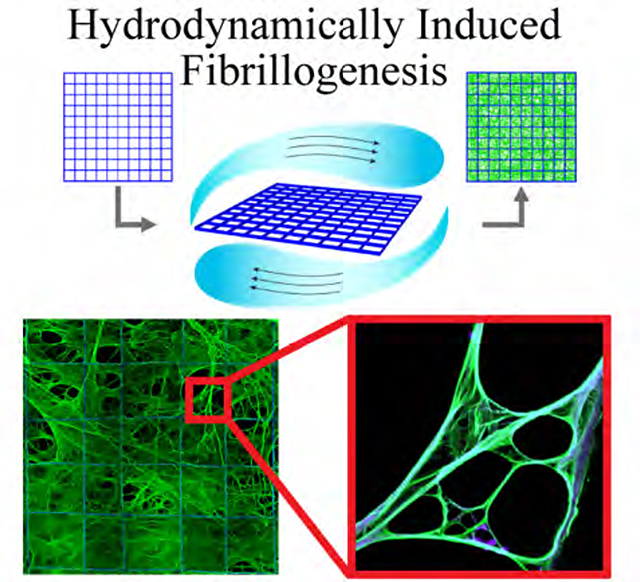
Large-scale, hydrodynamically induced fibrillogenesis on tessellated scaffolds
The cell-free synthesis of scalable, mechanically robust 3D networks of fFn suspended across a scaffold support would constitute a critical technological advance in engineered biomaterials systems. Cell-free fibrillogenesis has been previously observed by simple shearing of Fn solution, drawing individual fibers[12] or extracting fibrillar mats[13] from Fn solutions, or through the use of mechanical,[14] or interfacially active denaturants.[4, 14–15] However, these methods suffer from several limitations including manual positioning of protein fibers or networks,[12–13] low throughput,[12] or the need to chemically crosslink the protein to ensure fiber stabilization.[13b] In application to cell culture and cell analyses, these methods often lack (i) effective cell invasion and cell removal capabilities,[13a] (ii) transparency for microscopy techniques,[4, 12–13] (iii) a support where cells can deposit their own matrix or remain adhered after degrading the original protein matrix,[13a, 16] and (iv) sufficiently large areas (≥ 0.2 mm2) of 3D space free of synthetic material. When polymer scaffolds are used to mediate some of these shortcomings, the support material itself can act as a barrier for cell invasion and uninterrupted transport of nutrients.[2, 4, 12, 14–15] In contrast, we found that gentle shearing (angular velocity of 8 rpm) of a Fn solution at the three-phase contact line defined by the protein solution, air, and a support comprised of tessellated 500×500 μm square pores yields insoluble networks of fibrillar Fn, even in the absence of denaturants (Figure 1). Fibronectin used in this study was commercially available, purified Fn isolated from blood plasma. The tessellated microfiber scaffolds were fabricated from poly(lactide-co-glycolide) (PLGA) by 3D jet writing.[17] While the scaffold size, pore geometries, and thickness are tunable, all scaffolds used in this study spanned an area of 34.8 mm2 featuring square pores with a free volume of 96% and an overall thickness of 0.12 mm (Figure 1b). The thickness of the engineered fFn networks was chosen for optimal imaging results and to allow cells to perfuse in or out, while providing large volumes of freely-suspended protein matrix for cell growth in three dimensions. The fFn networks were mechanically robust for easy handling during culture, and readily implemented by standard cell culture techniques in a multi-well plate.
Figure 1. Hydrodynamically induced fibrillogenesis of fibronectin.

(a) Fluid shear is applied at the three-phase interface of a porous scaffold, air, and a fibronectin (Fn) solution resulting in the deposition of a network of insoluble Fn fibrils (fibrillar fibronectin, fFn) suspended across the scaffold, i.e., fFn networks. (b) Polymer microfiber scaffolds fabricated via 3D jet writing featuring tessellated square pores. Scale bars 500 μm (left) and 25 μm (inset). (c) High resolution scanning electron micrograph (SEM) of fFn freely-suspended within a pore of the scaffold. Scale bar 1 μm. (d) (left) Laser scanning confocal micrograph (LSCM) of fFn (green) suspended within the scaffold (blue) for comparison to (right) fibronectin deposited by human mammary fibroblasts cultured on glass and subsequently decellularized. Scale bar 25 μm. (e) NIH-3T3 mouse fibroblasts cultured three days on either (left) a fFn network or (right) fibronectin statically adsorbed onto a scaffold. Insets show representative images of the morphology and distribution of Fn (green) deposited on scaffolds (blue) either by hydrodynamic shearing at the three-phase interface (left inset) or static adsorption (right inset). Channels: blue, tessellated scaffold; green, fibronectin; cyan, cell nucleus; red, actin. Scale bars 500 μm.
Within 15 minutes, hydrodynamically induced fibrillogenesis resulted in emerging networks of insoluble Fn fibrils that extended across the entire scaffold at lengths approaching several millimeters (Figures 1c, S1b,c,d,f). The absence of a shear force (Figure S1f and insets of Figure 1e), or hydrodynamic shearing over a smooth, non-porous support (Figure S1a) did not result in fibrillar networks. We instead observed a conformal Fn coating. The total amount of Fn loaded onto a single scaffold by hydrodynamically induced fibrillogenesis was 12-fold higher than the amount of protein detected for conformally coated scaffolds (Figure S1e). The fFn networks displayed morphological similarities to matrix deposited by human fibroblasts (Figure 1d, S2a,b).
fFn networks proved to be insoluble in a 1% deoxycholate solution (Figure S3), another feature of biologically derived Fn matrix.[18] Importantly, fFn networks supported confluent cell culture that extended throughout the entire scaffold volume (Figure 1e, S4). In contrast, identical scaffolds that were conformally coated with Fn did not promote continuous cell coverage of the open pore areas (Figure 1e). A wide range of mammalian cell types including normal cells (fibroblast and endothelial cells), cancer cells (breast and pancreatic cancer cells), and mesenchymal stem cells[17] formed confluent 3D cell cultures in as little as three days (Figure 1e, S4, S5).
Fibrillar Fn networks, but not conformally deposited Fn, stained positively with an Fn-3 antibody indicating exposed cellular fibronectin after hydrodynamic fibrillogenesis (Figure S6, S2b). This resembled the staining of Fn deposited by human mammary fibroblasts (Figure S2a, stained after removal of the cells). In contrast, statically adsorbed Fn on either tessellated scaffolds or tissue culture polystyrene (TCPS) was not recognized by the Fn-3 antibody (Figure S2c,d). For further validation, an IST-9 antibody was used since its specificity to the alternatively spliced domain A (EIIIA or EDA), a cellular fibronectin variant within the type III region of fibronectin, is well documented.[19] When directly comparing engineered fFn networks to Fn statically deposited onto non-woven mats (a scaffold of randomly deposited PLGA fibers), we found that the Fn-3 and IST-9 antibodies only recognized the fFn networks and not the Fn conformally deposited onto non-woven mats (Figure S7).[19a] To definitively confirm the presence of EDA(+) Fn in our protein source, liquid chromatography mass spectrometry-based (LC-MS) proteomics was performed. As expected, the main constituent was Fn with 71 to 82% sequence coverage (Figure S8c). Annotated MS/MS spectra for tryptic EDA peptides GLAFTDVDVDSIK and IAWESPQGQVSR are shown in Figure S8a and S8b respectively. These EDA fragments are expected to be identified after trypsin digestion and indeed were identified with nearly complete sequencing, confirming the presence of EDA(+) Fn in our protein source. Although, plasma Fn isoforms are the main constituent of Fn in blood plasma, previous studies have reported that small fractions of cellular Fn can circulate in the blood of healthy patients.[20]
Collectively, these data suggests that EDA(+) Fn within our protein source is physically exposed only after hydrodynamic fibrillogenesis, and is not available for antibody binding when then the Fn is conformally adsorbed onto a surface. We note that role of the EDA peptide in Fn biology is a topic of ongoing investigation. While this variant is not believed to be a requisite for fibrillogenesis,[21] some studies have shown it plays an important role by leading to more robust fibrillar networks secreted by cells.[22] Additionally, while some report recombinant EDA(+) Fn coated onto a surface may enhance cell migration,[23] other studies have suggested that knockdown of EDA-Fn secretion impedes cell motility even on Fn coated substrates.[24] Missirlis et al. discuss potentially conflicting results researchers have reported with respect to the role of EDA-Fn in cell motility.[24] Finally, the mechanosensitivity of the EDA domain is not well known[25] and warrants further investigation. Hence, it is reasonable that changes in Fn binding activity may occur when Fn is presented in a fibrillar state compared to a conformal surface coating, which has not been directly investigated to our knowledge. These many unknowns underscore the need for a native-like fibrillar Fn cell culture platform. We thus concluded that our Fn fibril production and characterization results are consistent with a mechanism where interfacial shearing induces mechanical deformation of solute Fn that extends the protein, enabling self-polymerization and fiber formation. This is analogous to the mechanically induced unfolding of the fibronectin molecule during cell-driven,[9b, 26] as well as previously reported shear-driven fibrillogenesis[4, 8a, 27]. In contrast to the widely used conformal Fn pre-coatings, hydrodynamically induced fibrillogenesis not only results in a stable, fibrillar matrix readily applicable to 3D cell culture, but also provides access to ECM mimicries with biologically distinct properties.[10a, 28]
Fibrillar Fn networks improve mouse breast tumor engraftment efficiency
Recognizing the prominence of Fn in primary breast cancer ECM,[29] we tested the ability of fFn networks to enhance tumor engraftment of breast cancer cells in a murine model of epithelial breast cancer. We cultured AT-3 murine breast cancer cells that stably expressed firefly luciferase[30] onto fFn networks. After three days, cells had infiltrated the entire scaffold (Figure 2a,b, S9a,b), while still remaining in a proliferative state (Figure S9c).
Figure 2. Engineered fFn networks enhance tumor engraftment efficiency in a mouse breast cancer model.

(a) AT-3 mouse breast cancer cells formed 3D cell volumes approximately 70 μm thick in vitro after three days on fFn networks. Scale bar 25 μm. (b) Large scale view shows that AT-3 cells in (a) proliferated and filled the 3D space within the fFn network. Scale bar 500 μm. (a-b) Channels: cyan, cell nucleus; red, actin. (c) Bioluminescence image of tumor formation in immune-competent mice 21 days after AT-3 cells were orthotopically implanted (image exposure time 10 seconds). Mice on the left after implantation of fFn networks carrying about 30,000 AT-3 cells into the mammary fat pads indicated by arrows (group 1). The contralateral mammary fat pad received an injection of approximately the same number of cells suspended in a Fn solution as indicated by circles (group 2). Mouse on the right is a positive control having received the group 3 fFn network in the left mammary fat pad (arrow), and the group 4 injection in the right, each delivering 200,000 AT-3 cells (the minimum required for tumor formation by cell injection[30]). (d) Mason’s Trichrome staining of a group 1 tumor graft that formed after 21 days showing AT-3 cells invading the surrounding tissues. Scale bar: 25 μm. Quantification of the (e) CD29+/CD24+ population capable of self-renewal (P<0.05) and (f) CD29+/CD24+/CD90.2+ tumor initiating population in AT-3s cultured three days on TCPS, TCPS with fibronectin conformally adsorbed (Fn on TCPS), or fFn networks. Single star indicates that the fFn network is statistically different from TCPS and Fn on TCPS; double star indicates that TCPS and Fn on TCPS are statistically similar.
The fFn networks carrying 30,000 cells per scaffold were orthotopically implanted into immunocompetent C57BL/6 mice. Bioluminescence imaging revealed initial signs of tumorigenesis as early as two days after implantation (Figure S10). For comparison, the cell suspension of an equivalent number of AT-3 cells did not support tumor engraftment over 21 days (Figure 2c, S10). Histology of engrafted tumors confirmed natural invasion of cancer cells from the fFn networks into adjacent tissue (Figure 2d). Pre-seeded with 30,000 AT-3 cells, fFn networks showed robust tumor engraftment at cell numbers that were 85% lower than what is typically needed for successful engraftment.[30] This increased tumorigenicity is consistent with our in vitro findings that AT-3 cells cultured on fFn networks have increased subpopulations of tumor-initiating (CD29+/CD24+)[31] and metastatic (CD29+/CD24+/CD90.2+)[31c, 32] cells (Figure 2e,f). We further note that the invasive subpopulation (CD90.2+) was nearly 30-fold higher on fFn networks relative to Fn-coated tissue culture polystyrene (TCPS) (Figure S9d).
Fibrillar Fn networks enrich tumor-initiating cell populations of human triple negative breast cancer cells
We next evaluated if the increased tumorigenicity observed in mouse breast cancer cells would also be observed in human triple negative breast cancer cells. We selected the MDA-MB-468 cell line because less than 3% of the total population express the CD44+/CD24− tumor initiating phenotype.[33] After four days of expansion on fFn networks, MDA-MB-468 cells formed a dense three-dimensional cancer microenvironment over extended areas of about 10 square millimeters (Figure 3a,b, S11a). Compared to cells cultured on either TCPS or Fn-coated TCPS, the CD44+/CD24− subpopulation was significantly higher on fFn networks (Figure 3c). In addition, the CD44+/CD24−/ALDH+ subpopulation significantly increased on fFn networks relative to the control groups for all time points (Figure 3d). CD44+/CD24−/ALDH+ breast cancer cells have higher tumor-initiating potential than either CD44+/CD24− or ALDH+ populations (ALDH, aldehyde dehydrogenase).[34] Because the ALDH+ subpopulation showed no significant difference relative to the controls after four days in culture on fFn networks (Figure S11b), the data indicate that enrichment of CD44+/CD24−/ALDH+ cells is largely driven by an increase in the CD44+/CD24− subpopulation. These data are consistent with an increased level of epithelial to mesenchymal transitions (EMT) in MDA-MB-468 cells after expansion on fFn networks relative to the control groups (Figure 3c, S11, S12).
Figure 3. Engineered fFn networks increase the tumor-initiating population in MDA-MB-468 human breast cancer cells.

(a) MDA-MB-468 breast cancer cells cultured four days on fFn networks form cell-cell and cell-ECM contacts. Scale bar 25 μm. (b) MDA-MB-468 cells form large interconnected volumes throughout fFn networks after four days. Scale bar 500 μm. (a-b) Channels: green, fibronectin; orange, laminin; cyan, cell nucleus; red, actin. (c-d) Population of MDA-MB-468s on fFn networks (black solid line and square marker), TCPS (black dotted line and crisscross marker), or fibronectin adsorbed conformally onto TCPS (grey line and triangular marker) that are (c) CD44+/CD24− and (d) CD44+/CD24−/ALDH+ measured at different culture time points. Marker expression for cells on TCPS or Fn on TCPS is significantly lower than that of the fFn network at values nearing zero in (d). The starred time point is statistically different from the other three time points within the fFn network dataset.
Stem-like breast cancer cells can secrete extracellular laminin during matrix remodeling to improve self-renewal and tumorigenicity.[35] With the observed enrichment of CD44+/CD24− and CD44+/CD24−/ALDH+ in MDA-MB-468 cells after culture on fFn networks, we asked whether significant amounts of endogenous laminin had been incorporated into the ECM, influencing cell phenotype. After four days of MDA-MB-468 cell culture, fFn networks were decellularized[36] and co-stained for Fn and laminin. MDA-MB-468 cells had only secreted small and sporadic amounts of laminin, while the original fFn network remained intact (Figure S13). This experiment indicates that Fn is the predominant ECM factor for early EMT transitions on fFn networks.
Fibrillar Fn networks promote expansion of patient-derived cells and induce EMT
Facile, reliable expansion of patient-derived breast cancer cells would tremendously advance precision oncology, but still remains an elusive goal.[37] Mouse tumor xenografts have been used to develop avatars using primary human patient cells, but in breast cancer this approach suffers from poor cell engraftment and limited cell proliferation.[37b, 38] We therefore evaluated fFn networks as a platform to directly expand patient-derived cells from malignant pleural effusion or ascites samples from 14 women representing a range of ages and molecular subtypes of metastatic breast cancers (Table S1). We successfully cultured metastatic breast cancer cells on fFn networks from all 14 patient samples. Importantly, cell expansion on fFn networks did not require prior fractionation of the patient samples and resulted in a 36-fold enrichment of the cancer cell population (Figure S14a). The majority of patients (9 out of 13; 1 sample was of unknown receptor status) were estrogen receptor positive, a breast cancer subtype that typically is less likely to form tumor grafts.[33c, 37a] Cancer cells from all fourteen patient samples proliferated on fFn networks, but not on TCPS or Fn-coated TCPS, as represented in Figure 4c and S15b. Ki67 staining further indicated that cells were in a proliferative state on fFn networks, but senescent on TCPS[37a] (Figure 4c). Cells from patient E (patient samples arbitrarily named), which were first cultured on fFn networks for one day, then removed and re-seeded onto TCPS, proliferated for five passages before entering senescence (Figure S15a). This repeatedly observed finding was in stark contrast to the unsuccessful direct culture of patient cells on TCPS, suggesting the potential use of fFn networks as a preconditioning microenvironment to promote expansion of cancer cells from pleural effusions, an accessible, yet underutilized source of patient tumor cells.
Figure 4. Engineered fFn networks enable expansion of patient breast cancer cells and enrich the tumor-initiating cell population in an epithelial to mesenchymal transition.

(a-b) Four-day culture of heterogenous cell populations from Patient E on fFn networks. Channels: cyan, cell nucleus; red, actin; yellow, cytokeratin 5. (a) 3D cell structures form within the pores and along the microfiber walls of the scaffold. Scale bar 25 μm. (b) Patient E cells fill fFn networks at large scale across all square 500 μm-wide pores of the tessellated scaffold. Scale bar 500 μm. (c) Patient cell proliferation measured via mitochondrial activity is far increased on fFn networks (black solid line and square marker) relative to the little to no growth on Fn adsorbed conformally onto TCPS (grey line and triangular marker), or TCPS alone (black dotted line and crisscross marker). Additionally, the inset shows representative images of Ki67 staining of patient cells cultured on either TCPS or fFn networks. Darker color indicates that the cells are in a proliferative state on fFn networks but senescent on TCPS. (d) Flow cytometry measurement of CD24 and CD44 in Patient E cells where the original sample is shown in red and the six-day in vitro culture on fFn networks is shown in blue. Cell phenotype concentrated towards CD44+/CD24− status after culture on fFn networks. (e-h) Flow cytometry measurements of the percentage of lineage negative cells that are (e) CD44+/CD24− (f) EpCAM+ (epithelial cell adhesion molecule) (g) ALDH+ (aldehyde dehydrogenase) and (h) CD44+/CD24−/ALDH+ within samples from Patients E, F, and G. Grey bars represent the original patient sample and white bars indicate result after cells were cultured on engineered fFn networks for six days.
The total patient cell population increased by more than 7-fold relative to the original patient sample over the course of eight days of culture on fFn networks (Figure 4c). In general, diverse cell populations were maintained on fFn networks showing heterogeneity in morphology and expression of cytokeratin 5 (CK5), an epithelial marker (Figure 4a,b S16a–d). After five days of culture, CK5+ cells remained in small clusters. By day 10, CK5+ cells had expanded and spread out into the open pores of the scaffold (Figure S16d). Different cell types were often found to spatially segregate as seen in Figure S16a, where CK5+ cells formed tight clusters on the fFn while actin-rich cells of a spindle-like morphology adhered first to the scaffold microfibers and then proliferated outwards.
Because of their distinct receptor status, patient samples E, F, and G were selected for evaluation of cancer stem cell markers after a six-day culture on fFn networks. Patient breast cancer cells with a mesenchymal CD44+/CD24− stem cell phenotype were enriched relative to the percent in the original sample (Figure 4d,e, S14c). This finding corroborates earlier results with MDA-MB-468 cells (Figure 3c). The epithelial tumor-initiating subpopulation marked as ALDH+ (Figure 4g, S14b,e) and the highly tumorigenic overlapping immunotype CD44+/CD24−/ALDH+[39] (Figure 4h) were maintained on fFn networks compared to the original patient sample (having averages within 10%). Additionally, the percentage of cancer cells expressing the epithelial marker, EpCAM (epithelial cell adhesion molecule), disappeared after culture on fFn networks (Figure 4f, S14d). Upregulation of mesenchymal stem-like cancer cells combined with downregulation of EpCAM and maintenance of the ALDH+ subpopulation characterizes an epithelial to mesenchymal transition, suggesting that fFn networks can induce EMT in breast cancer patient cells cultured in vitro. Despite these observed phenotypic changes, the breast cancer receptor status of the original sample was maintained after the six-day culture on fFn networks (Figure S16e).
Engineered fFn networks represent a major advance toward the goal of using cancer patient cells to engineer tumor tissue that authentically represents the subtypes, heterogeneity, architecture, and patient-to-patient variability of breast cancer. This work further suggests that Fn may play a role in EMT.[40] Scaffolds functionalized with our scalable fFn technology produce suitable microenvironments for direct and efficient culture of unfractionated patient samples. In short-term in vitro culture, fFn networks resulted in the expansion of patient samples, where less than 5% of the initial population were cancer cells, to complex cultures or microtumors with cancer cells constituting 36% of the total cell population.
Our results suggest that engineered fFn networks can provide the next-generation 3D cell culture platform for expansion of both immortalized and primary cancer cells in vitro. Harnessing the known ability of fibronectin to form fibrils under shear forces, we directly suspended fFn across open-pore scaffolds resulting in a platform that is readily implemented in cell culture. These fFn networks allow cells to craft their own microenvironment while we observe the cell response, representing a transformative technology in fundamental cancer studies,[37b] precision medicine,[33c] pharmaceutical development[37b] and preclinical screening.[37a]
Experimental Section
See Supporting Information for Materials
Scaffold fabrication
Tessellated scaffolds were made via 3D jet writing as previously described[17]. Briefly, PLGA was dissolved into a 30 wt% solution of chloroform and N,N-dimethylformamide at a ratio of 93:7. The solution was pumped through a 20 gauge needle at 0.04 mL/hr with an applied voltage of 16 kV. As the fluid jet descends to the ground electrode, it passes through a copper ring at 9 kV. The jetted fiber is collected on the ground electrode, a stainless-steel plate, which is translated through x-y coordinates by computer-controlled motions to stack the depositing fiber onto itself in a desired pattern. Non-woven mats were made by random deposition of electrospun PLGA fibers onto a flat grounded electrode without use of the copper ring.
Hydrodynamically induced fibrillogenesis and other fibronectin substrates
The tessellated scaffold fabricated via 3D jet writing was first secured in a custom-built stainless-steel frame. The framed scaffold was then placed at the center of a 2-mL microcentrifuge tube containing 900 μL volume of a 111 μg/mL solution of human fibronectin such that the air, scaffold, and solution formed a three-phase interface when the tube was laid on its side. The tube was placed on a rotisserie rotator at 30°C and 8 rpm (for an interface velocity of 10.4 mm/s) for a period of 2 h. The protein-loaded scaffold was then washed three times in DPBS and stored at 4°C up to one week until use. For suspended collagen networks, 900 μL of collagen I dissolved in 33 mM acetic acid at 2.7 mg/mL was combined with 100 μL of DMEM and 140 μL of 0.34 N NaOH in a 2-mL microcentrifuge tube. The solution turned from yellow to pink with the addition of base. All collagen preparations were performed on ice. The framed scaffold was then secured in the 2-mL tube to form a three-phase interface and rotated for four hours at 27°C. Conformal fibronectin coatings onto tessellated scaffolds, smooth tissue culture polystyrene surfaces, or non-woven fiber mats were prepared by statically submerging the substrate in a 111 μg/mL fibronectin solution for 2 hours at 30°C and overnight at room temperature.
Protein mass measurements
A Micro BCA assay (Pierce Biotechnology, Rockford, IL) was used to measure the mass of protein deposited onto the scaffolds. First, the PLGA was degraded in a 0.9 M NaOH solution for 1 h, and then the solution was neutralized in a 0.9 M HCl solution. The mass of protein was determined by measuring the absorption at 562 nm after a 1 h, 60°C incubation, in the Micro BCA working reagent and referenced to a standard curve of known masses.
ICC staining
Immunocytochemical (ICC) analysis was performed as previously described[17]. Briefly, cells on the fFn networks were fixed with a 2% paraformaldehyde overnight. Cells were then permeabilized with a 0.1% Triton X-100 solution for 5 minutes. The fixed cells were then exposed to a 5% BSA blocking solution for 1 h before application of the antibodies. Cells were imaged using a Nikon A-1 confocal microscope (Nikon Corp., Minato, Tokyo, Japan). If only protein without cells was present on the fFn network, then Triton X-100 was not used.
SEM imaging
The tessellated scaffold was sputter coated with gold and visualized with an Amray FE 1900 scanning electron microscope (SEM) with an acceleration voltage of 5 kV. All other SEM micrographs were taken with a Helios 650 Nanolab SEM/FIB by FEI of Thermo Fisher Scientific at voltages ranging from 2 to 5 kV. Prior to SEM, samples of cells or proteins were dehydrated by soaking for 20 minutes in a series of ethanol solutions increasing in concentration. The sample was then transferred to a microcentrifuge tube and tertbutanol was added to cover. The tube was placed in liquid nitrogen for two minutes then lyophilized overnight.
Deoxycholate treatment
Fibrillar Fn networks that were subjected to deoxycholate treatment were submerged in 1% deoxycholate solution overnight on an orbital shaker. fFn networks that were left untreated for comparison to deoxycholate exposure were also rotated overnight in water on the orbital shaker. These fFn networks were prepared using fluorescent fibronectin that was first conjugated to DyLight-650 using a DyLight antibody labeling kit following manufacturer instructions.
Cell culture
Malignant pleural effusion and ascites samples from women with metastatic breast cancer were collected with informed consent under a protocol approved by the University of Michigan Institutional Review Board. These samples were collected for clinical indications, and this research used excess fluid samples that otherwise would be discarded. We concentrated total cells in samples by centrifugation without additional processing steps prior to seeding onto scaffolds. Breast cancer patient cell samples were seeded in 1 mL onto fFn networks with square scaffold pores 500 μm wide at a concentration of 2×106 cells/mL. Cancer cell lines were seeded in 100 μL at a concentration of 2×106 cells/mL for 4 hours, then the concentration was diluted to 2×105 cells/mL. Cells were cultured in DMEM containing 10% FBS, 1% NEAA, 2 mM L-glutamine, and 1% antibiotic-antimycotic. Cells were seeded and cultured in low adhesion 24 well plates (Corning Inc., Corning, NY) and maintained at 37°C in 5% CO2. Although not always passaged, passaging primary lineage negative cancer cells could be done by placing a cell free fFn network in contact overnight with another that had already grown confluent with cells.
Proteomics
Lyophilized fibronectin (Corning) was dissolved in DPBS as described above. The protein concentration was determined by Qubit fluorometry (Invitrogen). 10μg of the sample was processed by SDS-PAGE using a 10% Bis-Tris NuPage mini-gel with the MOPS buffer system. The mobility region was excised and processed by in-gel digestion using a robot (ProGest, DigiLab) using the following procedure: washed with 25mM ammonium bicarbonate followed by acetonitrile, reduced with 10mM dithiothreitol at 60°C followed by alkylation with 50mM iodoacetamide at RT, digested with sequencing grade trypsin (Promega) at 37°C for 4h, and quenched with formic acid. Then the supernatant was analyzed directly without further processing. Half of the gel digest was analyzed by nano LC-MS/MS with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher Q Exactive. Peptides were loaded on a trapping column and eluted over a 75μm analytical column at 350nL/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 70,000 FWHM and 17,500 FWHM for MS and MS/MS respectively. The fifteen most abundant ions were selected for MS/MS. Data were searched using a local copy of Mascot (Matrix Science) with the following parameters: Enzyme: Trypsin/P; Database: SwissProt Human (concatenated forward and reverse plus common contaminants); Fixed modification: Carbamidomethyl (C); Variable modifications: none; mass values: monoisotopic; peptide mass tolerance: 10 ppm; fragment mass tolerance: 0.02 Da; maximum. missed cleavages: 2. Mascot dat-files were parsed into Scaffold (Proteome Software) for validation, filtering and to create a non-redundant protein list. Data were filtered using 1% protein and peptide FDR and requiring at least two unique peptides per protein.
Proliferation assays
AT-3 mouse breast cancer cells and human breast cancer cell lines T47D and Sum159 all stably expressed firefly luciferase. Proliferation was tracked over time by administering 15 mg/mL D-luciferin to the cell medium at a ratio of 1:100 and incubating for 10 min before the relative light intensity was measured using a luminometer (Perkin Elmer MLD2300–000). The growth rates of the primary patient samples and the MDA-MB-468 cell line were evaluated by measuring changes in mitochondrial activity using a resazurin based assay kit (Tox-8, Sigma Aldrich, St. Louis, MO) following the manufacturer guidelines. The initial signal was recorded after seeding, and then subsequent mitochondrial activity measurements were normalized to the initial time point. Additionally, Ki67 staining was used to determine whether the primary patient samples were in a proliferative or senescent state.
Decellularization
If cells were removed from fFn networks to visualize the remaining protein matrix, samples were decellularized in a protocol adapted from Lu et. al..[36] Samples were washed with PBS, then DI water, and immersed in a solution of 0.1% Triton X-100 with 1.5 M KCl in 50 mM Tris buffer on a slow-moving shaker for two hours at 4°C. Samples were washed in 10 mM Tris buffer, followed by DI water for one hour each. The remaining protein matrix was fixed and stained via IHC protocol.
Animal protocol
All animal studies were approved by the University of Michigan Institutional Animal Care and Use Committee. Briefly, 6–8-week-old female C57BL/6J mice (Charles River Laboratories Inc., Wilmington, MA) were anesthetized using 1–2% isoflurane at a flowing oxygen rate of 1 L/min (EZ150 Isoflurane Vaporizer, EZ Anesthesia), and an incision was made to expose the mammary fat pad. A fFn network carrying AT-3 cells was then placed directly into the fat pad[30] and the incision was closed with wound clips. The number of AT-3 cells on the scaffold was verified using luminescence. 15 mg/mL D-luciferin was added to the cell medium at a ratio of 1:100 and incubated for 10 min before the relative light intensity was measured using a luminometer (Perkin Elmer MLD2300–000) and compared with a standard curve. Tumor engraftment was evaluated using bioluminescence imaging.[30]
Flow cytometry
Cells were stained following a previously established protocol.[41] Cells were removed using 0.25% trypsin, which was neutralized using a 3:1 volume of complete medium. Cells were then counted using a Luna-FL dual fluorescence cell counter (Logos Biosystems, Dongan-gu, Anyang-si, Gyueonggi-do, South Korea), and re-suspended in aldefluor buffer. Antibodies were added and incubated for 30 minutes, and then rinsed with aldefluor buffer. For primary patient sample analysis, the cells were first incubated in the lineage cocktail for 30 minutes, before staining for EpCAM, CD44, CD24, and ALDH activity. MDA-MB-468 cells were used to establish single color channels. Isotype controls were used to establish gating for the CD44, EpCAM, lineage negative cells, and CD24.[41] DEAB was used to establish gating for the aldefluor activity assay.[41b]
Histology
Tumor histology was evaluated by first fixing with 4% formalin for 24 h. Then the tissue was prepared for histology by dehydrating in ethanol, sealed with paraffin, and sliced for staining with Masson’s trichrome.
Statistics
Statistical analysis was performed using Minitab (Minitab Inc., State College, PA). One-way analysis of variance (ANOVA, p<0.05) was used to determine statistical significance. Tukey multi-comparison testing was performed to determine differences between groups. All data was reported as the mean ± standard deviation.
Supplementary Material
Acknowledgements
We acknowledge funding under U01CA210152 from the National Cancer Institute. We also thank the UM Center for Molecular Imaging for bioluminescence imaging instrumentation, the UM Microscopy and Imaging Laboratory for laser scanning confocal microscopy access, the Michigan Center for Materials Characterization for scanning electron microscopy access, Henriette A. Remmer and the University of Michigan Medical School Proteomics & Peptide Synthesis Core for fibronectin sequencing, and the lab of Diane M. Simeone for pancreatic cancer cells. S.J. acknowledges support from the National Science Foundation Graduate Research Fellowship Program: DGE 1256260. L.S. acknowledges support from the Department of Defense Breast Cancer Research Post-Doctoral Fellowship: W81XWH-10-1-0582. D.N. acknowledges NIH support from training grant T32 GM008353. J.H.J acknowledges the support of NIH’s Microfluidics in Biomedical Sciences Training Program: NIH NIBIB T32 EB005582. G.D.L. acknowledges NIH grants R01CA196018. Finally, we are thankful to Ayse Muniz for enlightening discussions.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Stacy Jordahl, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Luis Solorio, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Dylan Neale, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Sean McDermott, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Jacob H. Jordahl, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA
Alexandra Fox, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Christopher Dunlay, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Annie Xiao, University of Michigan, 1540 E. Hospital Drive, Ann Arbor, MI 48109.
Martha Brown, University of Michigan, 1500 E Medical Center Dr SPC 5916, Ann Arbor, MI 48109.
Max Wicha, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Gary D. Luker, University of Michigan, 1540 E. Hospital Drive, Ann Arbor, MI 48109
Joerg Lahann, Director of the Biointerfaces Institute, Professor of Chemical Engineering, Materials Science and Engineering, Biomedical Engineering, and Macromolecular Science and Engineering University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
References
- [1].Cantini M, Rico P, Salmeron-Sanchez M, in Biomimetic Approaches for Biomaterials Development, (Ed: Mano JF), Wiley-VCH Verlag GmbH & Co. KGaA., Weinheim, Germany: 2012, 189. [Google Scholar]
- [2].Lee J, Cuddihy MJ, Kotov NA, Tissue Engineering: Part B 2008, 14, 61. [DOI] [PubMed] [Google Scholar]
- [3].Boontheekul T, Mooney DJ, Curr. Opin. Biotechnol 2003, 14, 559. [DOI] [PubMed] [Google Scholar]
- [4].Ulmer J, Geiger B, Spatz JP, Soft Matter 2008, 4, 1998. [Google Scholar]
- [5].Wang JP, Hielscher A, J. Cancer 2017, 8, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS, Mol. Biol. Cell 2012, 23, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Nature 2005, 438, 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Mitsi M, Handschin S, Gerber I, Schwartlander R, Klotzsch E, Wepf R, Vogel V, Biomaterials 2015, 36, 66; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) To WS, Midwood KS, Fibrogenesis Tissue Repair 2011, 4, 21; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang K, Andresen Eguiluz RC, Wu F, Seo BR, Fischbach C, Gourdon D, Biomaterials 2015, 54, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Wang K, Seo BR, Fischbach C, Gourdon D, Cell. Mol. Bioeng 2016, 9, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Singh P, Carraher C, Schwarzbauer JE, Annu Rev Cell Dev Biol 2010, 26, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Cseh B, Fernandez-Sauze S, Grall D, Schaub S, Doma E, Van Obberghen-Schilling E, J. Cell Sci 2010, 123, 3989; [DOI] [PubMed] [Google Scholar]; b) Wierzbicka-Patynowski I, Schwarzbauer JE, J. Cell Sci 2003, 116, 3269; [DOI] [PubMed] [Google Scholar]; c) Baneyx G, Baugh L, Vogel V, PNAS 2002, 99, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lemmon CA, Weinberg SH, Sci Rep 2017, 7, 18061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Little WC, Smith ML, Vogel V, Matrix Biol. 2008, 27, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a)Ejim OS, Blunn GW, Brown RA, Biomaterials 1993, 14, 743; [DOI] [PubMed] [Google Scholar]; b)Brown RA, Blunn GW, Ejim OS, Biomaterials 1994, 15, 457. [DOI] [PubMed] [Google Scholar]
- [14].Ahn S, Deravi LF, Park S, Dabiri BE, Kim J, Parker KK, Shin K, Adv. Mat 2015. [DOI] [PubMed] [Google Scholar]
- [15].Pernodet N, Rafailovich M, Sokolov J, Xu D, Yang NL, McLeod K, J Biomed Mater Res A 2003, 64, 684. [DOI] [PubMed] [Google Scholar]
- [16].Deravi LF, Su T, Paten JA, Ruberti JW, Bertoldi K, Parker KK, Nano Letters 2012, 12, 5587. [DOI] [PubMed] [Google Scholar]
- [17].Jordahl JH, Solorio L, Sun H, Ramcharan S, Teeple CB, Haley HR, Lee KJ, Eyster TW, Luker GD, Krebsbach PH, Lahann J, Adv. Mat 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Fogerty FJ, Akiyama SK, Yamada KM, Mosher DF, J Cell Biol 1990, 111, 699; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bultmann H, Santas AJ, Peters DM, J Biol Chem 1998, 273, 2601; [DOI] [PubMed] [Google Scholar]; c) McKeown-Longo PJ, Mosher DF, J Cell Biol 1983, 97, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Liao YF, Wieder KG, Classen JM, Van De Water L, J Biol Chem 1999, 274, 17876; [DOI] [PubMed] [Google Scholar]; b) Borsi L, Carnemolla B, Castellani P, Rosellini C, Vecchio D, Allemanni G, Chang SE, Taylor-Papadimitriou J, Pande H, Zardi L, J Cell Biol 1987, 104, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Kanters SD, Banga JD, Algra A, Frijns RC, Beutler JJ, Fijnheer R, Diabetes Care 2001, 24, 323; [DOI] [PubMed] [Google Scholar]; b) Castellanos M, Leira R, Serena J, Blanco M, Pedraza S, Castillo J, Davalos A, Stroke 2004, 35, 1671. [DOI] [PubMed] [Google Scholar]
- [21].Leiss M, Beckmann K, Giros A, Costell M, Fassler R, Curr Opin Cell Biol 2008, 20, 502. [DOI] [PubMed] [Google Scholar]
- [22].Abe Y, Bui-Thanh NA, Ballantyne CM, Burns AR, Biochem Biophys Res Commun 2005, 338, 1640. [DOI] [PubMed] [Google Scholar]
- [23].Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K, J Cell Biol 1997, 139, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Missirlis D, Haraszti T, Kessler H, Spatz JP, Sci Rep 2017, 7, 3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zollinger AJ, Smith ML, Matrix Biol 2017, 60–61, 27. [DOI] [PubMed] [Google Scholar]
- [26].Mao Y, Schwarzbauer JE, Matrix Biology 2005, 24. [DOI] [PubMed] [Google Scholar]
- [27].Klotzsch E, Smith ML, Kubow KE, Muntwyler S, Little WC, Beyeler F, Gourdon D, Nelson BJ, Vogel V, Proc Natl Acad Sci U S A 2009, 106, 18267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Smith ML, Gourdon D, Little WC, Kubow KE, Eguiluz RA, Luna-Morris S, Vogel V, PLoS Biol 2007, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fernandez-Garcia B, Eiro N, Marin L, Gonzalez-Reyes S, Gonzalez LO, Lamelas ML, Vizoso FJ, Histopathology 2014, 64, 512. [DOI] [PubMed] [Google Scholar]
- [30].Fenner J, Stacer AC, Winterroth F, Johnson TD, Luker KE, Luker GD, Sci. Rep 2014, 4, 5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Cho RW, Wang X, Deihn M, Shedden K, Chen GY, Sherlock G, Gurney A, Lewicki J, Clarke MF, Stem Cells 2008, 26, 364; [DOI] [PubMed] [Google Scholar]; b) Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H, Delaloye J, Huelsken J, Nature 2012, 481; [DOI] [PubMed] [Google Scholar]; c) Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, Medina D, Tsimelzon A, Hilsenbeck S, Green JE, Michalowska AM, Rosen JM, Cancer Res. 2008, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lobba AR, Forni MF, Carreira AC, Sogayar MC, Cytometry A 2012, 81, 1084. [DOI] [PubMed] [Google Scholar]
- [33].a) Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL, J. Cell Mol. Med 2009, 13, 2236; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, BhatNakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H, Breast Cancer Res. 2006, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, Manuel CA, Edgerton SM, Harrell JC, Elias A, Sartorius CA, Breast Cancer Res. Treat 2012, 135, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a) Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S, Clin. Cancer Res 2009, 15; [DOI] [PubMed] [Google Scholar]; b) Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur M, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS, Cancer Res. 2009, 69, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chang C, Goel HL, Gao H, Pursell BM, Shultz LD, Greiner DL, Ingerpuu S, Patarroyo M, Cao S, Lim E, Mao J, McKee KK, Yurchenco PD, Mercurio AM, Genes Dev. 2015, 29, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lu H, Hoshiba T, Kawazoe N, Chen G, J. Biomed. Mater. Res. A 2012, 100, 2507. [DOI] [PubMed] [Google Scholar]
- [37].a) Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde J, Rebucci M, Tran-Perennou C, Sastre-Garau X, Sigal-Zafrani B, Delattre O, Dieras V, Poupon M, Clin. Cancer Res 2007, 13, 3989; [DOI] [PubMed] [Google Scholar]; b) DeRose YS, Wang G, Lin Y, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL, Nat. Med 2011, 17, 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M, Wong H, Fuqua SW, Contreras A, Gutierrez C, Huang J, Mao S, Pavlick AC, Froehlich AM, Wu M, Tsimelzon A, Hilsenbeck SG, Chen ES, Zuloaga P, Shaw CA, Rimawi MF, Perou C, Mills GB, Chang JC, Lewis MT, Cancer Res. 2013, 73, 4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G, Cell Stem Cell 2007, 1, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA, Cell 2008, 133, 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF, Proc. Natl. Acad. Sci. U.S.A 2003, 100, 3983; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G, Cell Stem Cell 2007, 1, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.