

Abstract
We describe the performance of MELD-accelerated Molecular Dynamics (MELDxMD) in determining protein structures in the NMR-data-assisted category in CASP13. Seeded from web server predictions, MELDxMD was found best in the NMR category, over 17 targets, outperforming the next-best groups by a factor of ~4 in Z-score. MELDxMD gives ensembles, not single structures; succeeds on a 326-mer, near the current upper limit for NMR structures; and predicts structures that match experimental Residual Dipolar Couplings (RDC) even though the only NMR-derived data used in the simulations was NOE-based ambiguous atom-atom contacts and backbone dihedrals. MELD can use noisy and ambiguous experimental information to reduce the MD search space. We believe MELDxMD is a promising method for determining protein structures from NMR data.
Keywords: CASP13, MELD, Protein Structure Prediction, Molecular Dynamics, NMR
1 |. INTRODUCTION
There are over 11,000 NMR protein structures and restraints in the Biological Magnetic Resonance Bank (BMRB) [1] and the Protein Data Bank (PDB) [2] – making NMR the most successful technique for protein structure determination after X-ray crystallography. NMR experiments take advantage of through sequence (COSY, TOCSY) or through space (NOESY) electron spin couplings[3] to identify distance restraints between pairs of atoms, which provides information to characterize the native state. Even so, determining structures from NMR data can often be troublesome. Information from NMR spectra are ambiguous (multiple atomic interactions can satisfy the same signal) and some signals can be missing or incorrect. Solving these problems can be rate-limiting for obtaining a protein structure using NMR experiments (Fig. 1 A). A further complication is that an increase in protein size leads to an increase of ambiguities, broadness of NMR peaks and peak overlaps. This limits state-of-the-art NMR protein structure determination to proteins smaller than 50–70 kDa [4]. Beyond that, it is usually not possible to generate an assignment informative enough to be exploited with current modeling tools. The field needs better ways for handling unassigned NMR spectra for larger proteins and their complexes.
Figure 1. MELDxMD handles ambiguous NMR data to generate near native structures.
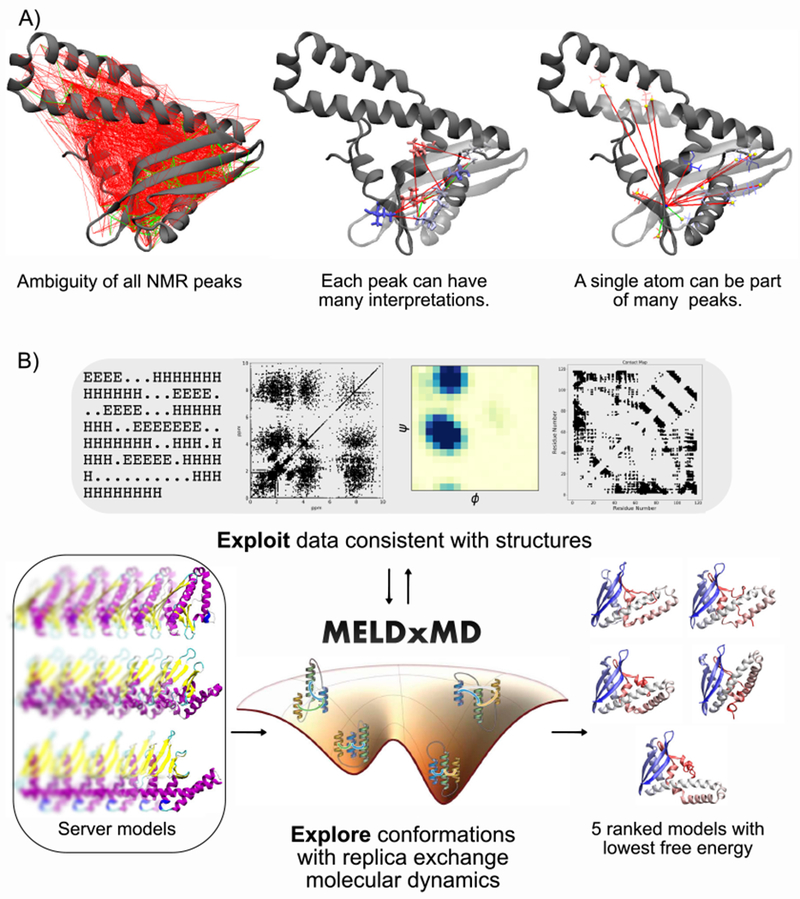
A) The ambiguity problem with unassigned NOESY data makes integrative modeling challenging. B) MELDxMD handles sparse, noisy, and ambiguous data from various sources such as secondary structure predictions, 2D NOESY peaks, NMR dihedrals from NMR chemical shifts, and contact predictions. For data-assisted CASP13, MELDxMD starting coordinates were 15 server models, these were seeded into Hamiltonian, Temperature replica exchange molecular dynamics that enforces restraints on subsets of data consistent with structure in the MELDxMD ensemble. The ensemble trajectory is clustered and representatives from the five most populated clusters are submitted to CASP. Funnel figure for MELDxMD adapted with permission from A. Perez, J. L. MacCallum, and K. Dill, Proc. Natl. Acad. Sci. U. S. A. 112, 11846 (2015). Copyright 2015 National Academy of Sciences, USA.
A method called MELD (Modeling Employing Limited Data)[5, 6] addresses some of these challenges. MELD can harness noisy and/or ambiguous data to accelerate physics-based molecular dynamics (MD) modeling. MELDxMD can take advantage of various types of information to limit the search-space of MD simulations; it does so using a Bayesian framework. MELDxMD retains the important advantage of physical MD that it satisfies detailed balance, and that it therefore computes populations and free energies. Using MELDxMD we can explore the different interpretations of NMR peak assignments and the different conformations that satisfy them, and exploit those conformations that are most compatible with both the data and the force field. MELD also permits the integration of multiple distinct sources of information in the same simulation, further reducing the search space.
Here, we tested MELDxMD on determining protein structures with NMR data, in the context of the CASP13 blind test. We show that: (1) MELDxMD NMR-sampling improves predictions as compared to ab initio MELDxMD CASP predictions (2) MELDxMD is currently the best performing tool to handle NMR data among the few groups that attempted this challenge (3) MELDxMD is an effective technique to predict protein structures from NMR data, where computationally cheaper machine learning or homology tools fail.
2 |. MATERIALS AND METHODS
2.1 |. MELDxMD leverages ambiguous NMR data for structure determination
MELD is an MD accelerator that can incorporate external information using Bayesian inference (Eq. 1) – an MD force field provides the prior, information the likelihood, and a posterior ensemble of structures:
| (1) |
where x is the structural ensemble, D is data, p(D|x) is the probability of D given x, p(x) is the probability of x from MD, and p(D) is a normalization term. Sources of information can be from protein heuristics, e.g. form a hydrophobic core, or from experimental data like NOESY peaks from NMR (Fig. 1 B). The information is used to apply restraints that, depending on the data, enforce atom-atom distance or backbone dihedrals. The amount of restraints enforced are based on the reliability of the information, and easily changed for different data. In addition, MELDxMD has the ability to take data from one or many sources, select subsets of data on-the-fly that are consistent with MD-generated structures, and populate low free energy ensembles that – given reliable data and accurate force field – predict native structures. MELDxMD simulations can start from a fully extended protein chain or from a template. During the CASP13 NMR data assisted predictions, the protocol used by our “Laufer” group (group code 431) seeded MELDxMD with templates from server predictions.
The best conformational searching must strike a balance between exploring (relatively undirected searching) and exploiting (downhill towards local minima in free energy). When NMR data has high accuracy, it can often identify a single conformational envelope (near-native ensemble), when interpreted correctly, that can be found by simple exploitation. But, as the number of identified peaks diminishes or are unassigned, the conformational possibilities explode combinatorially. Then, more effective methods are needed to explore and exploit.
MELD does this through a Hamiltonian-Temperature Replica Exchange Molecular Dynamics (H,T-REMD) protocol [6, 5], with an automated setup to read all the NMR information into simulations. In MELD’s H,T-REMD scheme, the high temperature replicas explore new conformations where no Hamiltonian modifications are applied, while the lower temperature replicas exploit conformations that satisfy subsets and interpretations of the NMR data. Inside each conformational envelope where the NMR peaks are satisfied, the restraint energy is null, so only the force field energy is used. This allows MELDxMD to identify low free energy structures, which are represented by high population conformational clusters.
2.2 |. NMR data processing
We were given four sources of information: (1) Residue Dipolar Coupling (RDC) data, (2) ambiguous NOE-based atom-atom contact lists, (3) sparse TALOS[7] dihedral-angle data and (4) evolutionary contacts. We used two of these four sources: the TALOS angles data and the NOESY peaks. We complemented the TALOS data with PSIPRED secondary structure predictions[8]. In addition, we calculated our own evolutionary information based on metagenomic libraries using jackhmmer[9] and GREMLIN[10]. The NOE-based contact lists came from ShiftX[11] chemical shifts simulated from CASP13 target X-ray crystal structures (except targets n1008 and N1008, which had real NMR data). The NOESY peaks were derived from the chemical shifts. Then ASDP (Automated Structure Determination of Proteins), software designed for automatic assignment of NOESY data [12, 13], was used to generate the ambiguous contact tables that contained possible atom-atom contacts for each peak. Some peaks had numerous atom-atom contacts (ambiguous data), while other peaks had only a single atom-atom contact (non-ambiguous). Noise was added by the organizers by removing some of possible assignments. As participants in the structure prediction competition, we did not know which peaks had missing interpretations and hence could be noisy (non-satisfiable peaks). Hence, from the user point of view the data was noisy and ambiguous. The NMR-derived ambiguous contact data contained distances in addition to atom-atom contacts. Prior to integrating the peaks list contact data in MELDxMD, it was filtered to remove peaks containing a local contact – defined as a contact between amino acids that are less than 4 residues apart in the sequence. Ambiguity from equivalent hydrogen atoms (e.g. methyl groups) was reduced by mapping onto the nearest heavy atom and increasing the distance by 1Å. Unambiguous hydrogen atoms (e.g. amide H) were left alone.
2.3 |. MELDxMD simulation protocol
We used the MELD OpenMM[14] plugin to carry out H,T-REMD with the ff14SBside force field[15] and GbNeck2 implicit solvent[16]. We seeded the replica exchange ladder with 15 predictions from the servers: Baker-Rosettaserver[17], Quark[18], Zhang-Server[19]. The server models were first minimized and then seeded to the replica ladder with each replica containing one of the models. Therefore, each server prediction was present between 6 and 7 times in the replica ladder. We used 100 replicas and ran for at least 1 μs or until the submission deadline of the given target.
Backbone dihedrals restraints came from secondary structure predictions (PSIPRED [8]) and from TALOS angle averages and standard deviations. Both were enforced at 60%. In cases where sequence homology queries against metagenomic libraries returned enough hits, we used co-evolutionary data produced by GREMLIN[10]. Each query sequence was iteratively searched against the May 2017 metaclust[20] database using jackhmmer[9], with bit score reporting and inclusion thresholds of 27, and maximum number of iterations set to 4. The output was filtered with HHfilter to include only 90% maximize pairwise sequence identity. GREMLIN residue-residue contacts that had at least 70% probability of being correct were used in MELD. Each residue-residue contact was treated as a MELD group of restraints between each pair of heavy atoms using a flat bottom harmonic restraint – 1 restraint had to be satisfied in each group. Those groups were inserted into a single collection, where 70% of the groups were enforced.
We incorporated NMR data by treating each NMR peak as one group of restraints. Inside each group only one restraint had to be satisfied at a given timestep during the MELD simulations – which restraint is satisfied depends on the conformation being sampled (see next). We arbitrarily decided to enforce NMR peaks at 90% accuracy for all targets since we were told to expect some noise. For target N1008, CASP provided real NMR data and mentioned to expect higher noise in the dataset – thus we enforced this data at 60%.
At no point is any information discarded. At each MD time step, MELD computes the energies of all possible restraints inside a group and chooses the one with lowest energy to represent that particular peak until the next time step. Then, at the same time step, all the peaks in the dataset are ranked according to their energy (that of the one restraint that MELD selected), and 90% of total peaks – those with lowest restraint energy – are enforced until the assessment occurs again at the next time step (4.5 femtoseconds). Each replica independently selects which restraints are active at a certain time step. The difference between the replicas is temperature and the strength of the restraints. Inside the replica exchange ladder, the strength of the restraints is controlled by k(α), where k is the force constant of the restraint and α is a scaling parameter that depends on the replica index. At the end points, k(α) is 0 at the highest replica (α =1) and k(α) is set to kmax (full force) at the lowest replica (α = 0). K(α) changes non-linearly in between. In this way no information is ever lost and detailed balance is always obeyed – vital for a physics based methodology. Restraints follow our previously developed MELD protocol[6, 5, 21].
Each simulation was started from server templates and ran for several days. To improve sampling and compensate for the low exchange probabilities in the REMD ladder, we scored the ensembles based on all the provided peaks that did not have ambiguous contacts. We selected structures that satisfied the most of these non-ambiguous contacts and seeded them into a new round of MELDxMD. This helped us to converge faster on the correct structures and allowed us to take advantage of experimental data, not only to guide the sampling, but also to score the sampled structures. After CASP we identified a bug in our code that made exchanges harder – post-CASP simulations were done resulting in structures of similar accuracy (see Fig. S1 and SI text for details).
2.4 |. Selecting 5 model structures to submit as CASP13 predictions
For each target, we clustered simulations at the lowest five temperatures and selected the five clusters with the highest population (lowest free energy). Clustering was done with a hierarchical agglomerative approach, using average-linkage to determine cluster distance and to stop clustering when minimum distance between clusters is greater than 2 Å. Clustering was done with CPPTRAJ[22] from AmberTools18. Our typical approach to select structures when making predictions is to use the representative structure from each of the top five clusters, but in this case, we also analyzed the ensembles for most non-ambiguous NOE-based contacts satisfied and lowest non-ambiguous contacts restraint energy. We then selected five structures based on those three criteria by visual inspection.
2.5 |. Measures of structure predictions and rankings.
The main measure of model quality used was GDT_TS (global distance test, total score) [23], which roughly represents the percentage of the structure within a certain cutoff to the native structure. The higher the score, the better the quality of the model. We used results reported by the CASP assessors for predictions submitted to CASP, and calculated the values for the post-CASP simulations in-house. TMscore[24] is another measure of structure quality, independent of protein size, and was also used to assess our predictions. The group rankings were done by z-score based on GDT_TS. In general, z-score is the difference between a sample and a mean divided by the standard deviation, which gives a relative measure of performance; positive z-scores are above average, z-scores of zero are average, and negative z-scores are below average. The group rankings were calculated according to the typical CASP procedure, by: 1) finding z-score using the best GDT_TS scores for each target 2) removing z-scores below 0.0 3) re-calculating z-scores 4) assigning z-scores of 0.0 or below to 0.0, and 5) summing z-scores across all targets to give a cumulative z-score for each group.
2.6 |. MELDxMD simulations without NMR data
We took part in the main CASP event (also as Laufer group 431), for which we made predictions based only on sequence knowledge. Instead of using experimental data, we used coarse physical insights (CPI) [5], evolutionary data when available [25], and seeded server predictions into MELDxMD. There were 8 assessment units for which we had both NMR data-assisted and regular CASP results.
3 |. RESULTS & DISCUSSION
3.1 |. MELDxMD successfully incorporates ambiguous and noisy NMR data
MELDxMD was developed to integrate many types of data from a variety of sources into physics-based simulations, which allowed us to include NMR data in the simulations without any custom MELD code modifications, only a script to pre-process the NMR data (see methods). The power of MELDxMD is its flexibility with external information and its ability to select subsets of noisy, sparse, and ambiguous data consistent with the structures sampled by the force field in MD to reduce conformational searching and populate ensembles of near-native structures. In CASP13, this approach had a higher cumulative z-score (Fig. 2A) than all other methods that used NMR data to build models, including the three baseline predictors that used ASDP. While in general a positive z-score value can be a result of many below average predictions and one very good prediction, our results show consistent enhancement over many targets (Fig. S2), asserting that our high z-score is, in fact, a consequence of regular improvements in prediction quality.
Figure 2. MELDxMD was the highest-ranked group in NMR data-assisted CASP13. It was also best-in-CASP for some individual targets.
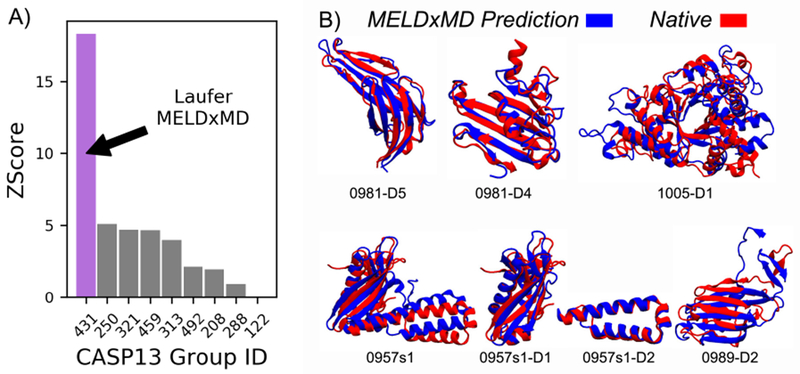
A) The MELDxMD method (group code 431) had the highest Z-score of all groups that competed in NMR data-assisted CASP13. B) MELDxMD predictions are aligned to reference structures, including 5 that were best in CASP, our best prediction (N0957s1-D2), and a large target that would be inaccessible by either MELDxMD or NMR alone (N1005-D1)
Below, we detail MELDxMD successes and failures from CASP13. We also discuss improving some explore and exploit strategies that we believe will improve the quality of NMR-informed physics-based simulations.
3.2 |. What went right?
MELDxMD made accurate predictions, including a 326-residue protein.
There were 14 targets released during CASP13 and we submitted 5 models for all 14 targets. Evaluators determined from those targets that there would be 17 evaluation units (domains). Of the 17 evaluation units, MELDxMD had the best NMR data-assisted prediction for 12/17 (see Fig. S2) (considering all 5 models, and 9/17 considering first model). Several of our predictions resulted in structures with GDT_TS values better than from any other predictor, including those from regular CASP13 (see Fig. 2B and 3). The targets ranged in size from 54 to 326 residues, and our GDT_TS from 26.22 to 86.11 (see Table S1 and Fig. S3). The two best MELDxMD predictions based on GDT_TS were domains D1 and D2 from target N0957s1, and both predictions for N0957s1-D1 and N0957s1 scored higher than all other CASP predictors (Fig. 2B, 3A, and S4). Comparing to methods that did not use NMR data, MELDxMD predicted structures with the highest GDT_TS scores for six targets: N0957s1-D1, N0981-D5, N0981-D4, N0957s1, N0989-D2, and N0981-D2 (Fig. 2B and 3).
Figure 3. MELDxMD was better with NMR data, better than server templates, and compared well against regular predictors.
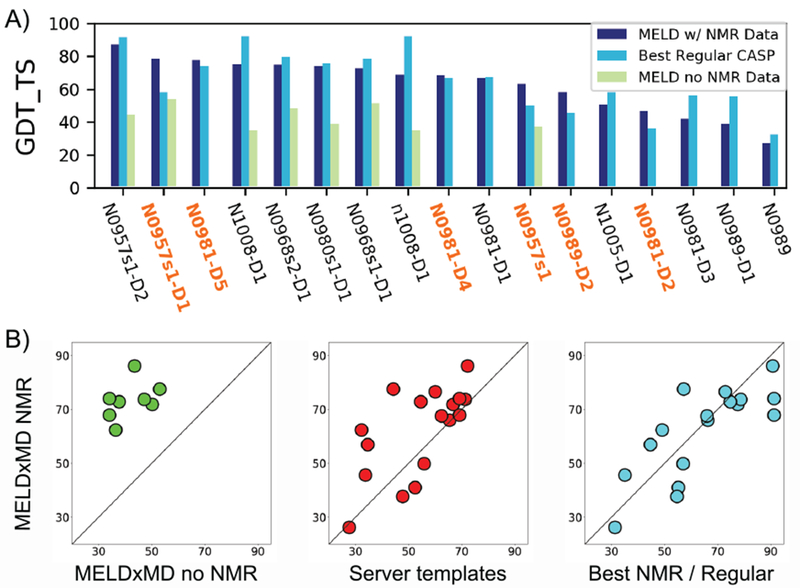
A) MELDxMD models that beat regular CASP predictions are highlighted in orange. The best MELDxMD model (dark blue) is compared to the best of any non-data-assisted prediction (light blue) and MELDxMD without NMR data (green) for all 17 evaluation units. Not all targets were run with MELDxMD without NMR data. B) The left panel shows 8 systems that were simulated by MELDxMD with and without NMR data. Using NMR data helped MELDxMD make better predictions. The middle panel shows MELDxMD with NMR data compared to the server predictions used to seed the MELDxMD replica ladder. MELDxMD generally improved on the server predictions. The right panel shows MELDxMD with NMR data compared to the best of other NMR predictors or regular category predictors (whichever was best for each target).
Target N1005-D1 was a challenge for MD + NMR methods, at 326 residues long. This size, ~40 kDa, is at the limits of NMR, and beyond the limits of MD. The largest protein previously folded by MD without data is just over 100 residues[26], and MELDxMD has only been able to fold proteins up to 240 residues, with the aid of experimental data[21]. There are only ~50 NMR structures currently in the PDB larger than this. However, MELDxMD was able to leverage the NMR data to explore near-native conformations and it generated 5 reasonable structures (TMScore 0.64–0.68). So, MELDxMD looks promising for getting NMR structures of these challengingly large proteins.
NMR data improved MELDxMD predictions.
Of the 17 NMR data-assisted evaluation units, MELDxMD was also run on 8 of them without incorporating NMR data – using only heuristics, evolutionary contacts, and server predicted starting templates (submitted as regular CASP targets). The NMR data improved GDT_TS by an average of 31.25 points (Fig. 3B, left panel), a larger improvement than any other group that submitted regular and data-assisted models.
MELDxMD with NMR predictions improved the best server predictions.
To reduce conformational searching compared to folding from a fully extended chain and exploit the ability of servers to quickly generate structures, the MELDxMD replica ladder was seeded with 15 server predictions. The server prediction templates were all 5 models from the Zhang-Server[19], Quark[18], and Baker-Rosettaserver groups[17], which were 3 of the top 4 ranked server groups in CASP13. MELDxMD improved the GDT_TS for 12 of 17 evaluation units an average of 13.70 points, i.e. for 12 targets the best MELDxMD model scored higher than the best of the 15 template models from the three servers (Fig. 3B, middle panel). Templates that scored higher than MELDxMD only did so by 6.04 GDT_TS, and were either high scoring predictions that servers simply outperformed (n1008-D1), or were hard targets that servers scored poorly (GDT_TS 27.64 to 55.83) but just slightly better than MELDxMD (N0981-D3, N0989, N0989-D1, and N1005-D1).
Without explicit instructions to do so, our structures agreed with RDC data.
In addition to Cα-based structural measures like GDT, the CASP assessors also identified how well predictions satisfied the given data. MELDxMD models had high RPF[12, 27] (recall, precision and F-measure for NOESY peak data) scores, and ranked near the top with the ASDP methods. A noteworthy result was that our models had good agreement with Residual Dipolar Couplings (RDC) data although we did not include RDC data to build our models. Finally, the sidechain rotamers that MELD explored were in good agreement with experiments, an attribute – and additional validation – of the force field.
MELDxMD worked as well on real data as on simulated data.
Targets N1008 (backbone only) and n1008 (backbone and sidechain) used real, rather than simulated, NMR data. These targets had more noisy data than the simulated ones – more unassigned and incorrectly assigned peaks – yet our best models had high GDT_TS scores of 74.03 and 67.87 (Fig. S5).
Improvements over CASP11-NMR.
CASP11 had previously challenged predictors to incorporate NMR data, and we had used MELD to successfully model some structures. One big difference between the two competitions is that, in CASP11, each NOESY peak had at least one correct atom-atom contact. In CASP13, we were told that after using ShiftX for ILVA methyl assignment, about 30% of the assignments would be deleted – hence some NOESY peaks might not be satisfied by the remaining assignments. Therefore, in CASP11 we tried to satisfy 100% of the NOESY peaks, while in CASP13 we enforce 90% – this increased the challenge in using CASP13 data: we considered CASP11 as ambiguous and CASP13 as ambiguous and noisy. Table S2 and Figure S6 show that the number of peaks and individual interpretations of the peaks was roughly similar in both CASPs, as were the overall quality of our resulting structures.
Hence, in CASP13, the sampling protocol and force field had to account for a larger success of the predictions. Furthermore, selection of structures based on agreement with NMR data was more challenging. In CASP11, every peak with only one interpretation had to be correct, so agreement with those peaks was a metric for selecting structures and success. In CASP13, due to the deleted assignments, some of those peaks did not necessarily have to be satisfied. Our combination of choosing structures based on cluster population and restraint energy penalty score proved useful to overcome this issue.
3.3 |. What went wrong?
Sometimes, replicas had poor exchanges.
The H,T-REMD scheme is meant to enhance sampling and expand the efficiency of MD exploration. Yet, we found that walkers (systems that move up and down the replica ladder by exchanging positions in the ladder with their neighbor) were not completing roundtrips between the highest and lowest temperatures as expected. Walkers favor exploration up the ladder, and exploitation down the ladder. Neighboring replicas (structures) exchange best when their energy overlap is high, and fail to exchange when large energy gaps occur. All of our prior experience has shown that 30 replicas is appropriate. However, we found that this was not the case with NMR data in this CASP. Figure S7 shows the exchange of a typical trajectory we sampled. As can be seen, exchanges are very infrequent and the lowest replica is only visited by four walkers. Hence, sampling and convergence is slow (in the limit of no exchanges this would be the equivalent of using independent MD trajectories).
To mitigate this problem during CASP and favor exchanges up and down the ladder, we resorted to two atypical interventions. The first was to increase the number of replicas from 30 to 100 in order to diminish the energy gap between the replicas and increase the probability of exchange and exploration. The second intervention consisted of periodically stopping the simulation, clustering structures, and identifying frames with low numbers of restraints violations (see below). Following this, new MELDxMD simulations were seeded using those frames capitalizing on the fact that starting with a set of similar structures reduces the energy gap and allows walkers to exchange more frequently.
NMR data led to energy landscapes that were difficult to explore.
An average protein had 200 NOESY peaks after pre-processing, and each of those peaks typically had six ambiguous interpretations, where only 90% of peaks were considered to be correct. These considerations result in 10218 possible subsets of restraints for an average protein, each of which restrains two atoms to distances between 5–7 Å. Therefore, the NMR restraints define a tight conformational envelope that creates a frustrated energy landscape that was difficult to navigate. To illustrate, Figure S8 shows ensemble fluctuations between simulations in which we used NMR data and those where we did not. The average root-mean-square fluctuation (RMSF) was smaller for simulations that used NMR data.
Sometimes, we used inaccurate co-evolutionary contacts.
The co-evolutionary contacts (ECs) we calculated were less precise than the ECs provided by CASP (See Fig. S9). These inaccurate contacts were enforced at 70% precision, as described in our protocol, which was too high given the true precision and guided the simulations to non-native structures. Post-CASP, we repeated simulations using the provided ECs and saw an overall improvement, with two proteins (N0968s2-D1 and N0957s1-D2) improving over 20 GDT points (see Fig. S10). Yet, the overall improvement from using the provided ECs was modest because, although more accurate, they were uninformative since they were mostly local contacts that occurred in secondary structure elements, areas where MELDxMD often does well on its own, and we expect that more long-range, informative ECs would further improve predictions.
Sometimes, our constraints favor structures that are too compact.
For some targets MELDxMD predicted the individual domains accurately, but failed in producing accurate models of the assembly. For example, our prediction for N0989-D2 was best in CASP, but our N0989 prediction was our worst (though this target challenged all regular and data-assisted predictors). Figure S11A shows N0989 to be made up of two domains interacting loosely with each other through a linker loop, whose flexibility was poorly modeled by MELDxMD, which tended to bring the domains together to form a more compact structure. We anticipate finding that some of the poor performance on entire proteins compared to domains could be explained by the tendency of the force field to make compact structures[15, 28, 29], thus, modeling each domain well but producing an overall structure that is more compact than true native. In addition, N0989 is a trimer but we only modeled the monomer, which could also explain the poor result. Previously, MELDxMD has shown the ability to rescue [26] force field errors given accurate external information, so more complete analysis will be done when all reference structures are published. We see two ways for improving on this issue in the future: (1) using RDC data to determine the relative orientations of the domains and (2) using NMR data for domain parsing (other groups in CASP identified that the NMR data showed contacts in one part of the protein and complete absence of peaks between domains). This domain parsing strategy could add enough informational restraints to prevent the force field from compacting domains together.
4 |. CONCLUSION
Determining protein structures from NMR data can sometimes be challenging because of ambiguities in assignments, or limitations or noise in the data. Some form of computer assistance is needed. Computational integrative modeling can be limited by the databases they require or by the assumptions required in using them, such as defining alignments. In principle, molecular dynamics using physical force fields can help, but brute force MD is usually computationally challenged, especially for large protein structures. CASP provides an excellent testbed for methods because it is blinded and time limited. Here, we find that MELDxMD can give a good solution to these problems. On the one hand, it substantially accelerates MD, and retains the benefits, such as an ability to compute ensembles and populations. On the other hand, it is able to handle troublesome ambiguities or noise or combinatoric choices in the data that it harnesses to speed up the MD. We have shown here that MELDxMD, seeded by servers, has significantly outperformed other methods in this CASP13 test, and that it has the capability of reaching for relatively large protein structures.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the support of the Blue Waters sustained-petascale computing project and NSF PRAC award ACI1514873. Blue Waters is supported by the National Science Foundation (Awards OCI-0725070 and ACI-1238993) and the State of Illinois. We also appreciate funding from NIH Grant GM125813, the Laufer Center and University of Florida start-up resources. We thank IRACDA NY-CAPS grant K12-GM-102778 for support to JCR. We also thank the CASP13 organizers, groups that contributed targets, and everyone involved in NMR assessment, led by Guy Montelione. We thank Thanh H. Nguyen, Anika Liu and Mark van Raaij for coordinates to target N0989 and Antonio Pichel and Mark van Raaij for coordinates of the domains in N0981.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- [1].Schulte CF, Tolmie DE, Maziuk D, Nakatani E, Akutsu H, Yao H, et al. BioMagResBank. Nucleic Acids Res 2007;36(suppl1):D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gilliland G, Berman HM, Weissig H, Shindyalov IN, Westbrook J, Bourne PE, et al. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Clore GM, and AG. Determination of three-dimensional structures of proteins and nucleic acids in solution by nuclear magnetic resonance spectroscop. Crit Rev Biochem Mol Biol 1989;24(5):479–564. [DOI] [PubMed] [Google Scholar]
- [4].Frueh DP, Goodrich AC, Mishra SH, Nichols SR. NMR methods for structural studies of large monomeric and multimeric proteins. Curr Opin Struc Biol 2013;23(5):734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Perez A, MacCallum JL, Dill KA. Accelerating molecular simulations of proteins using Bayesian inference on weak information. Proc Natl Acad Sci USA 2015;112(38):11846–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].MacCallum JL, Perez A, Dill KA. Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc Natl Acad Sci USA 2015;112(22):6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 2013;56(3):227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999;292(2):195–202. [DOI] [PubMed] [Google Scholar]
- [9].Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res 2018;46(W1):W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci USA 2013;110(39):15674–15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Neal S, Nip AM, Zhang H, Wishart DS. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J Biomol NMR 2003;26(3):215–240. [DOI] [PubMed] [Google Scholar]
- [12].Huang YJ, Powers R, Montelione GT. Protein NMR Recall, Precision, and F-measure Scores (RPF Scores): Structure Quality Assessment Measures Based on Information Retrieval Statistics. J Am Chem Soc 2005;127(6):1665–1674. [DOI] [PubMed] [Google Scholar]
- [13].Huang YJ, Tejero R, Powers R, Montelione GT. A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins 2006;62(3):587–603. [DOI] [PubMed] [Google Scholar]
- [14].Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput Biol 2017;13(7):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 2015;11(8):3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nguyen H, Roe DR, Simmerling C. Improved Generalized Born Solvent Model Parameters for Protein Simulations. J Chem Theory Comput 2013;9(4):2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Song Y, DiMaio F, Wang RYR, Kim D, Miles C, Brunette TJ, et al. High-Resolution Comparative Modeling with RosettaCM. Structure 2013;21(10):1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xu D, Zhang Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins 2013;81(2):229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 2010;5(4):725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Steinegger M, Söding J. Clustering huge protein sequence sets in linear time. Nat Commun 2018;9(1):2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Perez A, Morrone JA, Brini E, MacCallum JL, Dill K. Blind protein structure prediction using accelerated free-energy simulations. Sci Adv 2016;2(11):e1601274–e1601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 2013;9(7):3084–3095. [DOI] [PubMed] [Google Scholar]
- [23].Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res 2003;31(13):3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins 2004;57(4):702–710. [DOI] [PubMed] [Google Scholar]
- [25].Ovchinnikov S, Kamisetty H, Baker D, Roux B. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. eLife 2014;3:e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Robertson JC, Perez A, Dill K. MELDxMD Folds Nonthreadables, Giving Native Structures and Populations. J Chem Theory Comput 2018;14(12):6734–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang YJ, Rosato A, Singh G, Montelione GT. RPF: a quality assessment tool for protein NMR structures. Nucleic Acids Res 2012;40(W1):W542–W546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Roe DR, Okur A, Wickstrom L, Hornak V, Simmerling C. Secondary Structure Bias in Generalized Born Solvent Models: Comparison of Conformational Ensembles and Free Energy of Solvent Polarization from Explicit and Implicit Solvation. J Phys Chem B 2007;111(7):1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of Multiple AMBER Force Fields and Development of Improved Protein Backbone Parameters. Proteins 2006;65(3):712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Capraro DT, Roy M, Onuchic JN, Jennings PA. Backtracking on the folding landscape of the beta-trefoil protein interleukin-1beta? Proc Natl Acad Sci USA 2008;105(39):14844–14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Case D, Ben-Shalom I, Brozell S, Cerutti D, Cheatham T, Cruzeiro V, et al. AMBER 18. University of California, San Francisco: 2018;. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.