

Abstract
Aim
To evaluate the safety of efpeglenatide, a long‐acting glucagon‐like peptide‐1 receptor agonist (GLP‐1RA), and its effects on body weight management in adults without diabetes.
Materials and methods
In this phase II, randomized, placebo‐controlled, double‐blind trial, participants with a body mass index (BMI) ≥30 kg/m2 or ≥27 kg/m2 with comorbidity were randomized 1:1:1:1:1 to efpeglenatide (4 mg once weekly, 6 mg once weekly, 6 mg once every 2 wk, or 8 mg once every 2 wk; n = 237) or placebo (n = 60) in combination with a hypocaloric diet. The primary endpoint was body weight change from baseline after 20 wk of treatment, assessed using a mixed‐effect model with repeated measures with an unstructured covariance matrix over all post‐screening visits; treatment comparisons were based on least squares mean estimates.
Results
Over 20 wk, all doses of efpeglenatide significantly reduced body weight from baseline versus placebo (P < 0.0001), with placebo‐adjusted reductions ranging between −6.3 kg (6 mg once every 2 wk) and −7.2 kg (6 mg once weekly). Greater proportions of efpeglenatide‐treated participants had body weight loss of ≥5% or ≥10% versus placebo (P < 0.01, all comparisons). Efpeglenatide led to significant improvements in glycaemic variables (fasting plasma glucose and glycated haemoglobin) and lipid profiles (cholesterol, triglycerides) versus placebo. Rates of study discontinuations as a result of adverse events ranged from 5% to 19% with efpeglenatide. Gastrointestinal effects were the most common treatment‐emergent adverse events.
Conclusions
Efpeglenatide once weekly and once every 2 wk led to significant body weight reduction and improved glycaemic and lipid variables versus placebo. It was also well tolerated for weight management in adults without diabetes.
Keywords: clinical trial, drug development, GLP‐1, glycaemic control, obesity therapy, weight control
1. INTRODUCTION
Obesity is a major risk factor for a variety of metabolic diseases, including cardiovascular disease (CVD), diabetes, musculoskeletal disorders and some cancers.1, 2, 3, 4 Weight loss can lead to a reduction in obesity‐related complications, including the risk of developing diabetes.5, 6, 7 In overweight (body mass index [BMI] ≥25 kg/m2 and <30 kg/m2) or obese people (BMI ≥30 kg/m2) who have progressed to type 2 diabetes (T2D), weight loss is associated with reductions in glycated haemoglobin (HbA1c), improvements in CVD risk factors and reductions in mortality.8, 9, 10
Weight reduction through lifestyle interventions alone can be challenging.11 Options for effective pharmacological treatments are limited, and those available require daily dosing. There is an unmet need for effective weight loss treatment with less frequent dosing, which could help facilitate better adherence and sustainability of weight loss.12
Glucagon‐like peptide‐1 receptor agonists (GLP‐1RAs) are well‐established therapeutic options for T2D.13, 14 Another benefit of GLP‐1RA treatment is weight loss, which has been demonstrated in overweight or obese people with or without T2D.15 GLP‐1RAs reduce body weight by promoting satiety and reducing food intake, through peripheral and potentially through direct actions on the central nervous system (CNS).16, 17, 18 The anorectic effects of GLP‐1RAs appear to be independent of their effects on gastrointestinal (GI) motility and tolerability.19
Efpeglenatide (formerly HM11260C) is currently being developed to improve glycaemic control in people with T2D as a once‐weekly subcutaneous (s.c.) administration. Efpeglenatide is a long‐acting GLP‐1RA that is a single amino acid‐modified exendin conjugated to a fragment crystallizable region of human immunoglobulin 4 via a 3.4‐kDa mini‐polyethylene glycol linker.20, 21 The fragment crystallizable conjugation in efpeglenatide allows a long duration of action and a pharmacokinetic/pharmacodynamic profile that could support a flexible dosing frequency (once weekly, once every 2 wk and once monthly). Indeed, in a single ascending‐dose study in people with T2D, efpeglenatide demonstrated a half‐life of 5.6 to 7.5 d.22 In a previous phase II study (NCT01452451), once‐weekly or once‐monthly administration of efpeglenatide resulted in significant weight loss and glycaemic improvements compared with placebo in people with T2D on stable metformin monotherapy after 8 and 11 wk (three monthly doses) of treatment, respectively.23 Efpeglenatide once weekly (over 12 wk) and once monthly (over 16 wk) has been demonstrated to significantly improve glycaemic control and decrease body weight versus placebo in people with T2D in the phase II EXCEED 203 (NCT02057172)24 and LIBERATE 204 (NCT02081118) trials,25 respectively.
The objective of the present BALANCE phase II trial (NCT02075281) was to assess the safety and tolerability of s.c. efpeglenatide once weekly or once every 2 wk, in combination with a hypocaloric diet, and its potential to reduce body weight in obese or overweight people (with comorbidity), and without diabetes, over 20 wk of treatment.
2. METHODS
2.1. Trial design and participants
BALANCE was a multinational, double‐blind, randomized, placebo‐controlled, parallel‐group study that was initiated in April 2014 and completed in February 2015. The study comprised a screening period, a 2‐wk dose titration period (only the once‐every‐2‐wk doses were titrated) and an 18‐wk constant‐dose treatment period, with a 6‐wk follow‐up period (Figure S1). The study was designed and conducted in accordance with the International Conference on Harmonization Harmonized Tripartite Guideline for Good Clinical Practice and the Declaration of Helsinki. The protocol was approved by the institutional review boards or ethics committee at each study site, and each participant provided written informed consent.
Eligible participants were aged ≥18 y and <65 y at screening and in stable health, with a fasting plasma glucose (FPG) of <7 mmol/L (<126 mg/dL) and a BMI of ≥30 kg/m2. People with BMI ≥27 kg/m2 and <30 kg/m2 were also eligible if they had treated or untreated comorbidities, such as hypertriglyceridaemia, dyslipidaemia, hypertension, glucose intolerance (based on medical history) or sleep apnoea; these participants represent a population with obesity‐related risk factors, thereby allowing the evaluation of the potential for efpeglenatide to reduce the incidence of these conditions (in addition to reducing body weight). Participants were excluded from the study if they had a BMI of >42 kg/m2, drug‐induced obesity (also referred to as iatrogenic obesity), known diabetes or an HbA1c >48 mmol/mol (>6.5%; see Supporting Information for full details).
2.2. Interventions
On day 1, all eligible participants were randomized 1:1:1:1:1 via an interactive voice/web response system to efpeglenatide 4 mg once weekly, 6 mg once weekly, 6 mg once every 2 wk, or 8 mg once every 2 wk, or placebo. Participants, investigator staff, persons performing the assessments and data analysts were blinded to the treatment from the time of randomization until database lock. Participants received training on the injection method and self‐administered the study drug in the morning s.c. in the abdomen using a pre‐filled syringe. The first injection was on day 1 for participants in all treatment arms, with no period of adaptation to the injection device. All participants receiving efpeglenatide once every 2 wk started on 4 mg (day 1) and were titrated to their assigned dose in week 2 of the 2‐wk titration period; participants in the once‐weekly arms received their assigned dose without any titration during this period (Figure S1B). Participants were assessed at regular intervals over the treatment period, with three follow‐up evaluations post‐treatment (Figure S1B). Throughout the study, participants were instructed by study staff to adhere to a hypocaloric diet (~500 kcal/day deficit) and encouraged to increase physical activity; however, caloric intake and physical activity were not controlled.
2.3. Study endpoints
The primary efficacy endpoint was change from baseline (day 1 or last value observed prior to randomization if this was not available) in body weight to week 21 (ie, after 20 wk of treatment). Additional endpoints derived from body weight were per cent change in body weight from baseline, change in BMI from baseline and percentage of participants with ≥5% or ≥10% of body weight reduction from baseline to week 21. Change from baseline in waist circumference was also measured. A description of body weight and waist circumference measurements is provided in the Supporting Information. Glucose metabolism variables were measured at the central laboratory and included changes from baseline to week 21 in FPG and HbA1c levels. Lipid assessments included change from baseline to week 21 in total cholesterol, LDL cholesterol, VLDL cholesterol, HDL cholesterol and triglycerides.
Safety assessments included treatment‐emergent adverse events (TEAEs), vital signs, 12‐lead ECG variables, clinical laboratory assessments, injection‐site reactions and immunogenicity. Blood samples were collected to assess the development of anti‐efpeglenatide antibodies, which were determined by enzyme‐linked immunosorbent assay. Neutralizing antibodies were determined using a cell‐based assay. The incidence of symptomatic hypoglycaemia was reported by participants in study diaries (with an alert value of FPG ≤3.9 mmol/L [≤70 mg/dL]), rather than by a specific laboratory measurement. Severe symptomatic hypoglycaemia was indicated by a participant requiring assistance from another person to actively administer carbohydrates or glucagon, or take other corrective actions. Participants were instructed to record adverse events (AEs), in particular, symptomatic hypoglycaemia, nausea and vomiting, and injection‐site reactions in a daily diary provided to them.
2.4. Statistical analysis
Sample size was calculated assuming that the SD of weight change at week 21 would be 4.0 kg. A total of 270 participants (54 per group) would provide at least 85% power to detect a clinically relevant 3.0 kg difference (P = 0.05, two‐sided) in mean body weight between participants receiving efpeglenatide and placebo. Assumptions included a dropout rate of 40%.
Analysis of the primary efficacy endpoint was performed using a mixed‐effect model with repeated measures (MMRM) with an unstructured covariance matrix over all visits after screening, with visit, treatment group and their interaction as factors and baseline body weight as a covariate. Least squares mean (LSM) estimates (with two‐sided 95.1% confidence intervals [CIs]) were obtained for each treatment for each visit and for the difference between efpeglenatide and placebo. Statistical comparisons between efpeglenatide treatment groups were not performed. Further details regarding treatment of missing data and sensitivity analyses are provided in the Supporting Information.
For percentage of participants who lost ≥5% or ≥10% of body weight from baseline to week 21, Fisher's exact test was used to test whether the null hypothesis of equal proportions held or did not hold between treatment group and placebo. All other efficacy endpoints were summarized descriptively by treatment group and visit, and analysis was performed using the same method as for the primary efficacy analysis.
All efficacy endpoints were analysed using the full analysis set, which consisted of all participants who received the study drug and had at least one assessment recorded after dosing; participants were grouped according to their randomized treatment. For the primary efficacy analysis, supporting analysis was also performed on the per‐protocol population (all participants in the full analysis set who did not experience any major protocol violations that could have potentially impacted the primary efficacy endpoint). All safety endpoints were analysed using the safety set, which consisted of all participants who received any study drug, and study participants were grouped according to the treatment they actually received. An interim analysis was performed for administrative purposes, to aid in the planning of future studies in this developmental programme. The CI for the final endpoint was adjusted to 95.1% (α level of 0.049) to account for α spending for the interim analysis (α level 0.0031) based on the O'Brien–Fleming function. A list of important changes to methodology after trial initiation is provided in the Supporting Information.
3. RESULTS
3.1. Participant disposition and baseline characteristics
Overall, 297 participants were enrolled and randomized in the study, with 237 participants assigned to one of four efpeglenatide groups and 60 participants assigned to placebo (Figure S2). Study participation by country and by site is shown in Table S1. A total of 295 participants received the study drug (efpeglenatide overall, n = 235; placebo, n = 60) and were included in the safety set (note: two participants withdrew from the study prior to receiving the study drug and were not included in the safety set). Of the enrolled population, a total of 216 participants completed the 20‐wk treatment period (efpeglenatide overall, n = 168; placebo, n = 48) and 207 completed the follow‐up period (efpeglenatide overall, n = 160; placebo, n = 47 [Figure S2]). Discontinuation rates (for the safety set) over the 20‐wk treatment period were 25% to 31% with efpeglenatide and 20% with placebo. For the overall study period (20‐wk treatment and 6‐wk follow‐up), discontinuation rates (for the safety set) were 29% to 34% with efpeglenatide and 22% with placebo. Across all the efpeglenatide groups, AEs were the most common reasons for study discontinuation (efpeglenatide, 5%–19%; placebo, 5% [safety set]).
Demographic and baseline characteristics were similar across the treatment groups (Table 1). Overall, the majority of participants (95%) were obese (BMI ≥30 kg/m2) and 5% of participants were overweight (BMI 27 to <30 kg/m2 [safety set]).
Table 1.
Demographic and baseline characteristics (safety set)
| Placebo n = 60 | Efpeglenatide 4 mg once weekly n = 59 | Efpeglenatide 6 mg once weekly n = 59 | Efpeglenatide 6 mg once every 2 wk n = 59 | Efpeglenatide 8 mg once every 2 wk n = 58 | All N = 295 | |
|---|---|---|---|---|---|---|
| Age, years | 43.7 (11.8) | 42.9 (12.1) | 43.0 (13.0) | 43.3 (12.5) | 43.9 (9.2) | 43.4 (11.7) |
| Female, n (%) | 44 (73.3) | 41 (69.5) | 46 (78.0) | 43 (72.9) | 51 (87.9) | 225 (76.3) |
| Race, n (%) | ||||||
| White | 39 (65.0) | 41 (69.5) | 37 (62.7) | 45 (76.3) | 39 (67.2) | 201 (68.1) |
| Black or African‐American | 16 (26.7) | 12 (20.3) | 11 (18.6) | 8 (13.6) | 13 (22.4) | 60 (20.3) |
| Asian | 3 (5.0) | 4 (6.8) | 8 (13.6) | 5 (8.5) | 5 (8.6) | 25 (8.5) |
| Other | 2 (3.3) | 2 (3.4) | 3 (5.1) | 1 (1.7) | 1 (1.7) | 9 (3.1) |
| Ethnicity, n (%) | ||||||
| Hispanic or Latino | 7 (11.7) | 4 (6.8) | 10 (16.9) | 7 (11.9) | 6 (10.3) | 34 (11.5) |
| Not Hispanic or Latino | 53 (88.3) | 55 (93.2) | 49 (83.1) | 52 (88.1) | 52 (89.7) | 261 (88.5) |
| Weight, kg | 97.5 (12.1) | 100.8 (19.3) | 101.7 (19.4) | 99.5 (18.4) | 95.6 (13.3) | 99.0 (16.8) |
| BMI, kg/m2 | 34.9 (3.2) | 35.2 (4.5) | 36.3 (4.4) | 35.6 (4.8) | 35.2 (3.9) | 35.4 (4.2) |
| BMI 27 to <30 kg/m2, n (%) | 2 (3.3) | 5 (8.5) | 3 (5.1) | 2 (3.4) | 3 (5.2) | 15 (5.1) |
| BMI ≥30 kg/m2, n (%) | 58 (96.7) | 54 (91.5) | 56 (94.9) | 57 (96.6) | 55 (94.8) | 280 (94.9) |
| Waist circumference, cm | ||||||
| Women | 107.0 (10.1) | 107.1 (10.2) | 110.5 (13.4) | 106.8 (10.8) | 108.7 (11.1) | 108.1 (11.2) |
| n | 44 | 41 | 46 | 43 | 51 | 225 |
| Men | 115.8 (9.7) | 115.8 (14.3) | 116.9 (11.9) | 117.2 (11.3) | 110.3 (10.3) | 115.8 (11.7) |
| n | 16 | 18 | 13 | 16 | 7 | 70 |
| HbA1c, mmol/mol | 36.4 (4.4) | 36.1 (4.0) | 35.3 (4.7) | 36.9 (3.5) | 35.9 (4.2) | 36.1 (4.2) |
| HbA1c, % | 5.5 (0.4) | 5.5 (0.4) | 5.4 (0.4) | 5.5 (0.3) | 5.4 (0.4) | 5.5 (0.4) |
| FPG, mmol/L | 5.3 (0.6) | 5.2 (0.7) | 5.3 (0.6) | 5.4 (0.7) | 5.3 (0.5) | 5.3 (0.6) |
Data are mean (SD) unless otherwise stated.
Abbreviations: BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin.
3.2. Body weight and related variables
All doses of efpeglenatide led to significant reductions in body weight from baseline to week 21 (ie, after 20 wk of treatment) compared with placebo (Figure 1A,B and Table 2). Differences in LSM change from baseline to week 21 versus placebo with efpeglenatide 4 mg once weekly, 6 mg once weekly, 6 mg once every 2 wk and 8 mg once every 2 wk were −6.5 kg, −7.2 kg, −6.3 kg and −6.9 kg, respectively (P < 0.0001 for all comparisons; Figure 1B and Table 2); this difference was greatest in the 6‐mg‐once‐weekly group. Similar results were observed on analysis using the per‐protocol population, and on sensitivity analysis using last observation carried forward and pattern mixture model (Table S2).
Figure 1.
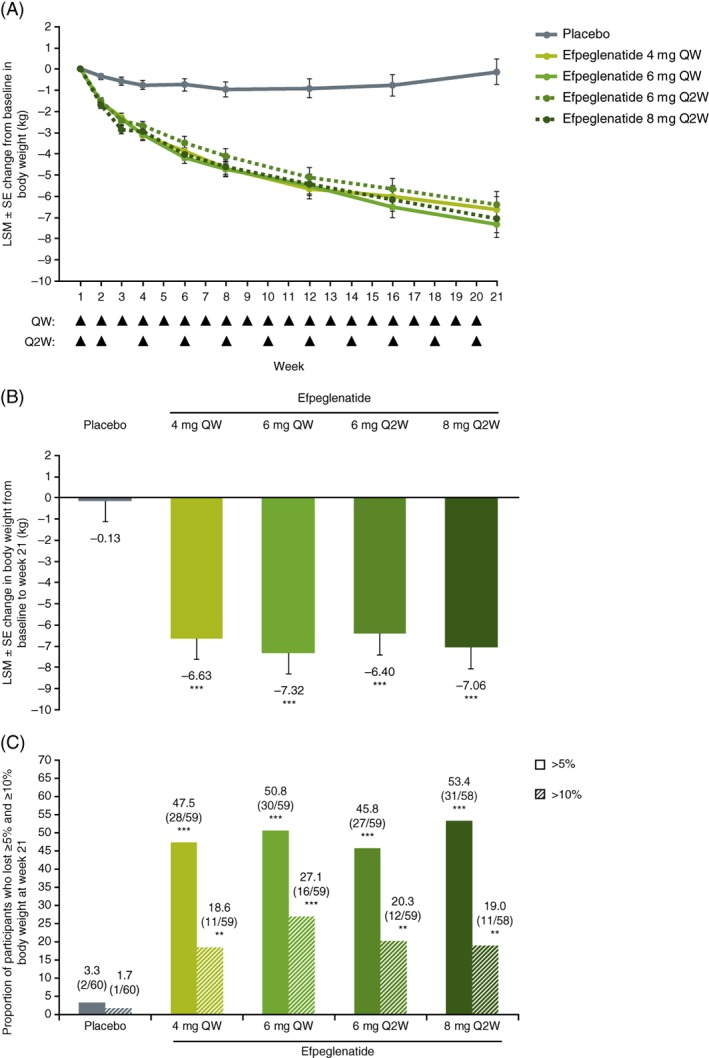
A, Least squares mean (LSM) change in body weight from baseline over the 20‐wk treatment period, B, Overall reduction at week 21 and C, Percentage of participants who lost at least 5% and 10% body weight at week 21 (full analysis set). Week 1 indicates baseline. Arrows indicate weeks of injection, which occurred at the beginning of the week. **P < 0.01 vs. placebo; ***P < 0.0001 vs. placebo. Abbreviations: Q2W, once every 2 wk; QW, once weekly
Table 2.
Treatment effects at week 21 (full analysis set)
| Efficacy endpoint | Placebo n = 60 | Efpeglenatide 4 mg once weekly n = 59 | Efpeglenatide 6 mg once weekly n = 59 | Efpeglenatide 6 mg once every 2 wk n = 59 | Efpeglenatide 8 mg once every 2 wk n = 58 |
|---|---|---|---|---|---|
| Body weight, kg | |||||
| Baseline | 97.5 (12.1) | 100.8 (19.3) | 101.7 (19.4) | 99.5 (18.4) | 95.6 (13.3) |
| Week 21 | 97.9 (12.2) | 93.6 (15.7) | 91.3 (16.3) | 93.2 (19.9) | 86.1 (12.6) |
| LSM (SE) change from baselinea | −0.1 (0.6) | −6.6 (0.6) | −7.3 (0.6) | −6.4 (0.6) | −7.1 (0.6) |
| n | 48 | 44 | 42 | 41 | 40 |
| LSM difference vs. placeboa (95.1% CI) | ‐ | −6.5 (−8.2, −4.8) | −7.2 (−8.9, −5.5) | −6.3 (−8.0, −4.6) | −6.9 (−8.7, −5.2) |
| P | ‐ | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
| Body weight, per cent change | |||||
| LSM (SE) per cent change from baselinea | 0.1 (0.6) | −6.7 (0.6) | −7.3 (0.6) | −6.7 (0.6) | −7.4 (0.6) |
| LSM difference vs. placeboa (95.1% CI) | ‐ | −6.8 (−8.4, −5.1) | −7.4 (−9.1, −5.7) | −6.7 (−8.4, −5.1) | −7.5 (−9.2, −5.8) |
| P | ‐ | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
| BMI, kg/m2 | |||||
| Baseline | 34.9 (3.2) | 35.2 (4.5) | 36.3 (4.4) | 35.6 (4.8) | 35.2 (3.9) |
| Week 21 | 34.8 (3.5) | 32.7 (3.8) | 33.0 (3.6) | 33.4 (5.5) | 32.0 (3.8) |
| LSM (SE) change from baselinea | 0.0 (0.2) | −2.4 (0.2) | −2.6 (0.2) | −2.3 (0.2) | −2.6 (0.2) |
| n | 48 | 44 | 42 | 41 | 40 |
| LSM difference vs. placeboa (95.1% CI) | ‐ | −2.4 (−3.0, −1.8) | −2.6 (−3.2, −2.0) | −2.3 (−2.9, −1.7) | −2.6 (−3.2, −2.0) |
| P | ‐ | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
| Waist circumference, cm | |||||
| Baseline | 109.3 (10.7) | 109.8 (12.2) | 111.9 (13.3) | 109.6 (11.8) | 108.9 (10.9) |
| Week 21 | 108.7 (10.3) | 104.1 (12.0) | 103.9 (11.4) | 103.0 (13.2) | 99.3 (10.0) |
| LSM (SE) change from baselinea | −0.9 (1.0) | −5.2 (1.0) | −6.7 (1.0) | −6.2 (1.0) | −8.3 (1.0) |
| n | 47 | 44 | 42 | 41 | 40 |
| LSM difference vs. placeboa (95.1% CI) | ‐ | −4.4 (−7.1, −1.6) | −5.9 (−8.6, −3.1) | −5.3 (−8.1, −2.5) | −7.5 (−10.3, −4.7) |
| P | ‐ | 0.0018 | < 0.0001 | < 0.0001 | < 0.0001 |
Data are mean (SD), unless otherwise stated.
Week 21 values correspond to 20 wk of treatment.
LSM difference vs. placebo is calculated as the study dose group – placebo in LSM change from baseline to week 21.
Abbreviations: CI, confidence interval; LSM, least squares mean; MMRM, mixed‐effect model with repeated measures.
From MMRM, using an unstructured covariance matrix; change from baseline as the outcome variable; baseline as a covariate; treatment group, visit and their interaction as factors.
Change from baseline in body weight to week 21 was also significant, with all efpeglenatide doses compared with placebo, when measured as per cent changes from baseline (differences in LSM changes from baseline vs. placebo were −6.8%, −7.4%, −6.7% and −7.5% with 4 mg once weekly, 6 mg once weekly, 6 mg once every 2 wk and 8 mg once every 2 wk, respectively; P < 0.0001 for all). At each visit through week 21, LSM decreases from baseline in body weight in each efpeglenatide group were greater compared with placebo, and were further decreased at each successive time point, whereas in the placebo group, the LSM change from baseline varied little (Figure 1A).
Significantly higher proportions of participants lost ≥5% or ≥10% of baseline body weight at week 21 across the efpeglenatide groups (45.8%–53.4% and 18.6%–27.1%, respectively) compared with placebo (3.3% and 1.7%, respectively, P < 0.0001 and P < 0.01 for all comparisons vs. placebo, respectively [Figure 1C]). Efpeglenatide 4 mg once weekly, 6 mg once weekly, 6 mg once every 2 wk and 8 mg once every 2 wk also led to significantly greater reductions in waist circumference from baseline versus placebo, with LSM differences from baseline versus placebo of −4.4 cm, −5.9 cm, −5.3 cm and −7.5 cm, respectively (P < 0.01 for all; Table 2). Reductions in BMI were also significant in all efpeglenatide groups compared with placebo (Table 2).
3.3. Glycaemic endpoints
At each visit through week 21, all efpeglenatide groups were associated with decreases from baseline in FPG, while increases from baseline were observed with placebo (Figure S3A; P < 0.01 for all comparisons of LSM changes vs. placebo at week 21 [Table S3]). Decreases in HbA1c were also observed in all efpeglenatide groups at every visit throughout the treatment period, but not with placebo (Figure S3B; P < 0.0001 for all comparisons of LSM changes vs. placebo at week 21 [Table S3]).
3.4. Lipid endpoints
At week 21, all doses of efpeglenatide were associated with significant reductions in total cholesterol from baseline compared with placebo (Table S4 and Figure S4A; P < 0.01 for all comparisons vs. placebo). All doses of efpeglenatide also led to significant reductions in LDL cholesterol and VLDL cholesterol levels versus placebo (P < 0.05 for all comparisons; Table S4 and Figure S4B,C). Efpeglenatide 6 mg once weekly, 6 mg once every 2 wk and 8 mg once every 2 wk led to significant reductions in HDL cholesterol at week 21 versus placebo (P < 0.05 for all); however, the difference in LSM change from baseline versus placebo with 4 mg once every 2 wk was not statistically significant (−0.06 mmol/L; P = 0.0520 [Table S4 and Figure S4D]). All doses of efpeglenatide led to significant reductions in triglycerides compared with placebo at week 21 (Table S4 and Figure S4E).
3.5. Safety assessments
3.5.1. Adverse events
Overall, 86.4% of participants (255/295) reported at least one TEAE; these rates were higher with efpeglenatide overall (88.1% [207/235]) compared with placebo (80.0% [48/60]) and similar across efpeglenatide dose groups (Table 3). Most TEAEs were considered to be mild or moderate.
Table 3.
Selected safety assessments (safety set)
| Placebo n = 60 | Efpeglenatide 4 mg once weekly n = 59 | Efpeglenatide 6 mg once weekly n = 59 | Efpeglenatide 6 mg once every 2 wk n = 59 | Efpeglenatide 8 mg once every 2 wk n = 58 | |
|---|---|---|---|---|---|
| Any TEAEa | 48 (80.0) | 51 (86.4) | 54 (91.5) | 51 (86.4) | 51 (87.9) |
| GI disorders | 28 (46.7) | 43 (72.9) | 49 (83.1) | 38 (64.4) | 44 (75.9) |
| Nausea | 11 (18.3) | 32 (54.2) | 35 (59.3) | 28 (47.5) | 36 (62.1) |
| Vomiting | 4 (6.7) | 13 (22.0) | 13 (22.0) | 10 (16.9) | 19 (32.8) |
| Diarrhoea | 12 (20.0) | 14 (23.7) | 12 (20.3) | 15 (25.4) | 16 (27.6) |
| Dyspepsia | 2 (3.3) | 12 (20.3) | 16 (27.1) | 9 (15.3) | 15 (25.9) |
| Constipation | 5 (8.3) | 10 (16.9) | 12 (20.3) | 9 (15.3) | 12 (20.7) |
| Severe GI‐related TEAEs | 0 | 2 (3.4) | 2 (3.4) | 1 (1.7) | 3 (5.2) |
| Vomiting | 0 | 2 (3.4) | 1 (1.7) | 0 | 2 (3.4) |
| Upper abdominal pain | 0 | 1 (1.7) | 1 (1.7) | 1 (1.7) | 1 (1.7) |
| Nausea | 0 | 1 (1.7) | 1 (1.7) | 0 | 2 (3.4) |
| Diarrhoea | 0 | 0 | 0 | 0 | 1 (1.7) |
| Dyspepsia | 0 | 0 | 0 | 0 | 1 (1.7) |
| Symptomatic hypoglycaemia | 0 | 0 | 1 (1.7) | 0 | 1 (1.7) |
| Injection‐site reaction | 13 (21.7) | 11 (18.6) | 7 (11.9) | 5 (8.5) | 8 (13.8) |
| Serious TEAEs | 0 (0) | 1 (1.7) | 3 (5.1) | 0 (0) | 2 (3.4) |
| Any TEAE leading to discontinuation | 4 (6.7) | 3 (5.1) | 11 (18.6) | 7 (11.9) | 10 (17.2) |
Data are n (%).
An adverse event was considered treatment‐emergent if it had a start date or increased in severity on or after the first dose of study drug.
Abbreviations: GI, gastrointestinal; TEAE, treatment‐emergent adverse event.
More than one event could be reported in a single participant.
The most frequently reported TEAEs were GI AEs, which occurred in 64.4% to 83.1% of participants in the efpeglenatide groups, and in 46.7% of participants in the placebo group (Table 3). Nausea was the most common GI AE in participants treated with efpeglenatide. Most cases of nausea and vomiting occurred within the first few injections and subsided over time (Figure 2). Among the participants receiving efpeglenatide who reported events of nausea, 53.4% experienced only one episode and 22.1% experienced two episodes; 17.6% experienced four or more episodes (Table S5). In the efpeglenatide groups overall, there were 15 severe GI AEs that were reported in a total of eight participants (Table 3). In participants receiving efpeglenatide, most TEAEs leading to discontinuation were GI disorders; none of these GI AEs were classified as serious AEs (Table S6).
Figure 2.
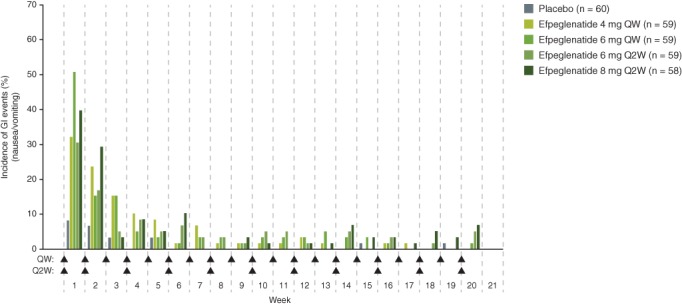
Incidence of nausea or vomiting events by study week (safety set). Arrows indicate weeks of injection, which occurred at the beginning of the week. Participants are included only once per study week, even if they experienced multiple events in that week. Abbreviations: GI, gastrointestinal; Q2W, once every 2 wk; QW, once weekly
Overall, 10 serious TEAEs were reported in six participants receiving efpeglenatide; of these, eight led to treatment discontinuation in four participants and were as follows: severe dehydration and severe acute renal failure (one participant; 6 mg once weekly), severe abdominal pain, moderate nausea and severe vomiting (one participant; 6 mg once weekly), moderate cholecystitis with moderate cholelithiasis (one participant; 8 mg once every 2 wk), and severe worsening of diverticulitis (one participant; 8 mg once every 2 wk). Two other participants experienced serious TEAEs that did not lead to discontinuation; these were prostate cancer (one participant; 4 mg once weekly) and erysipelas (one participant; 6 mg once weekly). Only severe dehydration and severe acute renal failure were considered definitely related to the study drug. No serious TEAEs were reported in the placebo group.
No cases of cardiovascular TEAEs, acute pancreatitis, thyroid cancer or cerebrovascular TEAEs were reported. Symptomatic hypoglycaemia was reported in one participant each in the efpeglenatide 6 mg once‐weekly (two events) and efpeglenatide 8 mg once‐every‐2‐wk groups (one event). No severe symptomatic hypoglycaemia events were reported.
3.5.2. Heart rate and blood pressure
Significant increases from baseline to week 21 in heart rate were observed in all the efpeglenatide groups (range of LSM change from baseline vs. placebo: 3.1–5.7 bpm), which were within the expected range for GLP‐1RAs (Table S7). Systolic (SBP) and diastolic blood pressure (DBP) decreased from baseline to week 21 within the expected range for GLP‐1RAs (range of LSM change from baseline vs. placebo: −0.2 to −6.0 mmHg [SBP], −0.2 to −1.7 mmHg [DBP]); this change was only significant for SBP with efpeglenatide 6 mg once weekly (−6.0 mmHg; Table S7).
3.5.3. Other safety assessments
During the treatment period, one participant in the placebo group had a high level of amylase (>300 U/L) with no sign of pancreatitis. High lipase values (>153 U/L) were recorded in 11 participants in the efpeglenatide group (4 mg once weekly: four participants; 6 mg once weekly: two participants; 6 mg once every 2 wk: three participants; 8 mg once every 2 wk: two participants).
At baseline, 2.6% of participants (6/235) in the efpeglenatide group overall were positive for anti‐efpeglenatide antibodies (Table S7). Treatment‐emergent antibodies, defined as treatment‐induced anti‐drug antibodies with any titre, or treatment‐boosted anti‐drug antibodies with a log 2‐expressed titre increase by ≥2 from pre‐existing levels, were observed in 21.7% of participants (51/235) receiving efpeglenatide. No participant receiving efpeglenatide or placebo was positive for in vitro anti‐efpeglenatide neutralizing antibodies at baseline, during treatment or follow‐up. Individual participant data suggest that the presence of anti‐efpeglenatide antibodies did not impact the efficacy of efpeglenatide with regard to body weight; however, the participant numbers were too low for formal statistical analysis.
Injection‐site reactions occurred for both efpeglenatide (8.5%–18.6%) and placebo (21.7%; Table 3).
4. DISCUSSION
In this trial, efpeglenatide administered once weekly (4 mg, 6 mg) and once every 2 wk (6 mg, 8 mg) led to significant reductions in body weight compared with placebo over 20 wk of treatment in obese or overweight participants (with comorbidity) without diabetes. These reductions were also associated with significant decreases in waist circumference and BMI. According to US Food and Drug Administration guidelines, a product can be considered effective for weight management if after 1 year of treatment the proportion of people who lose ≥5% of baseline body weight is at least 35% and is approximately double the proportion in the placebo‐treated group, and if the difference between groups is statistically significant.26 After 20 wk, nearly 50% of all efpeglenatide‐treated participants lost ≥5% of their baseline body weight (range across treatment groups: 46%–53.4%), and ~20% lost ≥10% of baseline body weight; these proportions were statistically significant versus the placebo group, in which <5% of participants met these targets. However, the long‐term effects of efpeglenatide on body weight reduction (eg, >1 y) have not yet been assessed.
Treatment benefits appeared to be related to dose. The mean reductions in body weight and BMI endpoints from baseline (vs. placebo) were greater with the higher doses of efpeglenatide once weekly and once every 2 wk compared with the respective lower doses of the same dosing regimen (Table 2). The higher doses of the efpeglenatide once‐weekly and once‐every‐2‐wk regimens were associated with similar reductions (vs. placebo) in body weight and BMI. Reductions in body weight and BMI endpoints (vs. placebo) were also comparable between the lower doses of efpeglenatide once weekly and once every 2 wk.
Treatment with efpeglenatide also led to significant improvements in other metabolic variables, including glucose metabolism and lipid profiles. Although the participants in this trial did not have diabetes, all doses of efpeglenatide significantly reduced FPG and HbA1c levels compared with placebo. Significant reductions in blood lipids, including total cholesterol, LDL cholesterol, VLDL cholesterol, HDL cholesterol and triglycerides were also observed with most doses of efpeglenatide versus placebo. Blood pressure tended to decrease with efpeglenatide, although the reduction was only significant with the 6‐mg once‐weekly dose. As the baseline blood pressure of the participants in this study was in the normal range, the potential for efpeglenatide to improve blood pressure in people with high blood pressure remains to be examined.
Efpeglenatide was generally well tolerated, and most TEAEs were mild or moderate in intensity. The most frequently reported TEAEs were GI disorders, consistent with the GLP‐1RA class.27 Most nausea and vomiting events occurred over the first few injections and subsided over time. As slow titration of GLP‐1RAs has been shown to mitigate GI side effects,27, 28, 29, 30 the tolerability of efpeglenatide should be assessed with a more optimal titration scheme than that used in the present study. It is worth noting that, while rates of GI disorders were high with efpeglenatide, they were comparable to those observed in phase II studies of semaglutide and liraglutide in people with obesity.31, 32 Increases in heart rate and decreases in blood pressure observed in the present study were within the expected range for GLP‐1RAs.27 While exendin‐based GLP‐1RAs are generally associated with high immunogenicity,33, 34 the immunogenicity of efpeglenatide was relatively low, with ~20% of efpeglenatide‐treated participants developing anti‐drug antibodies. While overall discontinuation rates with efpeglenatide appeared to be slightly higher in this phase II study compared with studies of other GLP‐1RAs in obesity, discontinuations attributable to AEs were similar.31, 32
Among the currently available GLP‐1RAs there is variability in their effects on weight loss in people with T2D.35 Although the exact mechanism underlying these differences is unclear, it is possible that they involve access to GLP‐1 receptors that mediate weight loss, some of which may be in the CNS.36, 37 It is unknown whether efpeglenatide crosses the blood–brain barrier; however, it is possible that the fragment crystallizable conjugation may prevent effective transport into the CNS. Nevertheless, the magnitude of weight loss in participants with obesity observed with efpeglenatide in the present study was comparable to that observed with liraglutide, a smaller GLP‐1RA that is approved for obesity as a once‐daily injection.38 In a 20‐wk, phase II clinical trial, liraglutide 3.0 mg once daily led to a placebo‐subtracted reduction in body weight of 4.4 kg (95% CI 2.9–6.0) in people with obesity and without diabetes.31 In BALANCE, the placebo‐adjusted per cent change in weight from baseline with efpeglenatide ranged between −6.7% and −7.5% after 20 wk of treatment (placebo‐adjusted mean weight loss: 6.3–7.2 kg). In people with obesity without diabetes, s.c. semaglutide once daily demonstrated a treatment difference in weight change from baseline versus placebo of between −4.6% and −15.7% at 52 wk.32 Although the observed reduction in weight by efpeglenatide once weekly and semaglutide once daily cannot be directly compared, especially owing to the difference in endpoint timings and treatment regimens, the 52‐wk reduction by semaglutide suggests the potential for treatment with GLP‐1RAs to demonstrate clinically significant weight reduction at 52 wk. The potential for clinically meaningful weight loss is particularly apparent if higher total weekly doses of GLP‐1RAs are used for weight reduction compared with the doses used to achieve glycaemic control, as demonstrated by semaglutide,32, 39 although this is yet to be determined for efpeglenatide. It is important to note that weight reduction with efpeglenatide did not appear to plateau at 20 wk, suggesting the potential for further weight loss at later time points.
Biochemical and preclinical studies suggest that efpeglenatide has unique receptor properties that may explain the greater maximal GLP‐1 receptor signalling and reduced desensitization seen with efpeglenatide versus other GLP‐1RAs following chronic exposure in vitro.21, 40 Efpeglenatide is associated with faster dissociation kinetics from the GLP‐1 receptor compared with other GLP‐1RAs, allowing efpeglenatide to function as a full agonist for glucose‐stimulated insulin secretion but with reduced agonist‐induced receptor internalization, resulting in more cell‐surface receptor availability with chronic exposure in vitro.21 Whether these unique receptor properties will translate to better weight loss effects of efpeglenatide compared with other GLP‐1RAs in patients remains to be investigated.
There were several limitations to this study. As this was a phase II study, the small sample size may have limited the statistical power needed to detect significance for some endpoints, and we did not control for multiplicity of P values. Although multiple analyses were used to handle missing data, it is important to note that the magnitude of weight loss described using these analyses in clinical trials may not reflect what occurs in the real‐world setting. The effect of study drug discontinuation on weight loss was not analysed in this study. Additionally, the relatively short duration of the study did not allow assessment of maximal weight loss or sustainability of weight loss, and the dose range tested was not sufficient to explore the full dose–response relationship. While participants were encouraged to reduce caloric intake and exercise, diet and physical activity were not actively controlled. Weight loss in the placebo group was minimal, suggesting that the guidance on diet and exercise may not have been effective in promoting weight loss. Finally, this study did not include a titration period for the once‐weekly doses, and it used a short titration period for the once‐every‐2‐wk doses. The safety profile, particularly the incidence of GI AEs, should be further investigated using optimized titration.
In conclusion, efpeglenatide once weekly and once every 2 wk was well tolerated, and treatment was associated with a significant and clinically meaningful reduction in body weight in obese or overweight people (with comorbidity) without diabetes. The weight loss effects were associated with improvements in glycaemic and lipid variables, consistent with an overall benefit with regard to metabolism. These findings support further development of efpeglenatide in obese people. Future studies are needed to assess the effects of higher doses as well as the long‐term safety and efficacy, and improvements in health outcomes including comorbidities associated with obesity.
4.1. Prior publication
This study has been previously presented as a poster at the 75th American Diabetes Association in Boston, Massachusetts, June 5–9, 2015 (Pratley RE et al. Diabetes. 2015;64[Suppl. 1]:LB‐303) and at the 51st European Association for the Study of Diabetes Annual Meeting in Stockholm, Sweden, September 14–18, 2015 (Pratley RE et al. Diabetologia. 2015;58[Suppl. 1]:1 Abstract 643).
CONFLICT OF INTEREST
R.E.P. has served as a consultant for AstraZeneca, Eli Lilly, Janssen Pharmaceuticals, Merck, Novo Nordisk, Pfizer and Sanofi, has received grants and research support from Eli Lilly, Janssen Pharmaceuticals, Lexicon, Ligand, Merck, Novo Nordisk and Sanofi‐Aventis US, and has received honoraria from Novo Nordisk; honoraria and fees for these activities are directed to a non‐profit organization. J.K. is an employee of Hanmi Pharmaceutical. M.E.T. is a consultant of ProSciento and a shareholder of Eli Lilly, and has received consulting fees from AstraZeneca, Intarcia and Servier. M.H. is an employee and shareholder of ProSciento. O.H. is an employee of Hanmi Pharmaceutical. J.S. and C.H.S. are employees and shareholders of Sanofi. S.J. has served as a consultant for AstraZeneca, Bayer, Berlin Chemie, Boehringer Ingelheim, Eli Lilly, Merck, Novo Nordisk, Orexigen, Roche and Servier, and has received honoraria from Abbott, AstraZeneca, Bayer, Berlin Chemie, Boehringer Ingelheim, Johnson & Johnson, Eli Lilly, Merck, Novartis, Novo Nordisk, Orexigen, Roche, Sanofi‐Aventis and Servier. K.‐H.Y. has received honoraria from AstraZeneca, Boehringer Ingelheim, Eli Lilly, Hanmi Pharmaceutical, Merck, Novo Nordisk, Sanofi and Takeda, and research support from AstraZeneca and Takeda.
AUTHOR CONTRIBUTIONS
R.E.P. and S.J. contributed data acquisition, data analysis/interpretation and critical revision of the manuscript for important intellectual content. J.K. contributed study conception and design, data analysis/interpretation and critical revision of the manuscript for important intellectual content. M.E.T., M.H., O.H., J.S., C.S.H. and K.‐H.Y. contributed data analysis/interpretation and critical revision of the manuscript for important intellectual content. All authors confirm that they meet the International Committee of Medical Journal Editors uniform requirements for authorship and that all authors have read, reviewed and agreed to the final version.
Supporting information
Table S1. Study participation by country and by site.
Table S2. Sensitivity and supportive analyses for primary endpoint.
Table S3. Glycaemic endpoints at week 21.
Table S4. Lipid endpoints at week 21.
Table S5. Frequency of nausea events.
Table S6. TEAEs leading to discontinuation by system organ class and preferred term.
Table S7. Heart rate and blood pressure change from baseline at week 21.
Table S8. Immunogenicity.
Figure S1. Study design and titration/administration scheme.
Figure S2. Participant disposition.
Figure S3. LSM change from baseline in FPG and HbA1c from baseline over the 20‐wk treatment period.
Figure S4. LSM change from baseline in total cholesterol, LDL cholesterol, VLDL cholesterol, HDL cholesterol and triglycerides over the 20‐wk treatment period.
ACKNOWLEDGMENTS
In November 2015, Sanofi obtained an exclusive license from Hanmi Pharmaceutical for the worldwide development and commercialization of efpeglenatide, an experimental, long‐acting diabetes treatment. The BALANCE 205 trial was conducted by Hanmi Pharmaceutical between April 2014 and February 2015.
Editorial assistance was provided by Namiko Abe, PhD, of Caudex (New York, NY, USA) and was funded by Sanofi. The authors thank Ike Ogbaa, MD, Sanofi, for help with preparing the manuscript, and the investigators and patients who participated in this study.
Pratley RE, Kang J, Trautmann ME, et al. Body weight management and safety with efpeglenatide in adults without diabetes: A phase II randomized study. Diabetes Obes Metab. 2019;21:2429–2439. 10.1111/dom.13824
Funding information The study was funded by Sanofi.
REFERENCES
- 1. World Health Organization . Obesity and overweight fact sheet, updated February 2018. http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed November 9, 2018.
- 2. Wilson PW, D'Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;162:1867‐1872. [DOI] [PubMed] [Google Scholar]
- 3. Manninen P, Riihimaki H, Heliovaara M, Makela P. Overweight, gender and knee osteoarthritis. Int J Obes Relat Metab Disord. 1996;20:595‐597. [PubMed] [Google Scholar]
- 4. Basen‐Engquist K, Chang M. Obesity and cancer risk: recent review and evidence. Curr Oncol Rep. 2011;13:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poirier P, Giles TD, Bray GA, et al; American Heart Association; Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association scientific statement on obesity and heart disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898‐918. [DOI] [PubMed] [Google Scholar]
- 6. Hamman RF, Wing RR, Edelstein SL, et al; for the Diabetes Prevention Program Research Group. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care. 2006;29:2102‐2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation. 2014;129:S102‐S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gummesson A, Nyman E, Knutsson M, Karpefors M. Effect of weight reduction on glycated haemoglobin in weight loss trials in patients with type 2 diabetes. Diabetes Obes Metab. 2017;19:1295‐1305. [DOI] [PubMed] [Google Scholar]
- 9. Wing RR, Lang W, Wadden TA, et al; Look AHEAD Research Group.Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34:1481‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williamson DF, Thompson TJ, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care. 2000;23:1499‐1504. [DOI] [PubMed] [Google Scholar]
- 11. Dombrowski SU, Knittle K, Avenell A, Araujo‐Soares V, Sniehotta FF. Long term maintenance of weight loss with non‐surgical interventions in obese adults: systematic review and meta‐analyses of randomised controlled trials. BMJ. 2014;348:g2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montesi L, El Ghoch M, Brodosi L, Calugi S, Marchesini G, Dalle Grave R. Long‐term weight loss maintenance for obesity: a multidisciplinary approach. Diabetes Metab Syndr Obes. 2016;9:37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drab SR. Glucagon‐like peptide‐1 receptor agonists for type 2 diabetes: a clinical update of safety and efficacy. Curr Diabetes Rev. 2016;12:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mearns ES, Sobieraj DM, White CM, et al. Comparative efficacy and safety of antidiabetic drug regimens added to metformin monotherapy in patients with type 2 diabetes: a network meta‐analysis. PLoS One. 2015;10:e0125879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vilsboll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon‐like peptide‐1 receptor agonists on weight loss: systematic review and meta‐analyses of randomised controlled trials. BMJ. 2012;344:d7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farr OM, Sofopoulos M, Tsoukas MA, et al. GLP‐1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP‐1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: a crossover, randomised, placebo‐controlled trial. Diabetologia. 2016;59:954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ten Kulve JS, Veltman DJ, van Bloemendaal L, et al. Endogenous GLP‐1 mediates postprandial reductions in activation in central reward and satiety areas in patients with type 2 diabetes. Diabetologia. 2015;58:2688‐2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Bloemendaal L, Ijzerman RG, Ten Kulve JS, et al. GLP‐1 receptor activation modulates appetite‐ and reward‐related brain areas in humans. Diabetes. 2014;63:4186‐4196. [DOI] [PubMed] [Google Scholar]
- 19. van Bloemendaal L, Ten Kulve JS, la Fleur SE, Ijzerman RG, Diamant M. Effects of glucagon‐like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol. 2014;221:T1‐T16. [DOI] [PubMed] [Google Scholar]
- 20. Hanmi Pharmaceutical Co. Ltd . Long Acting Protein / Peptide Discovery Platform Technology. 2018. http://www.hanmipharm.com/ehanmi/handler/Rnd-ProjectLapscovery. Accessed December 6, 2018. [Google Scholar]
- 21. Choi IY, Moon MJ, Trautmann ME, Hompesch M, Sorli CH. In vitro studies to evaluate the receptor kinetics of efpeglenatide versus other glucagon‐like peptide‐1 receptor (GLP‐1 R) agonists. Diabetes. 2018;67:1090‐P. [Google Scholar]
- 22. Kim YI, Kim SE, Choi IY, et al. Pharmacokinetics and pharmacodynamic effects of a single dose of the long‐acting GLP‐1R agonist LAPS‐Exendin‐4 in subjects with type 2 diabetes mellitus. Diabetes. 2012;61(Suppl. 1):A277. [Google Scholar]
- 23. Kang J, Choi S, Choi I, et al. HM11260C, a new generation long acting GLP‐1R agonist with a unique pharmacokinetic profile improves glucose control and GI tolerability: a phase IIa clinical trial in type 2 diabetes mellitus. Diabetes. 2013;62:LB16. [Google Scholar]
- 24. Rosenstock J, Kang J, Choi S, et al. Dose‐response improvements in glycemic control and body weight reductions with HM11260C, a once‐weekly GLP‐1 receptor agonist with liraglutide as reference, in type 2 diabetes (T2DM). Diabetes. 2015;64:A73. [Google Scholar]
- 25. Del Prato S, Kang J, Choi S, et al. Once a month treatment with HM11260C improves glycaemic control in type 2 diabetes mellitus: interim data from a 16‐week study. Diabetologia. 2015;58:S56. [Google Scholar]
- 26. Food and Drug Administration . Guidance for industry: developing products for weight management. https://www.fda.gov/downloads/Drugs/Guidances/ucm071612.pdf. Accessed December 6, 2018.
- 27. Raccah D. Safety and tolerability of glucagon‐like peptide‐1 receptor agonists: unresolved and emerging issues. Expert Opin Drug Saf. 2017;16:227‐236. [DOI] [PubMed] [Google Scholar]
- 28. Fineman MS, Shen LZ, Taylor K, Kim DD, Baron AD. Effectiveness of progressive dose‐escalation of exenatide (exendin‐4) in reducing dose‐limiting side effects in subjects with type 2 diabetes. Diabetes Metab Res Rev. 2004;20:411‐417. [DOI] [PubMed] [Google Scholar]
- 29. Gough SC, Bode B, Woo V, et al; NN9068‐3697 (DUAL‐I) Trial Investigators. Efficacy and safety of a fixed‐ratio combination of insulin degludec and liraglutide (IDegLira) compared with its components given alone: results of a phase 3, open‐label, randomised, 26‐week, treat‐to‐target trial in insulin‐naive patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2014;2:885‐893. [DOI] [PubMed] [Google Scholar]
- 30. Rosenstock J, Aronson R, Grunberger G, et al. Benefits of LixiLan, a titratable fixed‐ratio combination of insulin glargine plus lixisenatide versus insulin glargine and lixisenatide monocomponents in type 2 diabetes inadequately controlled on oral agents: the LixiLan‐O randomized trial. Diabetes Care. 2016;39:2026‐2035. [DOI] [PubMed] [Google Scholar]
- 31. Astrup A, Rossner S, Van GL, et al. Effects of liraglutide in the treatment of obesity: a randomised, double‐blind, placebo‐controlled study. Lancet. 2009;374:1606‐1616. [DOI] [PubMed] [Google Scholar]
- 32. O'Neil PM, Birkenfeld AL, McGowan B, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double‐blind, placebo and active controlled, dose‐ranging, phase 2 trial. Lancet. 2018;392:637‐649. [DOI] [PubMed] [Google Scholar]
- 33. Fineman MS, Mace KF, Diamant M, et al. Clinical relevance of anti‐exenatide antibodies: safety, efficacy and cross‐reactivity with long‐term treatment. Diabetes Obes Metab. 2012;14:546‐554. [DOI] [PubMed] [Google Scholar]
- 34. Fonseca VA, Alvarado‐Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE. Efficacy and safety of the once‐daily GLP‐1 receptor agonist lixisenatide in monotherapy: a randomized, double‐blind, placebo‐controlled trial in patients with type 2 diabetes (GetGoal‐Mono). Diabetes Care. 2012;35:1225‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trujillo JM, Nuffer W, Ellis SL. GLP‐1 receptor agonists: a review of head‐to‐head clinical studies. Ther Adv Endocrinol Metab. 2015;6:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baggio LL, Drucker DJ. Glucagon‐like peptide‐1 receptors in the brain: controlling food intake and body weight. J Clin Invest. 2014;124:4223‐4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Geloneze B, de Lima‐Junior JC, Velloso LA. Glucagon‐like peptide‐1 receptor agonists (GLP‐1RAs) in the brain‐adipocyte axis. Drugs. 2017;77:493‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Novo Nordisk . Saxenda: prescribing information. http://www.novo-pi.com/saxenda.pdf. Accessed November 9, 2018.
- 39. Food and Drug Administration . Ozempic: US prescribing information. https://www.novo-pi.com/ozempic.pdf. Accessed December 6, 2018.
- 40. Trautmann ME, Choi IY, Kim JK, Sorli CH. Preclinical effects of efpeglenatide, a long‐acting glucagon‐like peptide‐1 receptor agonist compared with liraglutide and dulaglutide. Diabetes. 2018;67:1098‐P. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Study participation by country and by site.
Table S2. Sensitivity and supportive analyses for primary endpoint.
Table S3. Glycaemic endpoints at week 21.
Table S4. Lipid endpoints at week 21.
Table S5. Frequency of nausea events.
Table S6. TEAEs leading to discontinuation by system organ class and preferred term.
Table S7. Heart rate and blood pressure change from baseline at week 21.
Table S8. Immunogenicity.
Figure S1. Study design and titration/administration scheme.
Figure S2. Participant disposition.
Figure S3. LSM change from baseline in FPG and HbA1c from baseline over the 20‐wk treatment period.
Figure S4. LSM change from baseline in total cholesterol, LDL cholesterol, VLDL cholesterol, HDL cholesterol and triglycerides over the 20‐wk treatment period.