

Summary
Inhibition of cell division is critical for viability under DNA‐damaging conditions. DNA damage induces the SOS response that in bacteria inhibits cell division while repairs are being made. In coccoids, such as the human pathogen, Staphylococcus aureus, this process remains poorly studied. Here, we identify SosA as the staphylococcal SOS‐induced cell division inhibitor. Overproduction of SosA inhibits cell division, while sosA inactivation sensitizes cells to genotoxic stress. SosA is a small, predicted membrane protein with an extracellular C‐terminal domain in which point mutation of residues that are conserved in staphylococci and major truncations abolished the inhibitory activity. In contrast, a minor truncation led to SosA accumulation and a strong cell division inhibitory activity, phenotypically similar to expression of wild‐type SosA in a CtpA membrane protease mutant. This suggests that the extracellular C‐terminus of SosA is required both for cell division inhibition and for turnover of the protein. Microscopy analysis revealed that SosA halts cell division and synchronizes the cell population at a point where division proteins such as FtsZ and EzrA are localized at midcell, and the septum formation is initiated but unable to progress to closure. Thus, our findings show that SosA is central in cell division regulation in staphylococci.
Staphylococcus aureus is a human pathogen and a model organism for cell division in spherical bacteria. We show that SosA is the DNA‐damage‐inducible cell‐division inhibitor in S. aureus responsible for cessation of the cell cycle prior to division plate completion. The extracellular domain of SosA appears essential for activity but is also a likely target for regulation by the CtpA protease. This represents the first description of a cell division inhibition process in coccoid bacteria.
![]()
Introduction
Bacteria multiply by coordinated and essential DNA replication and cell division events, two important biological processes that are valuable targets for antimicrobial therapy (Lock and Harry, 2008; Adams and Errington, 2009; Robinson et al., 2012; Sass and Brötz‐Oesterhelt, 2013; den Blaauwen et al., 2017). In the event of DNA damage, the SOS response is activated and ensures that cell division is delayed until the DNA is repaired. The SOS regulon is controlled by the conserved LexA repressor, which is inactivated in response to RecA, a sensor of DNA damage at stalled replication forks, binding to single‐stranded DNA (Kelley, 2006; Kreuzer, 2013; Baharoglu and Mazel, 2014). The SOS response has mostly been studied in Escherichia coli, a Gram‐negative rod‐shaped bacterium where the LexA‐regulated gene, sulA, encodes a cell division inhibitor. This inhibitor suppresses division by binding to FtsZ, which acts as a scaffold for the assembly of cell division components at the division site (Huisman and D'Ari, 1981; Huisman et al., 1984; Jones and Holland, 1985; Higashitani et al., 1995; Mukherjee et al., 1998; Cordell et al., 2003; Adams and Errington, 2009). In rod‐shaped bacteria, inhibition of cell division leads to filamentation as a consequence of lateral peptidoglycan synthesis. This phenotype is also observed in E. coli mutants lacking the Lon protease of which SulA is a substrate, further substantiating the role of SulA in cell division inhibition and filamentation (Mizusawa and Gottesman, 1983; Schoemaker et al., 1984).
SulA is poorly conserved in species outside Enterobacteriaceae. The α‐proteobacterium Caulobacter crescentus encodes an SOS‐induced cell division inhibitor SidA that does not show homology to SulA. In contrast to cytosolic SulA, SidA is a membrane‐anchored, small protein that does not interact with FtsZ but rather with later cell division proteins (Modell et al., 2011). In this species, yet another, possibly redundant, cell division inhibitor DidA is implicated in survival during DNA damage, while being independent of the SOS response (Modell et al., 2014). Hence, it appears that bacteria choose fundamentally different ways to orchestrate regulated cell division inhibition in response to DNA damage. For Gram‐positive, spherical cells such as Staphylococcus aureus, however, little is known of how SOS induction is coupled to cell division nor have specific posttranslational mechanisms for the negative regulation of cell division inhibition been identified.
S. aureus is a serious Gram‐positive human pathogen, notorious for being implicated in a wide range of infections and for being able to acquire resistance toward important antibiotic classes. It originally received its name from the grape‐like clusters of coccoid cells that result from the unique cell division process which occurs in three consecutive orthogonal planes (Tzagoloff and Novick, 1977; Turner et al., 2010; Pinho et al., 2013). In this organism, we and others have previously identified lexA and noted an open reading frame (designated sosA) that is divergently transcribed from lexA and controlled by the LexA repressor and the SOS response (Anderson et al., 2006; Cirz et al., 2007; Mesak et al., 2008; Cohn et al., 2011). The location of sosA adjacent to lexA indicated that it might encode a cell division inhibitor, as similar gene synteny has been observed for SOS‐induced cell division inhibitors encoded by Gram‐positive, rod‐shaped bacteria, namely Bacillus subtilis (Kawai et al., 2003), Bacillus megaterium (Buchholz et al., 2013), Listeria monocytogenes (van der Veen et al., 2007; 2010), Mycobacterium tuberculosis (Chauhan et al., 2006) and Corynebacterium glutamicum (Ogino et al., 2008) (Fig. 1A). The cell division inhibitors of these organisms display no similarity to SulA, and within the group they generally show little homology with the exceptions of YneA from L. monocytogenes and ChiZ from M. tuberculosis (Chauhan et al., 2006) that resemble B. subtilis YneA. Commonly, however, they have a single membrane spanning segment, a predicted extracellular C‐terminus and, except for DivS from C. glutamicum, a LysM domain, which is a ubiquitous peptidoglycan‐binding motif (Buist et al., 2008; Mesnage et al., 2014), suggesting that membrane and/or cell wall localization is a common feature characterizing Gram‐positive cell division inhibitors. In this line, it is interesting that M. tuberculosis ChiZ was reported to have cell wall hydrolase activity (Chauhan et al., 2006), which could be functionally important for LysM‐containing cell division inhibitors.
Figure 1.
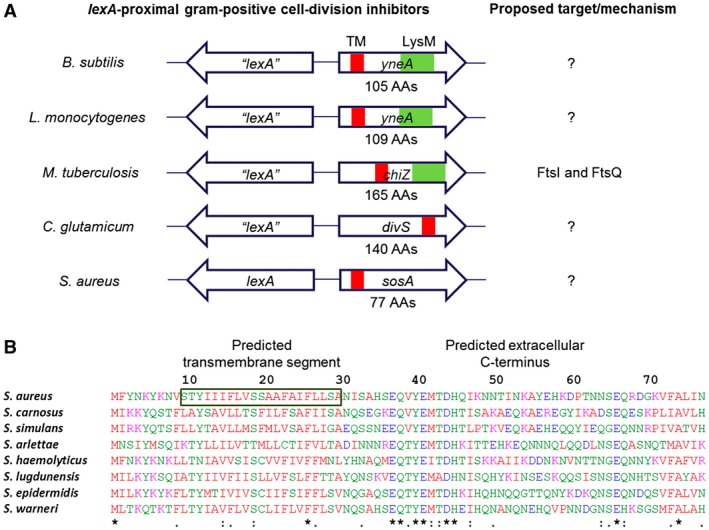
Gram‐positive SOS‐controlled cell division inhibitors. A. Schematic representation of genes encoding characterized Gram‐positive SOS‐regulated cell division inhibitors (not drawn to scale) including the uncharacterized sosA from S. aureus. Despite considerable sequence divergence, these genes are commonly chromosomally co‐localized with lexA homologous genes. The cell division inhibitors carry a single transmembrane domain (TM), and several proteins have an additional LysM domain. B. Alignment (CLUSTAL O[1.2.4]) of SosA sequences deduced from open reading frames next to lexA in S. aureus strain 8325‐4 (YP_499864) and seven Staphylococcus species: S. carnosus (CAL27889), S. simulans (AMG96201), S. arlettae (EJY94737), S. haemolyticus (YP_253482), S. lugdunensis (YP_003471776), S. epidermidis (YP_188489) and S. warneri (EEQ79882). The proteins are 77 amino acids long and are characterized by a predicted transmembrane segment at AAs 10–30 (for S. aureus SosA) and a predicted extracellular C‐terminal (TOPCONS [Tsirigos et al., 2015]) with considerable sequence conservation at the membrane‐proximal portion (‘*’ indicates fully conserved residues, ‘:’ indicates conservation of residues with highly similar properties).
The fundamental processes of staphylococcal cell division have been studied spatiotemporally using super‐resolution microscopy techniques (Turner et al., 2010; Monteiro et al., 2015), and recent efforts combining genetic approaches have unveiled the molecular mechanism in unprecedented detail (Lund et al., 2018; Monteiro et al., 2018). Here, we demonstrate that sosA encodes the SOS‐inducible cell division inhibitor in S. aureus and document its impact on cell division following treatment with DNA damaging agents. Moreover, we identify a possible mechanism for the proteolytic control of endogenous cell division inhibition. Thus, we further our insight into these basic biological phenomena, which could lead to the development of new antimicrobials targeting the cell division machinery in S. aureus.
Results
Conservation of sosA in staphylococci
In S. aureus, the open reading frame located adjacent to lexA (divergently transcribed) was named sosA and hypothesized to encode an inhibitor of cell division (Fig. 1A) (Cohn et al., 2011). The 77‐amino‐acid‐long product has homology and 25%–60% amino acid (AA) identity to proteins encoded by genes occupying the same chromosomal location in a range of staphylococci (Figs 1B and S1), while showing no clear homology to the known Gram‐positive cell division inhibitors. Of note, the C‐terminal part of SosA lacks a LysM domain, which is present in the cell division inhibitors YneA and ChiZ (Mo and Burkholder, 2010; Vadrevu et al., 2011). The N‐terminal half of SosA includes a putative transmembrane (TM) domain, and the C‐terminal part is predicted to be located extracellularly (TOPCONS server [Tsirigos et al., 2015]). Among the different staphylococcal species, a highly conserved sequence is located just C‐terminally of the predicted TM domain and, although less striking, some conservation also seems to exist in the extreme C‐terminus (Fig. 1B).
Genotoxic stress inhibits cell division in an SosA‐dependent manner
To analyze whether SosA serves as an SOS‐induced cell division inhibitor, we exposed 8325‐4, the common laboratory S. aureus strain, and JE2, a derivative of the clinically relevant USA300 lineage, to a lethal concentration of the DNA‐damaging agent Mitomycin C (MMC) and observed induction of SosA expression in both strains by western blot analysis (Fig. S2A). In contrast, no SosA was observed in deletion mutant derivatives but was expressed in a complemented strain (Fig. S2A). Upon exposure to the same lethal dose of MMC for a 2 h period, wild‐type (WT) cells showed 10–100‐fold greater survival than the mutant cells lacking SosA over the course of the experiment, whereas the optical densities of both cell‐type cultures were comparable (Fig. 2A). Although the killing kinetics of 8325‐4 and JE2 are different, the survival ratio of WT vs. mutant remained comparable, with WT showing 32‐ and 54‐fold greater survival at 80 min for the respective strain backgrounds. Flow cytometry measurements of cell size distributions revealed that treatment with MMC led to a three‐ and sixfold increase in forward light scatter (FSC‐A) values in 8325‐4 and JE2, respectively, while the cell size distribution of the mutants lacking sosA remained essentially constant (Fig. 2B). The constant cell size but increasing optical densities of the sosA mutant populations implies that during DNA damage these cells, in contrast to WT cells, continue to divide. Indeed, this striking phenotype was confirmed by time‐lapse microscopy, where WT cells increased in size over time, occasionally growing so large that lysis occurred, while the size of the mutant cells lacking sosA stayed the same and cell division continued (Fig. 2C and Movie S1). The phenotypes of the 8325‐4 sosA mutant were abrogated in a strain complemented with a chromosomal copy of sosA expressed from its native promoter (Fig. S2B and C). Thus, SosA seems to prevent or delay cell division; maintain viability of S. aureus under DNA damaging conditions, and as a consequence of continued activity of the peptidoglycan synthesis machinery cell size increases.
Figure 2.
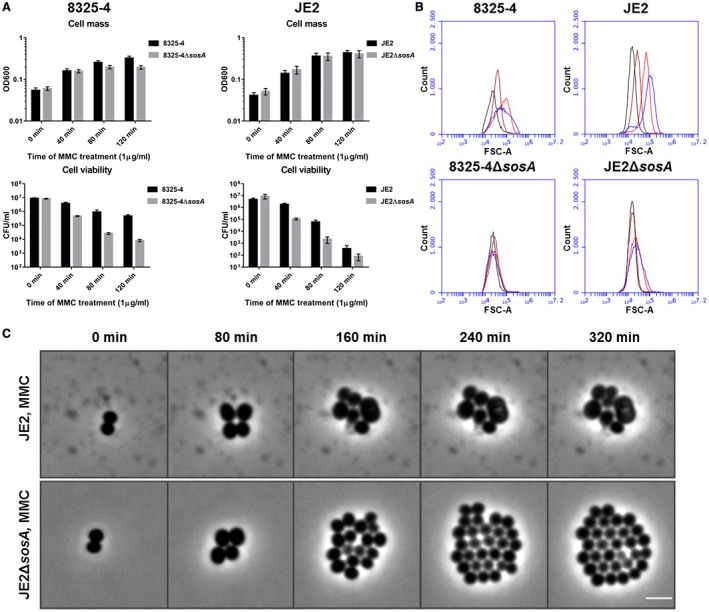
SosA supports survival of S. aureus subjected to lethal DNA damage and is involved in bacterial swelling. A. Culture optical density at 600 nm and cell viability of S. aureus strains 8325‐4 and JE2 in comparison with their respective ΔsosA mutants upon challenge with a lethal dose of mitomycin C (MMC, 1 µg ml−1) for 2 h. Error bars represent the standard deviation from three biological replicates. B. Cell size of 8325‐4 and JE2 WT and ΔsosA mutants exposed to mitomycin C estimated by flow cytometry (FSC‐A). Cells were grown exponentially prior to MMC addition at an OD600 of 0.05. Samples were taken after 0 (black), 40 (brown), 80 (red) and 120 (blue) min of incubation with MMC. C. Effect of MMC treatment (0.04 µg ml−1) on cell shape and cell number of JE2 WT and JE2ΔsosA as visualized by time‐lapse phase‐contrast microscopy. Scale bar represents 2 µm.
SosA overexpression inhibits cell division
If SosA is indeed a cell division inhibitor in S. aureus, it can be predicted that overexpression of the protein should be sufficient to inhibit colony formation even under conditions with no DNA damage. Hence, we expressed sosA episomally from an anhydrotetracycline (AHT)‐inducible promoter and found that while the control vector did not affect cell viability, the plating efficiency of cells carrying the sosA overexpression plasmid was markedly compromised under inducing conditions (Fig. 3A). When grown in liquid culture and compared to the control, the SosA overexpressing cells showed a two to threefold increase in forward light scatter values, reflecting that the size of the sosA‐expressing cells increased (Fig. 3B). Microscopy observations over time confirmed the slight increase in cell size and the reduction in the overall number of cells upon SosA overproduction (Fig. 3C and Movie S2). Interestingly, contrary to what was observed under DNA‐damaging conditions (Fig. 2B), we found that ectopic induction of SosA caused only a transient increase in cell size (compare time points 45 and 90–135 min in Fig. 3B), indicating the existence of a negative regulatory mechanism.
Figure 3.
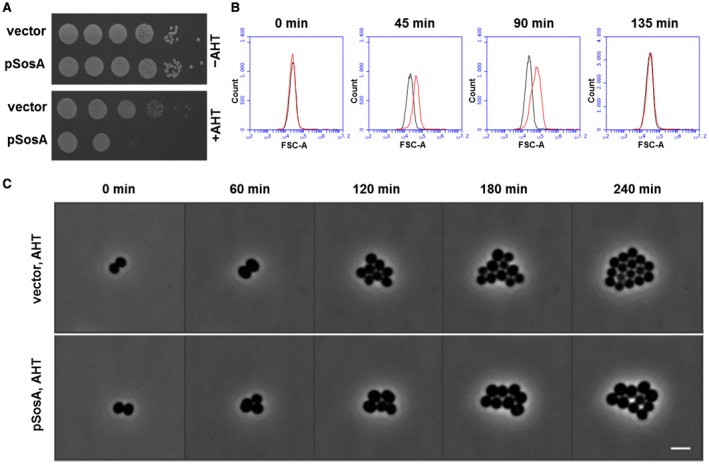
Expression of sosA alone interferes with S. aureus growth. A. The effect of a controlled expression of sosA on the ability to form colonies was assessed in S. aureus RN4220. Strains carrying either the vector control (vector) or sosA under an anhydrotetracycline (AHT)‐inducible promoter (pSosA) were grown exponentially to an OD600 of 0.5, serially 10‐fold diluted and plated on TSA plates in the presence or absence of TSA + 300 ng ml−1 of AHT. The plates were incubated overnight at 37°C and imaged. B. Evaluation of cell size distribution by flow cytometry (FSC‐A) of S. aureus RN4220 containing the control vector (black) or pSosA (red). Cells were grown exponentially prior to induction with 100 ng ml−1 of AHT. At the indicated time points, the cells were collected and analyzed by flow cytometry. C. Visualization of cell size increase and reduction of cell number of JE2/pRAB12‐lacZ (control) and JE2/pRAB12‐sosA in the presence of 200 ng ml−1 of AHT by time‐lapse phase‐contrast microscopy at 37°C. Scale bar represents 2 µm.
The C‐terminal part of SosA is functionally essential and possibly required for autoregulation
The C‐terminal part of SosA contains segments that appear somewhat conserved in staphylococci (Fig. 1B), and in silico it is predicted to be extracellular. First, we confirmed its extracellular location by constructing PhoA‐LacZ reporter fusions (Karimova and Ladant, 2017). Protein translocation to/across the membrane can be visualized using this system as LacZ remains enzymatically active only in the cytoplasm, whereas PhoA is active when in the periplasm. Here, a fusion of the reporter chimera to the C‐terminus of SosA conferred a phosphatase‐positive, β‐galatosidase‐negative phenotype in E. coli, indicative of translocation across the membrane (Fig. 4). Next, aiming at a functional characterization of its role in SosA activity, we constructed a series of C‐terminally truncated variants by the successive elimination of 10 AAs, resulting in four variants of SosA lacking between 10 and 40 of the C‐terminal AAs (Fig. 5A). Strikingly, we observed that truncation of the 10 C‐terminal AAs (and to some extent also removal of 20 C‐terminal AAs) strongly compromised plating efficiency even at low inducer concentrations (Fig. 5B). Time‐lapse microscopy revealed that cells producing SosAd10 (SosA missing the last 10 AAs) had a dramatic phenotype, with prominent cell swelling and a reduction of the cell cycle rate (Fig. 5C and Movie S3). The phenotypes from the plating assay correlated with the cell size measurements by flow cytometry (Fig. S2D). These results indicate that cells expressing the 10‐AA‐truncated proteins are stalled in a nondividing state leading to abnormal cell size, whereas those expressing full‐length SosA are only transiently halted or delayed in cell division (Fig. S2D and Movie S3). In contrast, deletion of 30 or 40 AAs from the C‐terminal part eliminated the ability of SosA to inhibit cell division, as cells expressing SosAd30 or SosAd40 gave comparable plating efficiency as those carrying the vector (Fig. 5B) and caused no effect on cell size upon induction (Fig. S2D). This prompted us to investigate the relevance of the highly conserved residues located in the membrane‐proximal part of the SosA C‐terminal part by performing alanine substitutions within the protein. To this end, we used the highly inhibitory SosAd10 variant as a scaffold and found that while point mutations at AA positions 37 or 38 (E and Q) had no effect, point mutations at positions 40 or 41 (Y and E) partially inactivated and the mutations at positions 44 and 45 (D and H) completely abolished the cell division inhibitory activity of the protein (Fig. 6A). The mutation at AA position 44 (D–A) was subsequently shown also inactivate the inhibitory activity of the full‐length protein (Fig. 6B). Importantly, the inactive variants SosAd40 and SosAd10 (44A) are still likely to be localized in the membrane, as are SosA and SosAd10, when evaluated by the PhoA‐LacZ translocation assay (Fig. 4). We conclude that conserved residues in the membrane‐proximal segment of the extracellular C‐terminal part of SosA are essential for inhibiting cell division, while truncation at the extreme C‐terminus of the protein augments activity.
Figure 4.
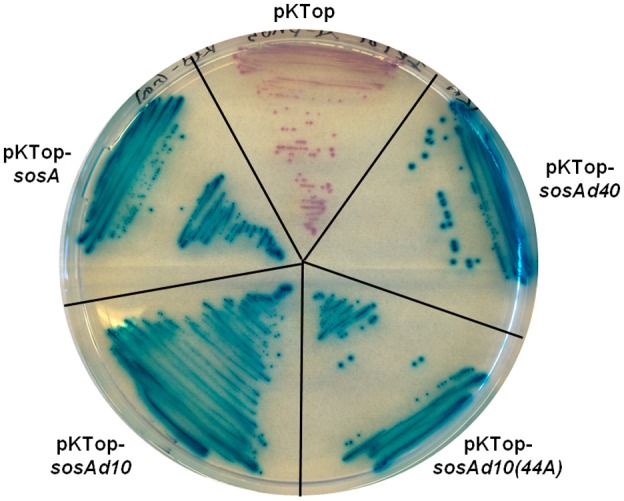
Evaluation of membrane localization of SosA and truncated variants SosAd10, SosAd10(44A) and SosAd40 by in frame fusion at the C‐terminus to PhoA‐LacZ in pKTop (the truncated variants are explained in Figs 5 and 6). E. coli IM08B cells carrying the constructs were streaked on a dual‐indicator plate containing LB agar plus 50 µg−1 of kanamycin, 1 mM IPTG, 5‐bromo‐4‐chloro‐3‐indolyl phosphate disodium salt (80 µg ml−1) and 6‐Chloro‐3‐indolyl‐β‐D‐galactopyranoside (100 µg ml−1). Cytoplasmic localization of the PhoA‐LacZ chimera is indicated by red/rose color development whereas translocation across the membrane is indicated by blue color development.
Figure 5.
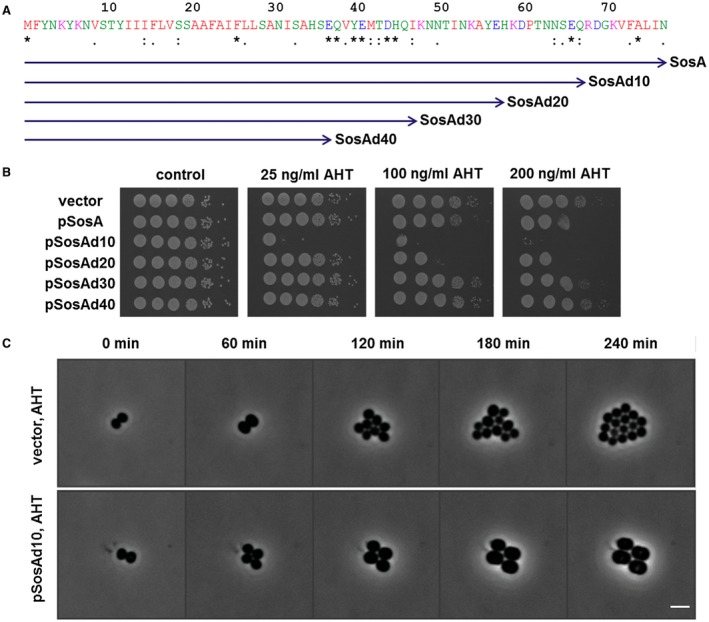
The effect of C‐terminal truncations of SosA on the inhibitory activity of the protein. A. Schematic representation of the different truncated SosA constructs. Full‐length SosA is a 77‐amino acid peptide. SosAd10 lacks the extreme C‐terminal 10 amino acids, while SosAd40 is SosA truncated of almost its entire extracellular C‐terminal part. Indicated conserved residues (*) originate from the alignment in Fig. 1. B. Activity of the constructs was assessed in S. aureus RN4220 and compared to the vector control (vector). Cells were grown exponentially to an OD600 of 0.5, serially 10‐fold diluted and plated on TSA (control) or TSA plus inducer (AHT) at indicated increasing concentrations followed by incubation overnight at 37°C. C. Visualization of the drastic cell size increase and the reduction in cell number of JE2/pRAB12‐lacZ (control) and JE2/pRAB12‐sosAd10 in presence of 200 ng ml−1 of AHT by time‐lapse phase‐contrast microscopy at 37°C. Scale bar represents 2 µm.
Figure 6.
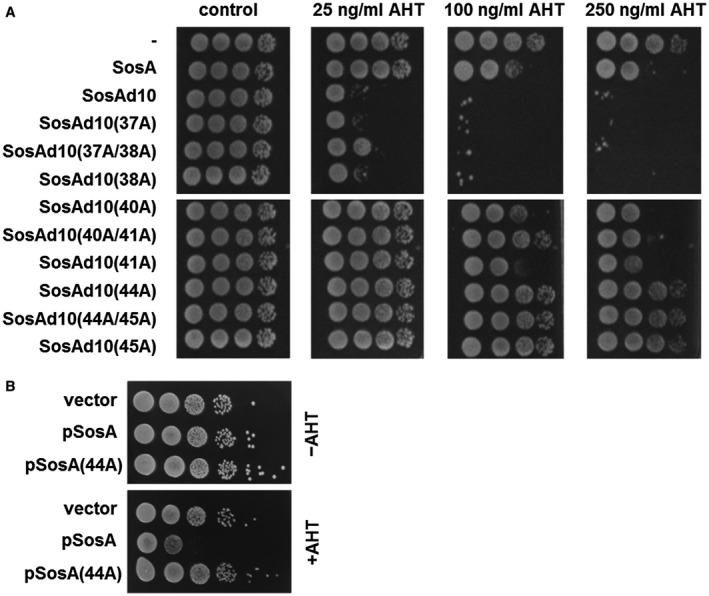
A. Assessment of the activity of SosAd10 variants with point mutations (alanine substitutions) at conserved residues (37/38, 40/41 and 44/45) within the C‐terminal part. Activity of the constructs was assessed in S. aureus JE2 and compared to the vector control (‐), SosA and SosAd10. Cells were grown exponentially to an OD600 of 0.5, serially 10‐fold diluted and plated on TSA (control) or TSA plus inducer (AHT) at increasing concentrations followed by incubation overnight at 37°C. B. Comparison of the activity of the point mutation protein SosA (44A) with WT SosA in S. aureus JE2. Cells were grown as above and plated on TSA or TSA plus 200 ng ml−1 AHT.
A CtpA protease mutant is hypersusceptible to SosA expression
We reasoned that one explanation for the severe impact of the 10‐AA‐truncated SosA on cell division could be that the extreme C‐terminus of SosA serves as an endogenous signal for proteolytic degradation, and its removal allows the mutant protein to escape degradation and accumulate. Indeed, we observed that the cellular accumulation of the truncated protein was higher than for the WT protein following 1 h of ectopic expression (Fig. S2E). In parallel, we observed the same pattern for cells expressing the 10‐AA‐truncated SosA variant with the 44A mutation that abolishes the cell division inhibitory activity suggesting that the phenotype of the mutation is not related to instability of the SosA variant (Fig. S2E). To identify a membrane localized protease that may be responsible for SosA turnover, we searched the Nebraska Transposon Mutant Library (Fey et al., 2013) for S. aureus protease mutant strains, and as a potential candidate, we identified the carboxyl‐terminal protease A, CtpA. The S. aureus CtpA protein is located at the cell membrane/cell wall fraction and is involved in stress tolerance and virulence in a mouse model of infection (Carroll et al., 2014). Importantly, overproduction of the WT SosA protein in cells lacking ctpA completely abolished cell viability, similarly to what was observed when the 10‐AA‐truncated SosA protein was expressed in WT S. aureus cells (Fig. 7A). In agreement with this result, we found that overproduction of SosA and CtpA in the ctpA mutant background alleviated the detrimental effect of SosA and that the degree of complementation depended on the relative expressional levels of the two proteins, where higher expression level of SosA was only partially complemented by CtpA expression (Fig. S3A). Further supporting that the plating phenotype of the ctpA mutant is a direct consequence of cell division inhibition by SosA, we observed that the cell size distribution of the mutant upon SosA expression completely phenocopies the distribution obtained when expressing SosAd10 in the WT (Fig. S3B). Substantiating the negative regulatory role played by CtpA with respect to SosA, we noted that the ctpA mutant was substantially more sensitive to MMC (measured by plating efficiency) than WT S. aureus, but only when encoding a functional sosA gene (Fig. 7B). Additionally, upon MMC‐induced DNA damage, higher levels of the SosA protein accumulated in the ctpA mutant when compared to the WT (Fig. 7C). Importantly, parallel experiments were recently reported showing that CtpA in B. subtilis is one of two proteolytic regulators of the cell division inhibitor YneA, with CtpA displaying direct proteolytic activity against the target in vitro (Burby et al., 2018). Altogether, based on these results, we propose that the extracellular C‐terminus of SosA is a substrate for CtpA proteolytic activity and that proteolysis relieves SosA‐mediated cell division inhibition allowing growth to resume once DNA stress has ceased.
Figure 7.
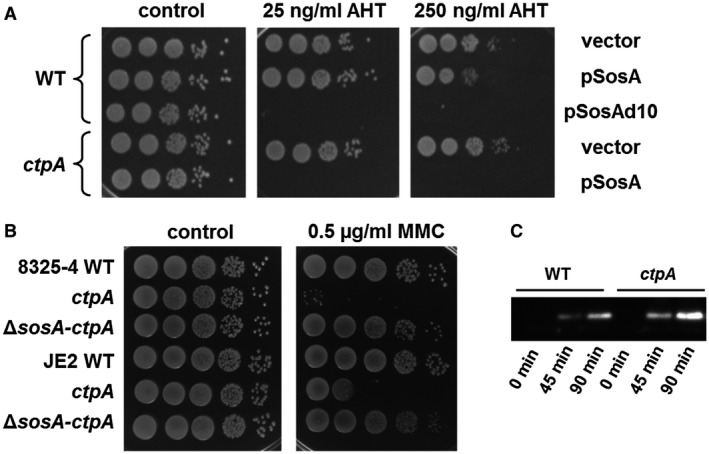
CtpA is a possible negative regulator of SosA. A. Hypersusceptibility of an S. aureus JE2 ctpA mutant to SosA‐mediated growth inhibition. Expression of sosA (pSosA) in WT S. aureus JE2 and the corresponding JE2‐ctpA mutant (ctpA) were compared and referenced to the vector control (vector) and expression of the hyperactive SosAd10 variant (pSosAd10) in the WT. Cells were grown exponentially to an OD600 of 0.5, serially 10‐fold diluted and plated on TSA (Control) or TSA plus inducer (AHT) at indicated concentrations. The plates were incubated overnight at 37°C and imaged. B. Assessment of mitomycin C susceptibility of a ctpA mutant by comparison of plating efficiency of S. aureus 8325‐4 and JE2 WT, ctpA and ΔsosA‐ctpA in presence of 0.5 µg ml−1 MMC. Cells were grown exponentially to an OD600 of 0.5 and serially 10‐fold diluted before plating and incubation at 37°C overnight. C. Western blot of accumulated SosA in S. aureus 8325‐4 WT and the ctpA mutant at indicated time points after the addition of 1 µg ml−1 of MMC to exponentially growing cells. The full blot is displayed in Fig. S5 and may serve as a control for equal loading and transfer by comparing intensities of nonspecific high molecular weight bands.
SosA is a late‐stage cell division inhibitor
To pinpoint at which step of the cell division process the SosA‐mediated inhibition is taking place, we employed fluorescence microscopy to monitor the cellular localization of FtsZ and EzrA, crucial early stage cell division proteins in S. aureus, upon induction of SosA overproduction. Neither the sosA overexpression plasmid in noninducing conditions (Fig. 8A and B) nor the control vector in non‐inducing or inducing conditions (Fig. S4A and B) changed the cell morphology or protein localization. As expected, SosA overexpression caused cell swelling but also it did not cause de‐localization of the cell division initiator FtsZ (Fig. 8A) or affected the placement of the membrane‐bound division protein EzrA (Fig. 8B), which is an early recruited protein connecting the cytoplasmic division components with the peptidoglycan‐synthesizing membrane‐bound complex (Steele et al., 2011). What was evident, however, was that whereas WT cells are heterogeneous with respect to FtsZ localization due to their presence in different phases of cell division, sosA‐expressing cells appear highly synchronized with FtsZ preferentially showing septal localization (compare uninduced and induced cells in Fig. 8A).
Figure 8.
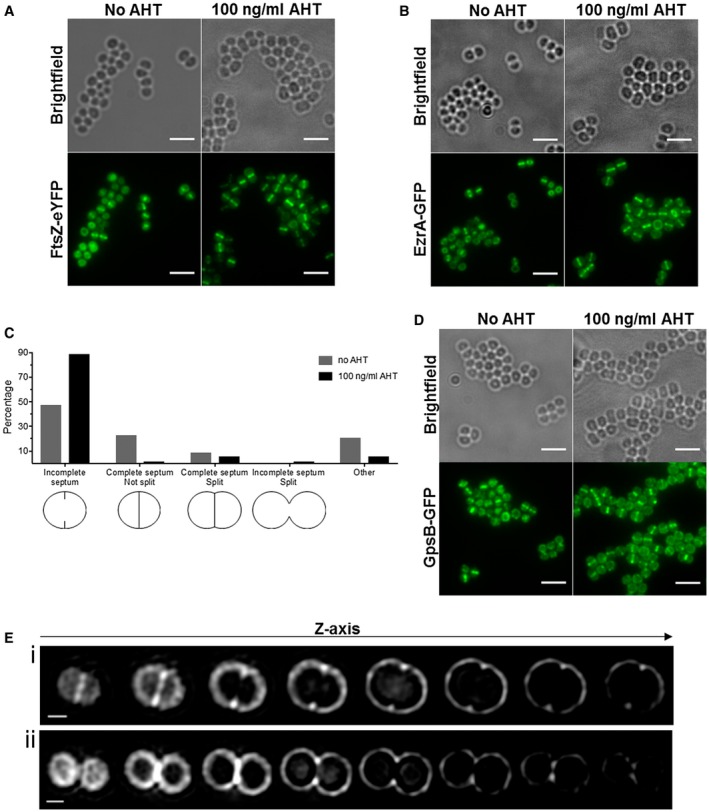
SosA does not impair the localization of cell division proteins and septum formation initiation. A. Localization of FtsZ‐eYFP in SJF4694 (JE2 pSosA pCQ11‐FtsZ‐eYFP) grown in the absence and presence of 100 ng ml−1 of AHT for 45 min. B. EzrA‐GFP localization in SJF4697 (JE2 pSosA ezrA‐gfp+) grown in the absence or presence of 100 ng ml−1 AHT for 45 min. C. Percentages of JE2 pSosA cells exhibiting incomplete, complete, complete split or incomplete split septa (n = 573 for no AHT, n = 607 for 100 ng ml−1 of AHT; organisms either isolated or co‐adhered in pairs; other represents staining in indistinct shape) after incubation with or without AHT for 45 min. D. GpsB‐GFP localization in SJF700 (JE2 pSosA gpsB‐gfp+) grown in the absence or presence of 100 ng ml−1 of AHT for 45 min. All fluorescence images (A, B and D) are average intensity projections and scale bars represent 3 um. E. 3D‐SIM Z‐stack images of JE2/pSosA grown with 100 ng ml−1 of AHT for 45 min and labeled with Alexa Fluor 647 NHS ester. (i) A cell with initiated septum formation and (ii) a cell splitting into daughter cells without finishing a septal disc. Scale bars represents 0.5 µm.
To further explore the cellular consequences of SosA‐mediated cell division inhibition, we examined the phenotypes of JE2/pSosA cells that were labeled for 5 min with HADA, marking regions of nascent peptidoglycan synthesis, and grown in the absence or presence of SosA induction (Fig. S4C). While in the absence of AHT, 47% and 23% of cells, respectively, showed a ring (incomplete septum) or line (complete septum) of nascent peptidoglycan synthesis, there was accumulation (89%) of cells with incomplete septa and a severe drop in cells with complete septa (1%) when SosA was overexpressed (Fig. 8C). Additionally, 1% of cells had an ‘hourglass’ shape after incubation with AHT, indicative of nonproductive, premature splitting taking place prior to septum completion. Next, whole cell walls of cells overproducing SosA were labeled with a fluorescent NHS ester and examined by 3D‐structured illumination microscopy (3D‐SIM). Microscopy visualization revealed that most cells had only signs of septation – a so‐called ‘piecrust’ (Turner et al., 2010) (Fig. 8Ei) – and showed that in an hourglass‐like cell, there was a gap in the septal peptidoglycan (Fig. 8Eii). The fact that SosA does not affect localization of early cell division proteins, FtsZ and EzrA, and the cell population becomes synchronized to a particular stage of the cell cycle (phase P2 [Monteiro et al., 2015]) shows that SosA does not hinder the initiation of septum formation but blocks the progression of septum completion – a phenotype similar to S. aureus DivIB‐depleted cells (Bottomley et al., 2014). Although DivIB has been shown to be dispensable for FtsZ and EzrA localization to the septum and piecrust formation, its absence results in inhibition of septum progression and completion and in the mislocalization of GpsB (Bottomley et al., 2014), a late cell division protein (Gamba et al., 2009; Bottomley et al., 2014). However, SosA overproduction did not impair the recruitment of GpsB to the midcell (Fig. 8D) even though visible cell enlargement occurred in this reporter strain (Fig. 8D) and not in the vector control (Fig. S4D). Altogether, these results indicate that SosA acts on (a) later cell division component(s) than FtsZ, EzrA and GpsB, and we conclude that SosA causes a characteristic halt in cell division by preventing progression through the morphological division stage P2, i.e. septum plate completion.
Discussion
Based on the results reported here, we propose the following model for regulation of cell division in the prolate spheroid bacterium S. aureus under DNA‐damaging conditions (Fig. 9). During normal growth, the expression of sosA and the entire SOS regulon is repressed by the LexA repressor. Under DNA‐damaging and SOS‐inducing conditions, LexA is inactivated through autocleavage, which results in the expression of sosA. In this process, the N‐terminal autocleavage product of LexA needs to be further proteolytically processed for sosA expression to be fully induced (Cohn et al., 2011), thus adding another layer of expressional control, suggesting tight regulation of the inhibitory protein. Upon derepression of LexA‐regulated genes, SosA accumulates to levels that inhibit cell division without causing FtsZ delocalization nor preventing the assembly of downstream components, EzrA and GpsB, which are representatives of early and later cell division components respectively. The inhibitor appears to affect the progression and completion of septum formation; consequently, the cells appear synchronized at the stage of septum formation initiation, while an ongoing off‐septal peptidoglycan synthesis leads to cell enlargement (Fig. S4C). Once conditions are favorable for continued division, sosA expression is repressed, the membrane protease CtpA degrades SosA and cell division resumes (Fig. 9).
Figure 9.
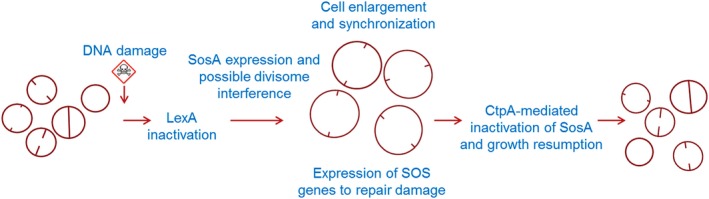
Proposed model for a regulated survival strategy for S. aureus upon DNA damage. As part of the SOS response, SosA is produced and, by a membrane‐localized activity, corrupts normal cell division activity, likely via an interaction with essential divisome components. In effect, cells are still able to initiate septum formation but are unable to complete it, leading to synchronization of the cell population. This provides a spatio‐temporal window for paralleled SOS response‐regulated DNA repair activity. At the same time, the cell size increases due to off‐septal activity of peptidoglycan‐synthesizing enzymes. As the SOS response diminishes, the cellular concentration of SosA is lowered, directly or indirectly, by the proteolytic activity of CtpA. At this stage, septa are allowed to complete and normal growth/division continues.
The model illuminates several remaining unanswered questions. It remains to be shown at which point and how SosA affects divisome activity. Presently, no direct interaction has been observed between the Gram‐positive cell division inhibitors and FtsZ, the scaffold for cell division components assembly at the division site. Fluorescence microscopy suggests that also SosA does not act at this early stage (Fig. 8). Though poorly characterized mechanistically, the Gram‐positive inhibitors may suppress cell division at later stages, potentially by interacting with FtsI/FtsQ (for M. tuberculosis ChiZ) as suggested by bacterial two‐hybrid analysis (Vadrevu et al., 2011) or via a pathway causing delocalization of FtsL and/or DivIC (for B. subtilis YneA) which was only suggested by indirect experimental data (Kawai and Ogasawara, 2006). These cell division inhibitors are characterized by the presence of a LysM peptidoglycan binding domain and for ChiZ also peptidoglycan hydrolase activity was suggested (Chauhan et al., 2006). Such characteristics could provide important clues to their division inhibitory activity. Somewhat uniquely, however, a LysM domain appears to be absent from SosA. Another distinguishing feature of SosA is that FtsZ shows preferential septal localization upon expression (as a likely cause of synchronization), whereas for B. subtilis YneA, M. tuberculosis ChiZ and also C. glutamicum DivS, FtsZ ring formation was reported to be compromised upon expression of the respective cell division inhibitors (Kawai et al., 2003; Chauhan et al., 2006; Ogino et al., 2008; Vadrevu et al., 2011). Our preliminary characterization by two‐hybrid analysis suggests that SosA is capable of interacting with several essential S. aureus cell division components (data not shown), which is a subject for future experimental verification. Our microscopy and localization data suggest that SosA acts at a decision point between septum initiation and septum completion. In this regard, it is interesting that cell division in S. aureus was recently reported to be a two‐step process characterized by an initial, slow FtsZ‐dependent activity followed by a faster process when the lipid II flippase, MurJ, arrives and directs peptidoglycan synthesis to the septum (in division stage P2 where SosA expressing cells are stalled) (Monteiro et al., 2018). More intriguingly, MurJ recruitment in S. aureus was reported to be mediated by the DivIB–FtsL–DivIC complex (Monteiro et al., 2018), and this complex may serve as a central regulatory hub in bacterial cell division (den Blaauwen and Luirink, 2019). We hypothesize that this divisional stage could constitute an important molecular cue for the inhibitory activity of SosA, while we keep in mind that the inhibitory activity of SosA could be multifactorial and further studies are required to delineate the exact function of the protein.
Analysis of the amino acid sequence of SosA indicated that the N‐terminal part contains a TM domain, while the C‐terminal is located extracellularly. The hypothesis that the TM domain promotes localization to the cellular membrane was supported by PhoA‐LacZ fusion to SosA and by various point and deletion mutants of the fusion protein (Fig. 4). The abrogation of activity by the 40‐AA truncation as well as by a single alanine substitution (D44A) suggests that this segment is essential for correct localization to the septum or, perhaps more interestingly, that the actual motif for cell division inhibition is localized at the exterior of the membrane. It should be noted that the PhoA–LacZ assay monitors protein translocation in E. coli and at present we cannot exclude that the (D44A) point mutation affects membrane translocation or stability within the membrane in S. aureus. Future work should reveal the extent to which this highly conserved residue is mechanistically important for SosA activity. If divisome interference indeed lies within this extracellular segment of SosA, it could provide a novel paradigm for interference with cell division beyond the transpeptidase‐inhibitory activity exerted by β‐lactam antibiotics.
Endogenously triggered cell division inhibition must be strictly regulated, as demonstrated by pioneering work showing that SulA of E. coli is degraded by the Lon protease (Mizusawa and Gottesman, 1983). Similarly, YneA from B. subtilis was originally reported to be regulated by proteolysis, although by an undefined mechanism, and a point mutation at its extreme C‐terminus generated a stabilized yet functional variant of the cell division inhibitor (Mo and Burkholder, 2010). In B. megaterium, the temporal expression of a YneA homologue has been suggested to be due to mRNA instability (Buchholz et al., 2013). We demonstrate that a minor truncation of the C‐terminus of SosA increases cell division inhibitory activity (Fig. 5), possibly as a consequence of accumulation of the protein (Fig. S2E). This led us to speculate that the extreme C‐terminus serves as a signal for proteolysis and we identified the CtpA protease‐deficient strain in which SosA appeared to accumulate and had an increased detrimental effect compared to the WT background. Bacterial carboxy‐terminal proteases are poorly characterized, particularly with respect to their substrate preferences; however, we propose that CtpA specifically regulates SOS‐induced cell division inhibition in staphylococci, likely via recognition of the C‐terminus of SosA. In support of this, Burby et al. (2018) recently disclosed that CtpA is involved in turnover of YneA in B. subtilis. Of note, carboxy‐terminal proteases are ubiquitous in bacterial genera, and homologs of CtpA are found in other Gram‐positive species, including also L. monocytogenes which share a homolog of YneA. Hence, the CtpA‐like proteases could represent functional homologs of the E. coli Lon protease, allowing negative regulation of membrane‐localized endogenous cell division inhibition more broadly among Firmicutes and beyond.
SosA may also prove to be a useful tool for scientists conducting studies of bacterial cell division, which have recently benefitted from the development of genetic tools and highly sophisticated microscopy imaging technologies. Most often bacteria grow as an unsynchronized population with individual cells in different phases of cell division, making it challenging to order the temporal events that occur during cell cycle progression, DNA replication and division. SosA expression is halting the S. aureus cell cycle at a specific morphological stage, e.g. following the piecrust formation, and we anticipate that the exploitation of this function may constitute a useful tool for the generation of synchronized staphylococcal cells for experimental purposes.
Apart from its basic biological interest, the bacterial cell division process may constitute an unexhausted source of novel therapeutic targets (Lock and Harry, 2008; Sass and Brötz‐Oesterhelt, 2013). We believe that SosA could be a natural scaffold, and that further studies on this protein can provide new therapeutically accessible targets that would interfere with an essential process in S. aureus and other staphylococci.
Experimental procedures
Bacterial strains and plasmids used in this work
The bacterial strains and plasmids used in this study are listed in Table S1. E. coli strains were grown in Luria–Bertani medium (Oxoid) or in LB agar (Oxoid). S. aureus strains were grown in Tryptic Soy Broth (TSB) or in Tryptic Soy Agar (TSA) (both Oxoid). Ampicillin (100 µg ml−1), chloramphenicol (10 µg ml−1) or erythromycin (5 µg ml−1) were added when appropriate. Anhydrotetracycline (AHT) (Sigma‐Aldrich) and Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) (Thermo Scientific) were used as inducers for protein expression.
Construction of strains and plasmids
All oligonucleotides used are listed in Table S1. S. aureus 8325‐4ΔsosA is a clean deletion of sosA (SAOUHSC_01334) in strain 8325‐4 obtained by allelic replacement using the temperature‐sensitive shuttle vector pIMAY (Monk et al., 2012). The 1 kb regions up‐ and downstream of sosA were PCR amplified (Phusion Hot Start II DNA Polymerase, Thermo Scientific) using primer pairs Up‐sosA_fw‐KpnI/Up‐sosA_rev and Dw‐sosA_fw/Dw‐sosA_rev‐SacI, respectively, and subsequently joined in a spliced overhang PCR using Up‐sosA_fw‐KpnI/Dw‐sosA_rev‐SacI. The resulting deletion fragment was cloned into pIMAY via KpnI/SacI, generating pIMAY‐ΔsosA, purified from E. coli DC10B and transformed into S. aureus subsequently maintained at 28°C. Chromosomal integration of the plasmid was performed at 37°C under selective conditions (chloramphenicol) followed by passage at 28°C without antibiotic selection and final plating on TSA containing 500 ng ml−1 of AHT for plasmid counterselection. Colonies were replica‐plated to select for sensitivity toward chloramphenicol and successful allelic exchange were screened for by PCR amplification using primer pairs Ctrl_dsosA_F/Ctrl_dsosA_R positioned outside sosA and Fwd_MCS/Rev_MCS targeting the vector respectively. The allelic replacement procedure was identical for S. aureus JE2 to create JE2ΔsosA.
The chromosomal transposon insertion mutation in ctpA conferring erythromycin resistance was obtained from the Nebraska Transposon Mutant Library (NTML, NE847) (Fey et al., 2013) and was moved by transduction (phage Φ11) to S. aureus JE2, JE2ΔsosA, S. aureus 8325‐4 and 8325‐4ΔsosA, resulting in JE2‐ctpA, JE2ΔsosA‐ctpA, 8325‐4‐ctpA and 8325‐4ΔsosA‐ctpA respectively. JE2‐ctpA(−erm), an erythromycin‐sensitive derivative, was generated by elimination of the transposon‐encoded ermB in JE2‐ctpA by allelic exchange using the pTnT vector using temperature‐mediated chromosomal integration and secY‐mediated counterselection as described before (Bose et al., 2013).
All plasmid constructs mentioned below were cloned in E. coli IM08B (Monk et al., 2015) from where they were transformed into S. aureus strains. For complementation of the sosA knockout, the sosA gene with its native promoter was PCR amplified from strain 8325‐4 using primers Up‐sosA‐promo_SalI and Dw‐sosA_EcoRI and cloned into designated restriction sites in plasmid pJC1112 (Chen et al., 2014). The resulting plasmid (pJC1112‐sosA) was transformed into the integration‐proficient strain RN9011, creating RN9011‐sosA‐compl. when selected with erythromycin. The chromosomally integrated plasmid was transduced into 8325‐4ΔsosA, resulting in 8325‐4ΔsosA‐compl. For sosA expression the sosA gene including its predicted ribosomal binding site was cloned into the BglII and EcoRI sites of pRAB12‐lacZ (Helle et al., 2011) using primers Up‐sosA_BglII/Dw‐sosA_EcoRI, at the same time eliminating the lacZ reporter gene. The resulting plasmid was called pSosA. Generation of plasmids encoding C‐terminally truncated variants of SosA was obtained by PCR amplification of DNA fragments using Up‐sosA_BglII and downstream primers Dw‐sosA(d10)_EcoRI to Dw‐sosA(d40)_EcoRI, all equipped with premature stop codons. The PCR products were ligated into pRAB12‐lacZ using BglII and EcoRI cut sites to create pSosAd10, pSosAd20, pSosAd30 and pSosAd40. Single and double amino acid substitutions (alanine) in the SosAd10 protein were obtained by cloning commercially synthesized DNA fragments (Twist Bioscience) into pRAB12‐lacZ using the same restriction sites. The mutation at AA position 44 (D to A) was introduced to the full length SosA protein via extension PCR by first using primers Up‐sosA_BglII/SosA_R‐long on the SosAd10(44A) template, then using that product as template for amplification with primers Up‐sosA_BglII/Dw‐sosA_EcoRI followed by cloning into pRAB12‐lacZ, thereby generating pSosA(44A). Plasmid pCtpA was constructed by cloning the ctpA gene behind the Pspac promoter in pSK9067 (Brzoska and Firth, 2013) using primer pair ctpA_F‐SalI/ctpA_R‐EcoRI. For membrane topology analysis using pKTop (Karimova and Ladant, 2017), sosA, sosAd10, sosAd10(44A) and sosAd40 were PCR amplified from respective expression constructs using primer SosA_F‐BamHI and primers SosA_R‐KpnI, SosAd10_R‐KpnI or SosAd40_R‐KpnI, respectively, and cloned in frame in front of the phoA‐lacZ chimeric gene using BamHI/KpnI restriction sites.
In order to construct JE2 strains producing a fluorescent fusion of FtsZ, EzrA or GpsB in the presence of the sosA expression plasmid, JE2 pSosA was transduced with a lysate from SH4665 (SH1000 pCQ11‐FtsZ‐eYFP), JGL227 (SH1000 ezrA‐gfp+) or JGL228 (SH1000 gpsB‐gfp+), resulting in SJF4694 (JE2 pRAB12‐lacZ pCQ11‐FtsZ‐eYFP), SJF4696 (JE2 pRAB12‐lacZ ezrA‐gfp+) and SJF4699 (JE2 pRAB12‐lacZ gpsB ‐gfp+) respectively. Control strains SJF4693 (JE2 pRAB12‐lacZ pCQ11‐FtsZ‐eYFP), SJF4696 (JE2 pRAB12‐lacZ ezrA‐gfp+) and SJF4699 (JE2 pRAB12‐lacZ gpsB‐gfp+) were constructed by a phage transduction of JE2 pRAB12‐lacZ with lysates from SH4665 (SH1000 pCQ11‐FtsZ‐eYFP), JGL227 (SH1000 ezrA‐gfp+) or JGL228 (SH1000 gpsB‐gfp+) respectively. To induce FtsZ‐eYFP production in SJF4693 and SJF4694, cells were grown in the presence of 50 μM IPTG.
Determination of OD600 and CFU after MMC treatment
Strains were grown overnight on TSA plates and used for inoculating TSB and allowed to grow to OD600 = 0.05 when Mitomycin C (MMC from Streptomyces caespitosus, Sigma‐Aldrich) was added. Cell density was monitored by OD600 measurements at intervals onward and samples were withdrawn to determine culture colony forming units by serial dilution in 0.9% w/v NaCl and plating on TSA.
Flow cytometry
Cell size distributions of cultures were arbitrarily quantified by flow cytometry using the forward scatter signal (FSC‐A) acquired on a BD Accuri™ C6 flow cytometer (BD Biosciences). Cell samples were diluted in 0.9% w/v NaCl to an approximate density of 106 cells ml−1 and sonicated briefly to allow acquisition of scatter signal from single cells predominantly. Sonication was performed with a Bandelin sonopuls HD2070/UW2070 apparatus (Bandelin electronics, Germany) fitted with the MS 73 probe. Ten pulses of 500 ms were given at 50% power. All flow cytometry experiments were independently repeated at least twice with similar results.
Plating assays
Spot dilution was used to evaluate plating efficiency of strains carrying various plasmid constructs. Strains were grown exponentially until an approximate OD600 of 0.5 under plasmid‐selective conditions. Strains were then 10‐fold serially diluted in 0.9% w/v NaCl and positioned as 10 µl spots on TSA containing selective antibiotics with/without inducer at indicated concentrations and incubated at 37°C overnight. All spot plating assays were independently repeated with similar results.
Labeling S. aureus with HADA
Cells grown to mid‐exponential phase (OD600 ~ 0.5) were incubated with 500 µM HADA at 37°C for 5 min. Cells were then washed by centrifugation and resuspension in PBS.
Labeling S. aureus with NHS ester
Labeling with NHS ester was performed as described before (Lund et al., 2018). Briefly, cells grown to mid‐exponential phase (OD600 ~ 0.5) were collected by centrifugation and growth medium was discarded. Cells were resuspended in PBS containing 8 μg ml−1 Alexa Fluor 647 NHS ester (Invitrogen) and incubated at room temperature for 5 min. Cells were washed by centrifugation and resuspension in PBS.
Fixing
Cells were fixed by incubation in 4% (w/v) paraformaldehyde at room temperature for 30 min.
Widefield epifluorescence microscopy
Fixed cells were dried onto a poly‐L‐Lysine coated slide and mounted in PBS. Imaging was performed using either a Nikon Ti Inverted microscope fitted with a Lumencor Spectra X light engine or a v4 DeltaVision OMX 3D‐SIM system (Applied Precision, GE Healthcare, Issaquah, USA).
3D‐structured illumination microscopy
A high‐precision cover slip (High‐precision, No. 1.5 H, 22 × 22 mm, 170 ± 5 µm, Marienfeld) was cleaned by sonicating in 1 M KOH for 15 min at room temperature. The coverslip was washed with water and incubated in 0.01% (w/v) poly‐L‐lysine solution (Sigma) for 30 min at room temperature. The coverslip was rinsed with water and dried with nitrogen. Fixed cells were dried onto the poly‐L‐Lysine coated cover slip and mounted on a slide with SlowFade Diamond (Invitrogen). 3D SIM visualization was performed using a v4 DeltaVision OMX 3D‐SIM system (Applied Precision, GE Healthcare, Issaquah, USA) equipped with a Plan Apo 60x, 1.42 NA oil objective, using 1.514 immersion oil, a 642 nm laser and a standard excitation/emission filter set (683/40). For each z‐section, a sample was imaged in five phase shifts and three angles. The z‐sections were 0.125 nm in depth. Raw data were reconstructed with the Softworx software (GE Healthcare) using OTFs optimized for the specific wavelength and oil used.
Time‐lapse microscopy
Strains were grown overnight in TSB medium at 37°C, then diluted 100 times in fresh TSB and grown until OD = 0.1. Cells were washed once with fresh TSB and spotted onto TSB–acrylamide (10%) pads previously incubated for 2 h in TSB medium supplemented, when appropriate, with 0.04 µg ml−1 mitomycin C (MMC) or 200 ng ml−1 anhydrotetracycline (AHT). Pads were placed into a Gene frame (Thermo Fisher Scientific) and sealed with a cover glass. Phase‐contrast images were acquired on a DV Elite microscope (GE healthcare) equipped with a sCMOS (PCO) camera and a 100x oil immersion objective. Images were acquired with 200 ms exposure time every 4 min for at least 6 h at 37°C using Softworx (Applied Precision) software. Images were analyzed using Fiji (http://fiji.sc).
Western blot analysis
Purified rabbit anti‐SosA antibody was obtained via GenScript Biotech (Netherlands). A custom peptide, CEHKDPTNNSEQRDGA, comprising AAs 57–70 of SosA was used for immunization. Bacterial cells were pelleted and frozen immediately at −80°C before being resuspended in PBS containing cOmplete™ Mini Protease Inhibitor Cocktail (Sigma‐Aldrich) and lysed by bead beating (Fastprep‐24™, MP Biomedicals). The protein concentration of the lysates was measured using the Qubit™ Protein Assay Kit (ThermoFisher Scientific) and samples were normalized to equal amounts of protein, 20 µg of total protein was separated on NuPAGE® 4‐12% Bis‐Tris gels using MES buffer and the XCell sure‐lock mini‐cell system (ThermoFisher Scientific), transferred to a polyvinylidene difluoride membrane (PVDF) and probed with primary antibody diluted 1:1000. Bound antibody was detected with the WesternBreeze® Chemiluminescent Kit, anti‐rabbit according to the instructions from the manufacturer (ThermoFisher Scientific). All western blots were independently repeated with similar results. Uncropped versions of blots are displayed in Fig S5.
Author contributions
M.S.B., K.W., P.K., M.T.C., S.J.F. and H.I. conceived and designed the study. Experiments were performed by M.S.B., K.W., C.G. and A.L.B. M.S.B. K.W., C.G., G.L., D.F., J.‐W.V., S.J.F. and H.I. contributed to analysis of data and drafting of the manuscript. All authors read and approved the final manuscript.
Supporting information
Acknowledgements
This work was supported by grants from the Danish Council for Independent Research – Technology and Production (1337‐00129 and 1335‐00772) to M.S.B., by the Danish National Research Foundation (DNRF120) to H.I. and by the Medical Research Council (MR/K015753/1 to the Wolfson Light Microscopy Facility at the University of Sheffield and MR/N002679/1). We would like to thank Prof Simon Jones (University of Sheffield) for the HADA dye.
References
- Adams, D.W. and Errington, J. (2009) Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nature Reviews Microbiology, 7, 642–653. [DOI] [PubMed] [Google Scholar]
- Anderson, K.L. , Roberts, C. , Disz, T. , Vonstein, V. , Hwang, K. , Overbeek, R. , et al. (2006) Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log‐phase mRNA turnover. Journal of Bacteriology, 188, 6739–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu, Z. and Mazel, D. (2014) SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiology Reviews, 38, 1126–1145. [DOI] [PubMed] [Google Scholar]
- den Blaauwen, T. and Luirink, J. (2019) Checks and balances in bacterial cell division. MBio, 10, e00149–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Blaauwen, T. , Hamoen, L.W. and Levin, P.A. (2017) The divisome at 25: the road ahead. Current Opinion in Microbiology, 36, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, J.L. , Fey, P.D. and Bayles, K.W. (2013) Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus . Applied and Environmental Microbiology, 79, 2218–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley, A.L. , Kabli, A.F. , Hurd, A.F. , Turner, R.D. , Garcia‐Lara, J. and Foster, S.J. (2014) Staphylococcus aureus DivIB is a peptidoglycan‐binding protein that is required for a morphological checkpoint in cell division. Molecular Microbiology, 94, 1041–1064. [DOI] [PubMed] [Google Scholar]
- Brzoska, A.J. and Firth, N. (2013) Two‐plasmid vector system for independently controlled expression of green and red fluorescent fusion proteins in Staphylococcus aureus . Applied and Environmental Microbiology, 79, 3133–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz, M. , Nahrstedt, H. , Pillukat, M.H. , Deppe, V. and Meinhardt, F. (2013) yneA mRNA instability is involved in temporary inhibition of cell division during the SOS response of Bacillus megaterium . Microbiology, 159, 1564–1574. [DOI] [PubMed] [Google Scholar]
- Buist, G. , Steen, A. , Kok, J. and Kuipers, O.P. (2008) LysM, a widely distributed protein motif for binding to (peptido)glycans. Molecular Microbiology, 68, 838–847. [DOI] [PubMed] [Google Scholar]
- Burby, P.E. , Simmons, Z.W. , Schroeder, J.W. and Simmons, L.A. (2018) Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery. PLoS Genetics, 14, e1007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, R.K. , Rivera, F.E. , Cavaco, C.K. , Johnson, G.M. , Martin, D. and Shaw, L.N. (2014) The lone S41 family C‐terminal processing protease in Staphylococcus aureus is localized to the cell wall and contributes to virulence. Microbiology, 160, 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, A. , Lofton, H. , Maloney, E. , Moore, J. , Fol, M. , Madiraju, M.V. and Rajagopalan, M. (2006) Interference of Mycobacterium tuberculosis cell division by Rv2719c, a cell wall hydrolase. Molecular Microbiology, 62, 132–147. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Yoong, P. , Ram, G. , Torres, V.J. and Novick, R.P. (2014) Single‐copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus . Plasmid, 76, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz, R.T. , Jones, M.B. , Gingles, N.A. , Minogue, T.D. , Jarrahi, B. , Peterson, S.N. and Romesberg, F.E. (2007) Complete and SOS‐mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. Journal of Bacteriology, 189, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, M.T. , Kjelgaard, P. , Frees, D. , Penadés, J.R. and Ingmer, H. (2011) Clp‐dependent proteolysis of the LexA N‐terminal domain in Staphylococcus aureus . Microbiology, 157, 677–684. [DOI] [PubMed] [Google Scholar]
- Cordell, S.C. , Robinson, E.J. and Lowe, J. (2003) Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proceedings of the National Academy of Sciences, 100, 7889–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey, P.D. , Endres, J.L. , Yajjala, V.K. , Widhelm, T.J. , Boissy, R.J. , Bose, J.L. , et al. (2013) A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio, 4, e00537‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba, P. , Veening, J.W. , Saunders, N.J. , Hamoen, L.W. and Daniel, R.A. (2009) Two‐step assembly dynamics of the Bacillus subtilis divisome. Journal of Bacteriology, 191, 4186–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helle, L. , Kull, M. , Mayer, S. , Marincola, G. , Zelder, M.E. , Goerke, C. , et al. (2011) Vectors for improved Tet repressor‐dependent gradual gene induction or silencing in Staphylococcus aureus . Microbiology, 157, 3314–3323. [DOI] [PubMed] [Google Scholar]
- Higashitani, A. , Higashitani, N. and Horiuchi, K. (1995) A cell division inhibitor SulA of Escherichia coli directly interacts with FtsZ through GTP hydrolysis. Biochemical and Biophysical Research Communications, 209, 198–204. [DOI] [PubMed] [Google Scholar]
- Huisman, O. and D'Ari, R. (1981) An inducible DNA replication‐cell division coupling mechanism in E. coli . Nature, 290, 797–799. [DOI] [PubMed] [Google Scholar]
- Huisman, O. , D'Ari, R. and Gottesman, S. (1984) Cell‐division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proceedings of the National Academy of Sciences, 81, 4490–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, C. and Holland, I.B. (1985) Role of the SulB (FtsZ) protein in division inhibition during the SOS response in Escherichia coli: FtsZ stabilizes the inhibitor SulA in maxicells. Proceedings of the National Academy of Sciences, 82, 6045–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova, G. and Ladant, D. (2017) Defining membrane protein topology using pho‐lac reporter fusions. Methods in Molecular Biology, 1615, 129–142. [DOI] [PubMed] [Google Scholar]
- Kawai, Y. and Ogasawara, N. (2006) Bacillus subtilis EzrA and FtsL synergistically regulate FtsZ ring dynamics during cell division. Microbiology, 152, 1129–1141. [DOI] [PubMed] [Google Scholar]
- Kawai, Y. , Moriya, S. and Ogasawara, N. (2003) Identification of a protein, YneA, responsible for cell division suppression during the SOS response in Bacillus subtilis . Molecular Microbiology, 47, 1113–1122. [DOI] [PubMed] [Google Scholar]
- Kelley, W.L. (2006) Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Molecular Microbiology, 62, 1228–1238. [DOI] [PubMed] [Google Scholar]
- Kreuzer, K.N. (2013) DNA damage responses in prokaryotes: regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harbor Perspectives in Biology, 5, a012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock, R.L. and Harry, E.J. (2008) Cell‐division inhibitors: new insights for future antibiotics. Nature Reviews Drug Discovery, 7, 324–338. [DOI] [PubMed] [Google Scholar]
- Lund, V.A. , Wacnik, K. , Turner, R.D. , Cotterell, B.E. , Walther, C.G. , Fenn, S.J. , et al. (2018) Molecular coordination of Staphylococcus aureus cell division. Elife, 7, e32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesak, L.R. , Miao, V. and Davies, J. (2008) Effects of subinhibitory concentrations of antibiotics on SOS and DNA repair gene expression in Staphylococcus aureus . Antimicrobial Agents and Chemotherapy, 52, 3394–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnage, S. , Dellarole, M. , Baxter, N.J. , Rouget, J.B. , Dimitrov, J.D. , Wang, N. , et al. (2014) Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nature Communications, 5, 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusawa, S. and Gottesman, S. (1983) Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proceedings of the National Academy of Sciences, 80, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo, A.H. and Burkholder, W.F. (2010) YneA, an SOS‐induced inhibitor of cell division in Bacillus subtilis, is regulated posttranslationally and requires the transmembrane region for activity. Journal of Bacteriology, 192, 3159–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell, J.W. , Hopkins, A.C. and Laub, M.T. (2011) A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes & Development, 25, 1328–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell, J.W. , Kambara, T.K. , Perchuk, B.S. and Laub, M.T. (2014) A DNA damage‐induced, SOS‐independent checkpoint regulates cell division in Caulobacter crescentus . PLoS Biology, 12, e1001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk, I.R. , Shah, I.M. , Xu, M. , Tan, M.W. and Foster, T.J. (2012) Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis . MBio, 3, e00277‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk, I.R. , Tree, J.J. , Howden, B.P. , Stinear, T.P. and Foster, T.J. (2015) Complete bypass of restriction systems for major Staphylococcus aureus lineages. MBio, 6, e00308‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro, J.M. , Fernandes, P.B. , Vaz, F. , Pereira, A.R. , Tavares, A.C. , Ferreira, M.T. , et al. (2015) Cell shape dynamics during the staphylococcal cell cycle Nature . Communications, 6, 8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro, J.M. , Pereira, A.R. , Reichmann, N.T. , Saraiva, B.M. , Fernandes, P.B. , Veiga, H. , et al. (2018) Peptidoglycan synthesis drives an FtsZ‐treadmilling‐independent step of cytokinesis. Nature, 554, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, A. , Cao, C. and Lutkenhaus, J. (1998) Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli . Proceedings of the National Academy of Sciences, 95, 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino, H. , Teramoto, H. , Inui, M. and Yukawa, H. (2008) DivS, a novel SOS‐inducible cell‐division suppressor in Corynebacterium glutamicum . Molecular Microbiology, 67, 597–608. [DOI] [PubMed] [Google Scholar]
- Pinho, M.G. , Kjos, M. and Veening, J.W. (2013) How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nature Reviews Microbiology, 11, 601–614. [DOI] [PubMed] [Google Scholar]
- Robinson, A. , Causer, R.J. and Dixon, N.E. (2012) Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Current Drug Targets, 13, 352–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass, P. and Brötz‐Oesterhelt, H. (2013) Bacterial cell division as a target for new antibiotics. Current Opinion in Microbiology, 16, 522–530. [DOI] [PubMed] [Google Scholar]
- Schoemaker, J.M. , Gayda, R.C. and Markovitz, A. (1984) Regulation of cell division in Escherichia coli: SOS induction and cellular location of the sulA protein, a key to lon‐associated filamentation and death. Journal of Bacteriology, 158, 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele, V.R. , Bottomley, A.L. , Garcia‐Lara, J. , Kasturiarachchi, J. and Foster, S.J. (2011) Multiple essential roles for EzrA in cell division of Staphylococcus aureus . Molecular Microbiology, 80, 542–555. [DOI] [PubMed] [Google Scholar]
- Tsirigos, K.D. , Peters, C. , Shu, N. , Käll, L. and Elofsson, A. (2015) The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Research, 43, W401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, R.D. , Ratcliffe, E.C. , Wheeler, R. , Golestanian, R. , Hobbs, J.K. and Foster, S.J. (2010) Peptidoglycan architecture can specify division planes in Staphylococcus aureus . Nature Communications, 1, 26. [DOI] [PubMed] [Google Scholar]
- Tzagoloff, H. and Novick, R. (1977) Geometry of cell division in Staphylococcus aureus . Journal of Bacteriology, 129, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadrevu, I.S. , Lofton, H. , Sarva, K. , Blasczyk, E. , Plocinska, R. , Chinnaswamy, J. , et al. (2011) ChiZ levels modulate cell division process in mycobacteria. Tuberculosis (Edinb), 91, S128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen, S. , Hain, T. , Wouters, J.A. , Hossain, H. , de Vos, W.M. , Abee, T. , et al. (2007) The heat‐shock response of Listeria monocytogenes comprises genes involved in heat shock, cell division, cell wall synthesis, and the SOS response. Microbiology, 153, 3593–3607. [DOI] [PubMed] [Google Scholar]
- van der Veen, S. , van Schalkwijk, S. , Molenaar, D. , de Vos, W.M. , Abee, T. and Wells‐Bennik, M.H. (2010) The SOS response of Listeria monocytogenes is involved in stress resistance and mutagenesis. Microbiology, 156, 374–384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials