

Abstract
Malignant tumours show a marked degree of morphological, molecular and proteomic heterogeneity. This variability is closely related to microenvironmental factors and the location of the tumour. The activation of genetic alterations is very tissue‐dependent and only few tumours have distinct genetic alterations. Importantly, the activation state of proteins and signaling factors is heterogeneous in the primary tumour and in metastases and recurrences. The molecular diagnosis based only on genetic alterations can lead to treatments with unpredictable responses, depending on the tumour location, such as the tumour response in melanomas versus colon carcinomas with BRAF mutations. Therefore, we understand that the correct evaluation of tumours requires a system that integrates both morphological, molecular and protein information in a clinical and pathological context, where intratumoral heterogeneity can be assessed. Thus, we propose the term ‘tissunomics’, where the diagnosis will be contextualised in each tumour based on the complementation of the pathological, molecular, protein expression, environmental cells and clinical data.
Keywords: genomics, histopathology, proteomics, tumour heterogeneity
General Background of Tumour Pathology
Despite constant progress in cancer research, mortality rates remain high.1, 2 Five‐year survival is greater than 80% for carcinoma of the breast, prostate and bladder, cutaneous melanoma and testicular and thyroid tumours, but less than 10% in pancreatic, liver, oesophageal, lung and stomach tumours and glioblastoma. If we focus on tumour stage, we observe that in luminal breast cancer, expected survival is greater than 90% in patients without metastases at diagnosis and less than 25% in patients with disseminated disease.3 With this in mind, early detection, histological typing and extent of dissemination are critical for treatment success. Furthermore, the more than 250 tumour types and hundreds of subtypes, on a background of thousands of genetic, epigenetic and microRNA alterations,4, 5, 6 highlight the importance of precise tumour sample evaluation in deciding the most appropriate clinical treatment.
Inter‐ and intratumour heterogeneity (ITH)7, 8 act as a critical barrier to improving therapeutics: most of the targets and biomarkers approved by the United States Food and Drug Administration are not expressed uniformly throughout tumour tissue; for example, the human epidermal growth factor receptor 2 (HER2) in gastric adenocarcinoma,9, 10 progesterone and oestrogen receptors in breast tumours (considered positive when detected in 1% of tumour cells)11 and EML4–ALK translocation in lung adenocarcinoma (threshold of 15%),12 among others.13, 14
Current Situation and Challenges
Why Are Malignant Tumours Heterogeneous?
The paradigm of cancer as a genetic disease has existed since the early 1970s. For the last 50 years, cancer has been explained according to the somatic mutation theory (SMT),15 which states that genetic mutations accumulate in cells by inducing clonal expansion. Following Darwinian‐type evolution, an initial clone accumulates multiple mutations, resulting in malignant transformation characterised by growth independence and resistance to apoptosis. These clones have trunk and branch mutations, and may also have driver and passenger mutations.16, 17, 18 During tumour progression and after chemotherapy or radiotherapy, genetic alterations accumulate to select for the best‐adapted clone. Molecular heterogeneity has been described in renal adenocarcinoma, breast cancer and glioblastoma, among others.19 It is thought that these clones with greater proliferative advantages and resistance to local stresses then predominate.20, 21, 22, 23, 24
Other theories include the tissue organisation field theory (TOFT), which postulates that neoplasia arises from a local tissue disorder and may be due to a defect in interaction between cells and other tissue components,15, 25, 26, 27 where epigenetic or genetic changes affect both epithelial and stromal cells in multiple locations. Grouping together the evidence for these theories, it has been demonstrated that (i) many spontaneous somatic mutations accumulate in cells during their lifetime, (ii) oncogenes and tumour suppressor genes play a critical role in cancer development and (iii) environmental factors trigger cancer onset and progression.15 Regarding field cancerisation, some authors have proposed that cancer is a tissue disorder with similarities to abnormal embryonic development,15, 25 while others propose that cancer is a consortium of clones and local factors.28 It is understood that a single clone cannot harbour all the genetic alterations required for the invasive and metastatic tumour phenotype and that several clones acquire malignant properties that, synergistically, allow cells to grow, invade and metastasise. This consortium of tumour cell clones and microenvironment cells includes leucocytes, lymphocytes, fibroblasts and endothelial cells. A minimum number of cells is also required for a clone to be biologically viable – a phenomenon known as the Allee effect. This clonal cooperation has been proposed in pancreatic and breast cancer and melanoma and for circulating tumour cell (CTC) clusters.29, 30, 31, 32, 33
Some Oncogenic Alterations Are Tissue‐Specific
Certain genetic alterations are only observed in the context of specific cell or tissue types;34 for example, alterations in the APC gene in colorectal cancer, CDH1 (E‐cadherin) in gastric carcinoma, the VHL gene in renal carcinoma or BCR/ABL in leukaemia.35, 36 These specific genetic alterations demonstrate that tissue type plays a role, which is currently not well understood and is under intensive investigation. The higher incidence of certain somatic mutations in some cell types might be explained by a cell‐ or tissue‐specific chromosome disposition during interphase. This, along with epigenetic imprinting, may serve as a mechanism of gene regulation and allow differential access of carcinogens to certain DNA regions, resulting in a higher probability of mutations and specific rearrangements in some cells but not others. Similarly, the accessibility of certain chromosomal regions might explain cell‐type‐specific chromosomal translocations such as BCR/ABL in leucocytes or VHL mutations in renal cell carcinoma.37, 38 Some mutations and genetic alterations are uniformly present in the entire organism, such as germline mutations, BRCA1 and BRCA2, VHL in Von Hippel Lindau syndrome and as many as 30 others, but they are usually only associated with tumours in specific tissues, such as BRCA1/2 in breast and ovarian tumours.39 This clearly indicates that driver, trigger or passenger mutations only exert an effect in certain tissues.
Some Histopathological Features Suggest Oncogenic Alterations
The specific morphological patterns observed in human tumours have helped to identify distinct genetic changes: for example, characteristic chromosomal translocations have been identified in desmoplastic round‐cell tumours, clear‐cell sarcoma, synovial sarcoma and rhabdoid tumours.40, 41, 42 Moreover, a simple haematoxylin and eosin stain can indicate the presence of a number of hereditary tumours with specific genetic alterations, such as medullary thyroid carcinoma, multiple endocrine neoplasia (MEN),43, 44 the cribriform‐morular variant of papillary thyroid carcinoma,45 familial adenomatous polyposis,46 medullary breast carcinoma, familial breast cancer associated with specific BRCA1 mutations,47 renal carcinoma, associated with SDHB mutations which shows flocculent cytoplasmic vacuoles,48, 49 hereditary leiomyomatosis and renal cell cancer syndrome,50 and sebaceous lesions associated with Muir–Torre syndrome in colon cancer.51, 52 Thus, hereditary cancer predisposition syndromes can be identified by immunohistochemistry, which has emerged as a cost‐effective strategy (see Table 1).53
Table 1.
Molecular alterations detected by immunohistochemistry (IHC) analysis in tumour samples
| Molecular alterations in tumors | Principles of immunohistochemical staining in tumoral cells | Examples |
|---|---|---|
| Chromosomal translocation | Overexpression of protein encoded by one of the genes involved in fusion or chimeric protein encoded by the two genes involved in fusion. Positive immunostaining in tumour cells | BCL2 expression in follicular lymphoma |
| Cyclin D1 expression in mantle cell lymphoma | ||
| ALK expression in different tumour cells (anaplastic lymphoma, NSCLC) | ||
| NUT expression in NUT midline carcinoma | ||
| ERG expression in prostate carcinoma | ||
| ROS1 expression in NSCLC | ||
| Gene mutation | Aberrant subcellular localisation of antigen or protein | Nuclear translocation of B‐catenin in (CRC, desmoid fibromatosis, cribriform morular variant of PTC) |
| Cytoplasmic expression of nucleophosmin in AML | ||
| Cytoplasmic expression of BRCA1 in BC. | ||
| Stabilisation and strong expression of the protein encoded by the gene mutated | P53 in different tumour types (high grade serous ovary carcinoma) | |
| Mutation specific antibodies, the antibody do not recognize the WT form of the protein (gene) | IDH1 R132H expression in gliomas | |
| EGFR L858R expression in NSCLC | ||
| EGFR Del 19 expression in NSCLC | ||
| BRAF V600E expression in different tumor types (Melanoma, PTC, CRC, others.) | ||
| Presence or lack of mutation of certain genes give an overexpression of certain surrogate markers | LCC without IGH mutation is associate with overexpression of ZAP‐70 | |
| Gene deletion or loss of function | Inactivating mutation, deletion or promoter hypermethylation of gene gives a loss of protein expression of the encoded genes | Loss of E‐cadherin staining in lobular breast carcinoma |
| Loss of INI‐1 staining in rhabdoid tumor, and epithelioid sarcoma | ||
| Loss of staining for any MMR proteins (MLH1, MSH2, MSH6, PMS2) in HNPCC with MSI | ||
| Loss of staining for parafibromin in parathyroid Carcinoma | ||
| Loss of staining for Rb in spindle cell /pleomorphic lipoma | ||
| Lack of staining for SDHB in hereditary paragangliomas and gastrointestinal stromal tumor | ||
| Loss of PTEN expression in endometrial cancer with PTEN mutation and Cowden Syndrome | ||
| Gene amplification | Increase in copy number of gene gives an overexpression of the encoded protein | HER2 strong staining in breast and gastric cancer associated with HER2 amplification |
| Strong staining for MDM2 or CDK4 associates with 12q14‐15 amplification in liposarcoma or well‐differentiated osteosarcoma. | ||
| Strong MET expression in NSCLC |
Adapted from Chan et al.53 .
In contrast, other molecular alterations have been associated with tumours whose morphological characteristics are strikingly distinct, such as (i) the ETV6–NTRK3 translocation, detected in diverse tumour types including infantile fibrosarcoma, cellular mesoblastic nephroma and secretory breast carcinoma;54 (ii) translocation affecting the ALK gene, in anaplastic large‐cell lymphoma, lung adenocarcinoma, inflammatory myofibroblastic tumour and others;55 (iii) EWSR1–CREB1 translocation in clear‐cell sarcoma and angiomatoid fibrous histiocytoma;56 and (iv) BRAF mutations and translocations in benign nevi, malignant melanoma, colon adenocarcinoma, glioblastomas and pilocytic astrocytoma, as well as others.57
Thus, the association between genetic alterations and specific tumour types is not clear, but some histopathological patterns suggest certain genetic alterations and hereditary tumour syndromes.
Problems and Barriers
Bias and Limitations of Molecular Classification and Somatic Theory
Intratumour heterogeneity and pitfalls in the interpretation of oncogenic factors in small biopsies
Intratumour heterogeneity at the morphological, molecular and proteomic level is seen in most tumours. In melanoma, intratumour heterogeneity of BRAF mutations is described in more than 10% of cases11, 13, 14, 58 so, depending on the zone biopsied within a tumour, the determination of BRAF mutations may be wild‐type or mutant, with clear clinical implications (see Figure 1). This example can be extended to other targets and biomarkers, such as EGFR mutations in non‐small cell lung cancer (NSCLC),59 ALK rearrangement and others.60
Figure 1.
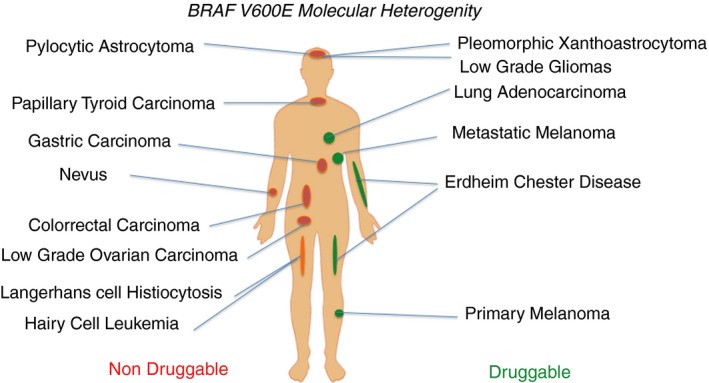
Genetic alterations such as BRAF mutations can be detected in many different tumours. Importantly, the biological meaning and the response to specific BRAF inhibitors depend on tumour type and tissue localisation.
Molecular and preneoplastic changes in the context of the adjacent tissue: field cancerisation
Until now, tumour diagnosis has focused on histological and molecular features, yet it is well known that the surrounding cells and other areas of the tissue may display premalignant histological changes such as dysplasia; furthermore, many genetic alterations occur without cytological changes. These environmental changes can affect tumour progression by exerting evolutionary pressure on tumour cells and ultimately determining which mutations are selected.27 Examples of preneoplastic histological lesions include dysplasia (cytological atypia, large hyperchromatic nuclei or nucleoli) and in‐situ carcinomas (with no basement membrane invasion), which are observed in most carcinomas. It is noteworthy that some early neoplastic lesions such as colon adenomas and skin naevi are polyclonal. Progression to cancer probably involves the accumulation of multiple field cancerisation driver mutations among synergistically acting groups of mutations.27 In this regard, molecular studies of peritumour areas in normal ducts of the breast or prostate may show a high number of genetic alterations and driver mutations.61 The clinical meaning of these molecular changes remains unknown, and further large studies are needed to establish their correlation with tumour relapse. Nonetheless, two important conclusions can be drawn: (i) the study of non‐tumour tissue can be relevant in predicting prognosis and risk of second or multiple neoplasia and (ii) we must be very cautious making a diagnosis of malignancy based only on molecular data, as not all mutations in, for example, driver genes, are directly linked to cancer development.
Mixed tumours as an example of the relevance of integrating molecular features into their morphological context
Mixed tumours provide the most extreme examples of intratumour heterogeneity. They are composed of two or more histological components, each with a different natural history, and are occasionally detected in every organ of the human body.
In most cases, the two biologically distinct tumour components are clonally related, and it is thought that the more aggressive component arises from the more indolent one and is usually responsible for the aggressiveness of the tumour. The amount of each component is also closely related to outcome.
In a small proportion of cases the two components are not clonally related: they are not true mixed tumours, but collision tumours in which two distinct neoplasms have developed independently, with a different natural history.
In a high proportion of mixed tumours, morphological appearance correlates with molecular diversity. For example, mixed endometrioid and serous carcinomas of the endometrium contain elements with the microscopic characteristics of endometrioid and serous carcinoma, and molecular analysis of microdissected tumour tissue shows that the two components have a different mutational profile. Other mixed tumours are ambiguous, and different microscopic features develop in tumour components with similar molecular alterations, probably as a result of interaction with the microenvironment.62
In some mixed tumours the microscopic diversity is extreme. This is the case in biphasic synovial sarcoma, carcinosarcomas and some mesotheliomas. In carcinosarcomas (named differently in different organs: metaplastic carcinoma, sarcomatoid carcinoma, carcinosarcoma) the malignant epithelial component gives rise to mesenchymal elements, and may even contain fully developed sarcomatoid features, such as malignant skeletal muscle, cartilage or bone elements. Integration of the molecular features into the appropriate microscopic context is essential to understand these tumours and accurately assess prognosis.
Cancer‐type‐independent molecular classification of therapeutic targets
Currently, precision oncology is based on the existence of alterations in certain specific therapeutic targets regardless of tumour location or type. Tumours with somatic mutations such as BRAF, EGFR or gene rearrangements such as ALK are considered therapeutic options regardless of tumour type or location, and tumours may even be characterised as BRAFomas or ALKomas. However, some clinical data have shown that, while BRAF inhibitors are effective in melanoma, the same inhibitors of the same BRAF mutations in colorectal cancer induce feedback loops in signalling pathways that can have a detrimental effect.63, 64 Cell signalling pathways are also highly regulated, often dependent on numerous positive and negative feedback loops, which are specific to the cell type and tumour environment. For this reason, BRAF inhibition in colorectal cancer indirectly results in EGFR receptor activation and subsequent PI3K–Akt pathway activation. Similarly, ALK alterations and their inhibitors do not have the same clinical response in lung tumours as in neuroblastomas.65 Further examples are inhibitors of EGFR, HER2 and HER3, depending on the tumour type and location. In a recent basket clinical trial, not all tumours were affected by the same HER mutations, and response to the HER2 inhibitor (neratinib) varied in different tissues: for example, colorectal or bladder cancer with HER2 mutations did not respond.66 In summary, specific drug treatment depends on the tissue context.34
Assessment and validation of molecular subtypes by protein expression
Molecular classification based on mRNA expression profiling of gene groups is a frequent approach to identify and classify molecular subtypes of tumours. One of the major achievements of molecular classification has been the subcategorisation according to prognosis, and it is instrumental for the identification of new genetic alterations. However, mRNA‐based expression studies are often limited by a low reproducibility, depending on mRNA stability. Furthermore, single‐cell sequencing approaches are not feasible in routine diagnostics. Currently, samples are sequenced as the bulk of tumour cells and therefore do not provide information on ITH. In addition to these technical issues, monitoring gene expression at the mRNA level does not necessarily correlate with the function of a protein within a cell, as many post‐transcriptional modifications determine whether or not mRNA finally gives rise to a functional protein. Importantly, however, the different molecular subtypes based on enormous mRNA expression matrices can be validated according to protein or gene expression using just a few in‐situ hybridisation or IHC probes in most molecular cancer subtypes (see Table 2).
Table 2.
Molecular signatures detected by IHC and FISH analysis in FFPE tumour samples
| Tumour type | Molecular signature Classification | Immunohistochemistry | FISH |
|---|---|---|---|
| Breast cancer | Luminal A | ER+, PR+, GATA+, FOXM+, Ki67 <15% | |
| Luminal B | ER+, PR+, GATA+, FOXM+, Ki67 >15% | ||
| HER2 enriched | ER−, PR−, HER2+ | ERB2 ampl. | |
| Basal‐like | ER−, PR−, HER2−, CK5+, EGFR+ | ||
| Claudin low | ER−, PR−, HER2−, CD44+, Snail+ | ||
| Normal breast‐like | ER−, PR−, HER2−, CD36+ | ||
| Colon cancer | CMS1 (immune) | FRMD6−, HTR2B−, ZEB1−, CDX2+ | |
| CMS2 | FRMD6−, HTR2B−, ZEB1−, CDX2+ | ||
| CMS3 | FRMD6−, HTR2B−, ZEB1−, CDX2+ | ||
| CMS4 (mesenchymal) | FRMD6+, HTR2B+, ZEB1+ CDX2− | ||
| Glioblastomas | Proneural | IDH mut, p53mut, OLIG2 | PDGFRA ampl, CDK4 ampl, CDK6 ampl |
| Mesenchymal | CD44, NF1 | EGFR ampl | |
| Classical | EGFR vIII mut, p53− | EGFR ampl, loss of PTEN and CDKN2A | |
| Neural | – | ||
| Medulloblastoma | WNT‐activated | β‐Catenin nuclear, GAB1−, YAP1+, Filamin A+ | Monosomy chr. 6 |
| SHH‐activated, TP53‐WT | β‐Catenin cyto, GAB1+, YAP1+, Filamin A+ | GLKi1 ampl, PTCH1 del | |
| SHH‐activated, TP53‐mut | β‐Catenin cyto, GAB1+, YAP1+, Filamin A+, p53+ | MYCN ampl, CLI2 ampl, 17p loss | |
| Group 3 (Gabaergic) | β‐Catenin cyto, GAB1−, YAP1−, Filamin A− | 17q ampl | |
| Group 4 (Glutaminergic) | β‐Catenin cyto, GAB1−, YAP1−, Filamin A− | MYC ampl, CDK6 ampl | |
| Endometrial | Ultramutated (POLE) | ||
| Hypermutated (MSI) | MMRd (MLH1−) | ||
| Copy number low | |||
| Copy number high | P53+ |
IHC, Immunohistochemistry; FISH, Fluorescence in‐situ hybridisation; FFPE, Formalin‐fixed paraffin‐embedded.
Breast carcinoma
According to the mRNA expression profile, six molecular subtypes are described: luminal A, luminal B, HER2+, basal‐like, claudin‐low and normal breast‐like.67, 68 However, RNA signatures can vary dramatically between patients with the same molecular subtype69 and, with the exception of the basal subtype, are not highly reproducible for diagnosis on microarray analysis.67, 70, 71 A high degree of ITH has been described, as assessed by immunohistochemical (IHC) studies,72 array‐CGH studies71 and massive parallel sequencing technologies.73 Given these problems in the exact diagnosis of breast cancer subtype by molecular characterisation, IHC‐based characterisations are recommended as a diagnostic approach in most of the consensus oncology guidelines.74 Additional IHC staining helps to distinguish luminal A and B subtypes, such as GATA3 and FOXM.75 CK5 and EGFR expression define the basal subtype of breast cancer,76 and CD44 and Snail expression determine the claudin‐low subtype.77 Finally, the normal breast‐like subtype is defined by low expression of the above‐mentioned markers and expression of factors such as CD36 in a low‐grade histological tumour.78
Colorectal carcinoma
Molecular subtypes of colorectal carcinoma have been distinguished by genomic, epigenetic and transcriptomic parameters.79, 80 Four different colorectal carcinomas, CMS1–4, have been described according to immunohistochemistry. The molecular subtypes can be identified as mesenchymal type (CMS4), which has the worst prognosis, immune type (CMS1) and epithelial subtypes (CMS2/3), based on four proteins: CDX2, FRMD6, HTR2B and ZEB1. In all subtypes, the drivers and actionable genes must be studied according to approved protocols (e.g. RAS, BRAF).81, 82
Lung carcinoma
Three molecular subtypes have been described based on DNA methylation [high, intermediate or low CpG island methylator phenotype (CIMP)], transcriptomics and RNA array: proximal proliferative, proximal inflammatory and terminal respiratory unit.83 Recently, using an integrative analysis with all available omics data, six different molecular subtypes have been described.84 Protein markers to distinguish those subtypes are still under way and that classification is not used in clinical oncology guides.
Nevertheless, druggable genetic alterations can be observed in the different molecular subtypes, and current oncology guidelines follow a reasonable scheme based on druggable genes, combining polymerase chain reaction (PCR)‐based methods to identify EGFR and BRAF mutations, and in‐situ hybridisation or immunohistochemistry for the expression of BRAF V600E, ALK, ROS, HER2, RET and MET.
Glioblastomas
Four molecular subtypes have been described based on gene expression profile: proneural, mesenchymal, mixed and unknown,85 and up to six based on methylation arrays, recurrent driver genes and transcriptomics, by the DKFZ (Deutsches Krebsforschungs‐zentrum).86, 87, 88, 89 These molecular subtypes are used currently in clinical oncology guides. In brief, according to the most recent World Health Organisation (WHO) classification,90 grade IV gliomas are classified as IDH‐mutant GB, IDH‐wt GB and midline glioma with H3 K27M mutation. In order to discriminate between subtypes the following can be analysed using IHC, fluorescence in‐situ hybridisation (FISH) or PCR: IDH1/IDH2, ATRX, TP53, FGFR, NF1, H3.1 and H3.3 mutations, EGFR and PDGFR amplifications, BRAF V600 mutation or translocations and CDKN2A/CDKN2B deletion.
Medulloblastomas
Medulloblastomas have four molecular subtypes with clear clinical differences (group 1 WNT activated; group 2 SHH activated; group 3 or GABAergic; and group 4 or glutaminergic). These subtypes can be assessed by immunohistochemistry or FISH, studying β‐catenin, GAB1, YAP1, Filamin A, p53, Glki1, 17q+ (group 3), MYC and CDK6 amplification.91, 92, 93, 94
Endometrial carcinoma
Two types of endometrial carcinoma have been described based on a combination of epidemiological, clinical, histological and molecular genetic data, distinguishing type I endometrioid endometrial carcinoma (EEC) and type II non‐endometrioid endometrial carcinoma (NEEC).95 A molecular classification of EC has been proposed based on The Cancer Genome Atlas Research Network (TCGA) study, published in 2013. TCGA analysis revealed four tumour groups:96 group 1, EEC with somatic inactivating mutations in POLE exonuclease and very high mutation rates (hypermutated) (7%); group 2, EEC with microsatellite instability (MSI), frequently with MLH‐1 promoter hypermethylation and high mutation rates (28%); group 3, EEC with low copy number alterations (39%), also called EEC with no specific molecular profile; and finally, group 4 (serous‐like or copy‐number high) (26%), with a low mutation rate but frequent TP53 mutations. These four types show different clinical, pathologic and molecular features.
Urothelial bladder carcinoma
Different molecular classifications are currently being explored. One of them, based on RNA genomewide studies, has characterised two main molecular groups of bladder cancer with prognostic and predictive impact.97 Group 1 basal subtype includes a subgroup of claudin‐low and is characterised by expression of the basal cytokeratins CK5, CK14 and CD44. Group 2 luminal subtype has mutations of the fibroblast growth factor receptor 3 (FGFR3) gene and urothelial differentiation markers such as CK20, GATA3 and uroplakins. The luminal subtype has a more favourable prognosis than the basal subtype.97 Recently, a prospective multicentre validation study with a 12‐gene signature progression score in non‐muscle‐invasive bladder cancer was demonstrated to have independent prognostic power beyond clinical and histopathological risk factors, and could help in stratifying patients to optimise treatment and follow‐up.98 Other molecular alterations have also been described, such as FGFR3 mutations in 80%,99 PI3KCA mutations in approximately 20% and mutations in the core promoter region of the human telomerase reverse transcriptase (TERT) gene in up to 85% of bladder tumours. Interestingly, FGFR3 and PIK3CA mutations have been demonstrated as biomarkers on liquid biopsy for disease surveillance for bladder cancer.100 Different cell cycle genes may be altered in invasive urothelial carcinoma, mainly tumour suppressor genes, including TP53, p16 and Rb.101 Finally, combinations of TP53, p21, p27 and pRb proved to be more precise in classifying bladder cancer patients into risk groups.102
Gene expression heterogeneity and environmental cells
mRNA expression versus protein expression
Molecular tumour subtypes based on mRNA expression profiles often show discordance with protein expression levels, depending on the RNA stability and the area sampled. It is well known that RNA studies are problematic regarding reproducibility, and that protein expression shows the real functional status of the targets. A classic example is breast cancer, in which mRNA profiles can define at least six different molecular categories. While this can be used as a general classification scheme, only IHC or FISH analysis of proteins in tumour cells within the individual subgroup can provide the exact diagnosis to guide treatment and prognosis. Some reports67 describe discord, when comparing mRNA and protein expression levels, of up to 50% of cases in HER2 status, and in more than 15% of cases when distinguishing between luminal A and luminal B.
Tissue‐specific protein expression profiles
The relation between tissue context and protein expression has recently been reported in an extensive gene expression study of 17 different tumour types in more than 8000 patients.103 The study encompassed massive studies of sequencing, mRNA expression, protein expression and engaged databases from the TCGA and the Human Protein Atlas. The authors also carried out in‐depth bioinformatics and systems biology analysis. Interestingly, more than 2700 genes correlated with prognosis, showing opposite prognostic effects depending on tumour type and location. Within the genes associated with worse prognosis, two groups were described. The first group comprised genes related to cell proliferation, of which more than 190 were identified (of the 314 described related to the cell cycle). This gives an idea of the great redundancy of gene alterations, especially the level of expression that may exist depending on tumour type. The second group comprised genes associated with tissue differentiation and their prognostic correlation. This is a point that has always been described in pathology, distinguishing tumours with a low degree of malignancy, i.e. histologically better differentiated, from those with a high degree of malignancy, i.e. poorly differentiated with a higher degree of cellular atypia. The study also found that more than 50% of the genes associated with prognosis were not related to Hanahan and Weinberg's classic hallmarks.6 Therefore, one of the conclusions was the enormous variation in gene expression profiles, both at an inter‐ and intratumour level. They described new sets of genes that, according to their expression, were associated with prognosis in the different tumour types, underlining the clinical importance and the validity of protein expression versus mRNA or genetic alterations. For example, in lung carcinoma, they proposed a prognostic correlation with the expression of the genes ERO1A, S100A16, S100A6, MKi67, SLC2A1, TACC3, ANLN, and CADM1.103, 104, 105
Assessment of intratumour heterogeneity based on protein expression
Currently, the most robust readout for ITH assessment comes from IHC. A classic example is ITH of oestrogen receptors, which may be positive in just 1% of cells; in fact, this ITH has been reported to be associated with long‐term risk of fatal breast cancer.106 Other examples are the heterogeneous expression of cyclin D1 in mantle cell lymphomas, which otherwise carry a homogeneous CCND1–IGH translocation107 and the subtype of HER2+ breast or gastric tumours that can range from 10% to 100% positive cells.9, 108
Within an entire tumour, protein expression often depends on local factors such as hypoxia, nutrient starvation or oxidative stress, independently of the cell's genetic background. These environmental cues can alter entire signalling pathways downstream of well‐established oncogenic alterations.109 Many tumours may have activating mutations in the most upstream elements of certain signalling cascades, such as RAS, EGFR and HER2, but phosphorylation of downstream factors such as MAP‐kinases or mTOR may be inhibited by environmental factors;110 therefore, even if specific driver genetic alterations are inhibited, no effect will be observed in cells in which these signalling pathways are inhibited by such factors.109, 111
Evaluation of heterogeneity in immune checkpoints and inflammatory cells
Primary tumours have heterogeneous epithelial cell content, and form an ecosystem with the environmental cells, including stromal cells, endothelial cells, leucocytes, lymphocytes, histiocytes and their secreted factors. In a pathological context, we can quantify and evaluate the expression of the immune checkpoints programmed cell death ligand 1 (PD‐L1) and programmed cell death‐1 (PD‐1) in epithelial and inflammatory parts of the tumour and use this information for clinical correlation studies. Similar to cell signalling pathways, key components of immune checkpoints, such as PD‐L1, can also have heterogeneous expression throughout the tumour.112 The PD‐L1 protein is expressed in a wide range of cell types, including lymphocytes, macrophages and dendritic cells, and is stimulated by variable and complex mechanisms. In contrast to other immune checkpoint components, PD‐L1 is rarely expressed in normal tissue but is induced in tumour‐associated tissue, is a tumour biomarker and is also an attractive drug target.113, 114 As many current treatment schemes are based on immunotherapy, pathological assessment should include immune checkpoint proteins. Furthermore, the heterogeneous and dynamic expression of factors such as PD‐L1 in basal conditions and in response to different treatments presents a challenge to predicting which patients will respond to anti‐PDL1 therapies. The assessment of CD4+, CD8+ cells, and tumour mutational burden (TMB) can also be helpful in clinical decision‐making. TMB has recently been proposed as a new biomarker to quantify the potential tumoral neoantigenicity, with the aim of predicting patients who will benefit from immunocheckpoint inhibitors. Nevertheless, different technologies and different thresholds have been proposed to define high versus low TMB in different tissue samples.115, 116, 117
Finally, the role of the microenvironment in carcinogenesis is gaining importance, although in small biopsies it is impossible to integrate the role of leucocytes, stromal cells and lymphocytes and the assessment of immune checkpoints.
Discussion
The Solution: To Integrate Clinical, Radiological, Molecular and Expression Data Within a Tissue Context
We postulate that cancer formation is a consortium of tumour cell clones, microenvironmental cells and local stressors (such as hypoxia) that alter protein expression and enhance tumour heterogeneity. Therefore, we believe that cancer must be considered within the context of tissue type. The correct diagnosis of a tumour should assimilate the clinical, radiological, histopathological, molecular and proteomic data.
As intratumour protein and molecules are highly heterogeneous and diagnostic biopsies are small, we envision that correlation with radiological imaging and nuclear medicine will be crucial to select areas that are more representative of the whole tumour.
Determination of the exact histological type is crucial for an exact diagnosis; for example, in lung carcinoma there is a huge difference in prognosis and treatment between diagnoses of small‐cell carcinoma, adenocarcinoma or large‐cell carcinoma. Similarly, in ovarian carcinoma, prognosis and survival varies widely between types: high‐ or low‐grade serous carcinoma, endometrioid carcinoma, clear‐cell carcinoma or mucinous adenocarcinoma. Most tumours can be diagnosed according to histological criteria as high‐ or low‐grade. The distinction is based on the degree of cell differentiation, the intensity of cellular atypia and cell proliferation. A proliferation index, such as Ki67, can be used to quantify the percentage of replicative cells within a tumour. This simple protein can be detected regardless of which genetic alterations are inducing the proliferation. The above‐mentioned classical factors have all been validated by RNA and molecular data.103
Identification of master genes and funnel factors
As described above, cellular stress can alter gene expression in tumour cells at several levels modifying biochemical signalling pathways and feedback loops,111 and protein expression can be heterogenous. While these biochemical changes are restricted to certain areas within the tumour, funnel factors such as p4E‐BP1 and peIF4E are usually expressed evenly throughout the tumour, regardless of the upstream oncogenic alterations and local environmental changes. The search for cellular targets expressed in most areas of tumours is becoming crucial to overcome tumour heterogeneity.
Because of the huge amount of data and complex positive and negative feedback regulating the final signal and the biological effect, systems biology is becoming essential for the understanding and identification of the real nodes or factors which are drivers in specific tumours in individual patients. The proto‐oncogene myc in Burkitt lymphoma and the canonical nuclear factor kappa B heterodimer in diffuse large B cell lymphoma are classic examples of so‐called master genes.118, 119, 120
Advances in digital pathology
Advances in digital pathology (whole slide imaging) now allow interpretation of digital slides on computer stations and integration of molecular features into the microscopic context. The differential expression of genes in different areas of the tumour can be interpreted precisely, and haematoxylin and eosin‐stained microscopic sections can be merged with multiple immunohistochemical stains for different proteins. Digital pathology also allows tumour annotation for microdissection for different tumour components, with appropriate cellularity assessment of tumour elements and the non‐neoplastic microenvironment. Digital pathology is essential to establish computerised algorithms to correlate molecular alterations with tumour phenotype.
Tissue‐specific multiplex technology
Given the numerous alterations in genes, protein expression and mRNA, it is clear that we will have to implement multiplex technologies that will allow us to study multiple factors simultaneously on the same histological sections. With this approach we can minimise the amount of tissue needed for molecular and protein studies and for quantification of the expression of different factors and their inter‐relationship. Different platforms are emerging, with different quantitative multispectral fluorescence approaches, or with mass spectrometry in situ, to allow the study and quantification of several dozen markers. Also, new next‐generation sequencing technologies are being implemented in routine testing combining DNA and RNA analysis from the same slide reducing the amount of tissue and also the ITH.121, 122
An interpretative and holistic data assessment with ‘deep learning’ (artificial intelligence with automatic learning)
The main objective for the use of deep learning in pathology is to improve existing algorithms for biomarker quantification and to develop smart and robust algorithms. It can be based on three main areas or objectives: (i) tumour type classification, (ii) tumour prognosis (prediction system, learning from experience) and (iii) personalised treatment allocation system (also a reinforcement learning system). In fact, deep learning is already being applied in various tumour types, including breast, lung, melanoma and glioblastoma.123, 124, 125, 126 The integration of such information and algorithms into everyday clinical practice can help to minimise subjective evaluations and improve the precise diagnosis for each tumour and patient (see Figure 2).
Figure 2.
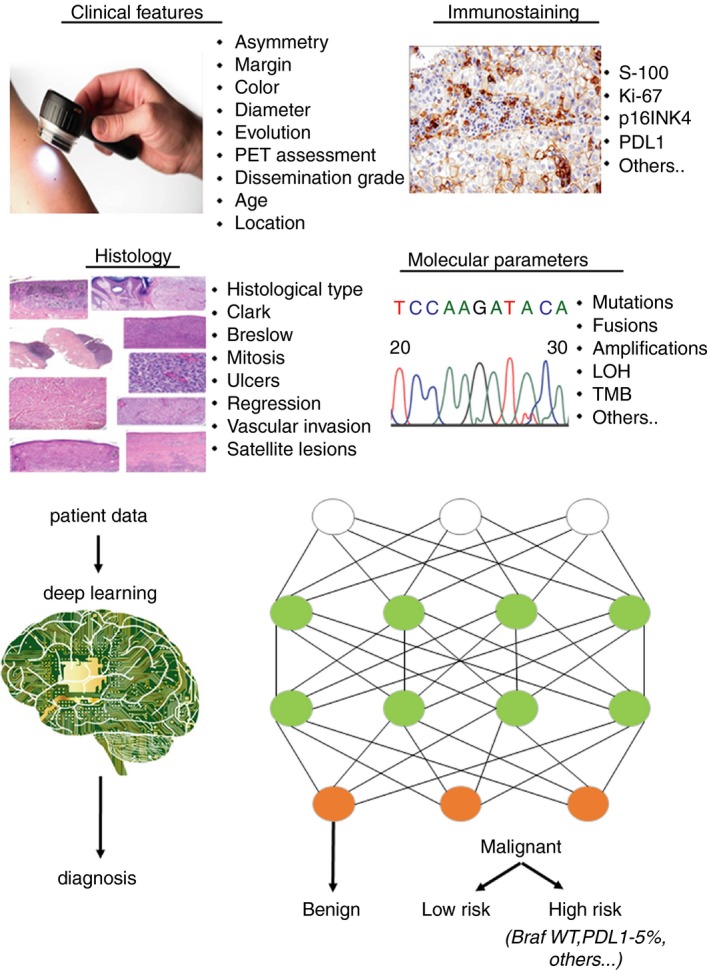
Schematic representation of the potential use of artificial intelligence (AI) in the diagnosis of melanoma. All information (clinical features, histology, molecular alterations, other parameters) are analysed through AI establishing different algorithms resulting in a more precise diagnosis.
Systems biology encompasses tools that hold great promise for deciphering the vulnerabilities of the tumour ecosystem as a whole. Studies are based on the premise that multiple oncogenic events converge on a number of cellular networks that may contain essential or synthetically lethal clinically targetable hubs or factors,127, 128 such as the eIF4F complex. Nonetheless, many contemporary systems biology approaches do not consider intratumour heterogeneity. This is relevant because key functional nodes within the metabolic, signal transduction and gene expression networks responsible for supporting the tumour phenotype are critically dependent on the heterogeneity of the tumour. These data can also help to classify tumours according to histopathological, biochemical and genomic features and thus help to tailor the diagnosis and clinical management to a patient's biological tumour profile. Accordingly, multidimensional molecular and gene expression data, which are associated with the response to antitumour treatments and clinical progress, are thought to facilitate the selection of patients who are more likely to respond to targeted or ‘precise’ therapies.129, 130, 131 The availability of systemwide data in a variety of cancers is facilitating the development of approaches that go beyond the classic, reductionist paradigms that associate single genes with cellular phenotypes and functions. Systems biology approaches consider the interplay between multiple molecular factors that underpin phenotype development. Accordingly, rather than a genetic disease, cancer is now perceived as a disease of ‘networks’,131 and to fully grasp the complexity of the tumour ecosystem the networks driving cancer need to be mapped and their dynamics and evolution over time deciphered. Emerging data show that these cancer networks are constantly rewired in part by clonal interactions, changing microenvironments and the acquisition of novel molecular alterations that are largely induced by anticancer treatments.127, 132 Therefore, minor subpopulations that are not readily detectable in bulk tumours or that can adapt to hostile environments may emerge following treatments that specifically target cancer‐driving mutations present in the predominant tumour subpopulations. Thus, in the coming years, systems biology implementation may be crucial for (i) detailed, clinically oriented subclassification of cancer types, (ii) mapping of oncogenic networks and identification of the critical nodes to overcome the effects of ITH and (iii) informing future preclinical research and design of Phase I clinical trials by anticipating therapy response in silico and predicting the best targets for each patient and tumour (personalised cancer therapy or precision medicine).
Liquid biopsies
As biopsies are associated with a degree of invasiveness and cannot always be repeated, new approaches have been proposed, such as the analysis of circulating tumour cells (CTCs), plasma‐derived cell‐free tumour DNA (cfDNA) or RNA and, more recently, DNA from circulating tumour exosomes in blood.133, 134, 135 This strategy enables a global assessment of the constellation of somatic genetic alterations in a tumour irrespective of its anatomical location, and is becoming a good alternative for the study of a tumour mutational landscape in situations where biopsies or surgical samples are difficult to obtain. Hence, liquid biopsy appears to be a very relevant tool for the identification of potential actionable genomic alterations and therapeutic targets, monitoring treatment response, predicting disease progression before clinical and radiological confirmation and identifying mechanisms of resistance in the particular context of patients with early‐stage and metastatic disease.136, 137 Furthermore, liquid biopsy provides additional cues on ITH, as reported in a recent pilot study in patients with early‐stage NSCLC.138
Conclusion
Tumours show such a degree of morphological, molecular and proteomic heterogeneity, and a close relationship with microenvironmental factors, that for the accurate pathological evaluation of tumours an integrated approach seems essential: all genomic and proteomic data should be evaluated taking into account the heterogeneity and tissue type (Figure 3). We propose the integration of all such factors, along with the classical pathological factors, within the context of the tissue; we call this approach tissunomics.
Figure 3.
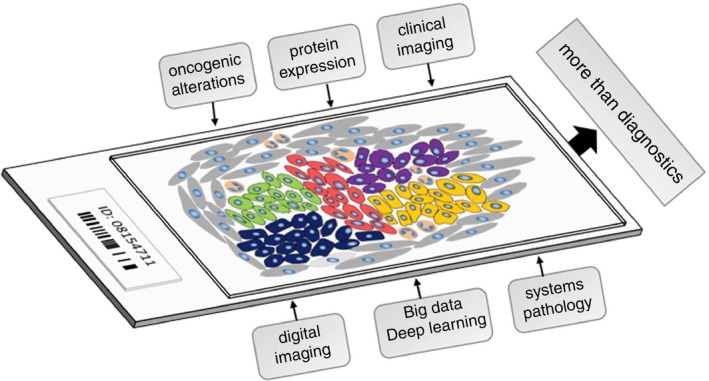
Schematic drawing of a tumour section for histopathological analysis. Heterogeneity within a tumour is depicted by different colours of the tumoral cells. Stromal cells and infiltrating cells of the immune system are coloured in grey and orange, respectively. Variables considered for the evaluation of the tumour sample are indicated in the boxes above the slide. New methodologies, proposed to improve the diagnosis, are shown in the boxes below.
Conflicts of interest
All authors declare no conflicts of interest.
Acknowledgements
S.R.Y.C. acknowledges support from Fondo de Investigaciones Sanitarias (FIS; PI17/02247 and PI14/01320), Centro de Investigación Biomédica en Red de Cáncer (CIBERONC; CB16/12/00363) and Generalitat de Catalunya (AGAUR; 2017 SGR 1799 and 2014 SGR 1131).
Ramón y Cajal S, Hümmer S, Peg V, Guiu X M, De Torres I, Castellvi J, Martinez‐Saez E & Hernandez‐Losa J (2019) Histopathology 75, 4–19 10.1111/his.13828 Integrating clinical, molecular, proteomic and histopathological data within the tissue context: tissunomics
References
- 1. Global Burden of Disease Cancer Collaboration , Fitzmaurice C, Allen C, Barber RM et al Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability‐adjusted life‐years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017; 3; 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018; 68; 7–30. [DOI] [PubMed] [Google Scholar]
- 3. Yong M, Jensen AO, Jacobsen JB, Norgaard M, Fryzek JP, Sorensen HT. Survival in breast cancer patients with bone metastases and skeletal‐related events: a population‐based cohort study in Denmark (1999–2007). Breast Cancer Res. Treat. 2011; 129; 495–503. [DOI] [PubMed] [Google Scholar]
- 4. Rosai J, Ackerman LV. Rosai and Ackerman's surgical pathology. 10th ed. Maryland Heights, MI: Mosby, 2011. [Google Scholar]
- 5. DeVita VT Jr, Lawrence TS, Rosenberg SA et al eds. Hellman and Rosenberg's cancer: principles and practice of oncology. Philadelphia: Wolters Kluwer, 2015. [Google Scholar]
- 6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144; 646–674. [DOI] [PubMed] [Google Scholar]
- 7. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015; 27; 15–26. [DOI] [PubMed] [Google Scholar]
- 8. Rybinski B, Yun K. Addressing intra‐tumoral heterogeneity and therapy resistance. Oncotarget 2016; 7; 72322–72342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gomez‐Martin C, Concha A, Corominas JM et al Consensus of the Spanish Society of Medical Oncology (SEOM) and Spanish Society of Pathology (SEAP) for HER2 testing in gastric carcinoma. Clin. Transl. Oncol. 2011; 13; 636–651. [DOI] [PubMed] [Google Scholar]
- 10. Bartley AN, Washington MK, Colasacco C et al HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: guideline from the College of American Pathologists, American Society for Clinical Pathology, and the American Society of Clinical Oncology. J. Clin. Oncol. 2017; 35; 446–464. [DOI] [PubMed] [Google Scholar]
- 11. Hammond ME, Hayes DF, Dowsett M et al American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010; 28; 2784–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abe H, Kawahara A, Azuma K et al Heterogeneity of anaplastic lymphoma kinase gene rearrangement in non‐small‐cell lung carcinomas: a comparative study between small biopsy and excision samples. J. Clin. Oncol. 2015; 10; 800–805. [DOI] [PubMed] [Google Scholar]
- 13. Eckel‐Passow JE, Lachance DH, Molinaro AM et al Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 2015; 372; 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodon J, Saura C, Dienstmann R et al Molecular prescreening to select patient population in early clinical trials. Nat. Rev. Clin. Oncol. 2012; 9; 359–366. [DOI] [PubMed] [Google Scholar]
- 15. Bertolaso M. Philosophy of cancer: a dynamic and relational view. New York: Springer, 2016. [Google Scholar]
- 16. Gerlinger M, Rowan AJ, Horswell S et al Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012; 366; 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Bruin EC, McGranahan N, Mitter R et al Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014; 346; 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J, Fujimoto J, Zhang J et al Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014; 346; 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawrence JE, Patel AS, Rovin RA et al Quantification of protoporphyrin IX accumulation in glioblastoma cells: a new technique. ISRN Surg. 2014; 2014; 405360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerlinger M, Horswell S, Larkin J et al Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014; 46; 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res. 2016; 76; 3136–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016; 352; 169–175. [DOI] [PubMed] [Google Scholar]
- 23. Almendro V, Kim HJ, Cheng YK et al Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014; 74; 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alderton GK. Tumour evolution: epigenetic and genetic heterogeneity in metastasis. Nat. Rev. Cancer 2017; 17; 141. [DOI] [PubMed] [Google Scholar]
- 25. Soto AM, Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory BioEssays. News Rev. Mol. Cell. Dev. Biol. 2011; 33; 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dotto GP. Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J. Clin. Invest. 2014; 124; 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018; 18; 19–32. [DOI] [PubMed] [Google Scholar]
- 28. Ramon YCS, Capdevila C, Hernandez‐Losa J et al Cancer as an ecomolecular disease and a neoplastic consortium. Biochim. Biophys. Acta 2017; 186; 484–499. [DOI] [PubMed] [Google Scholar]
- 29. Chapman A, Fernandez del Ama L, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 2014; 8; 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheung KJ, Ewald AJ. A collective route to metastasis: seeding by tumor cell clusters. Science 2016; 352; 167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheung KJ, Padmanaban V, Silvestri V et al Polyclonal breast cancer metastases arise from collective dissemination of keratin 14‐expressing tumor cell clusters. Proc. Natl Acad. Sci. USA 2016; 113; E854–E863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aceto N, Bardia A, Miyamoto DT et al Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014; 158; 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maddipati R, Stanger BZ. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 2015; 5; 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schneider G, Schmidt‐Supprian M, Rad R, Saur D. Tissue‐specific tumorigenesis: context matters. Nat. Rev. Cancer 2017; 17; 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polishchuk LA, Telegeev GD. Role of the hybrid Bcr/Abl kinase in the pathogenesis of chronic myeloid leukemia lacking C‐Abl and CXCR4 proteins. Exp. Oncol. 2014; 36; 138–143. [PubMed] [Google Scholar]
- 36. Marega M, Piazza RG, Pirola A et al BCR and BCR‐ABL regulation during myeloid differentiation in healthy donors and in chronic phase/blast crisis CML patients. Leukemia 2010; 24; 1445–1449. [DOI] [PubMed] [Google Scholar]
- 37. Stevens TJ, Lando D, Basu S et al 3D structures of individual mammalian genomes studied by single‐cell Hi‐C. Nature 2017; 544; 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tjong H, Li W, Kalhor R et al Population‐based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc. Natl Acad. Sci. USA 2016; 113; E1663–E1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene 2004; 23; 6445–6470. [DOI] [PubMed] [Google Scholar]
- 40. Chang F. Desmoplastic small round cell tumors: cytologic, histologic, and immunohistochemical features. Arch. Pathol. Lab. Med. 2006; 130; 728–732. [DOI] [PubMed] [Google Scholar]
- 41. Nielsen TO, Poulin NM, Ladanyi M. Synovial sarcoma: recent discoveries as a roadmap to new avenues for therapy. Cancer Discov. 2015; 5; 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guerreiro Stucklin AS, Ramaswamy V, Daniels C, Taylor MD. Review of molecular classification and treatment implications of pediatric brain tumors. Curr. Opin. Pediatr. 2018; 30; 3–9. [DOI] [PubMed] [Google Scholar]
- 43. Castinetti F, Moley J, Mulligan L, Waguespack SG. A comprehensive review on MEN2B. Endocr. Relat. Cancer 2018; 25; T29–T39. [DOI] [PubMed] [Google Scholar]
- 44. Guilmette J, Nose V. Hereditary and familial thyroid tumours. Histopathology 2018; 72; 70–81. [DOI] [PubMed] [Google Scholar]
- 45. Francesco C, Paolo T, Del Vecchio MT, Monica G, Guilia M, Aniello SG. Correspondence re: Cameselle‐Teijeiro J, Chan JKC: cribriform‐morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis‐associated thyroid carcinoma? Mod. Pathol. 2000; 13; 363–365. [DOI] [PubMed] [Google Scholar]
- 46. Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, de Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathology 2018; 50; 49–59. [DOI] [PubMed] [Google Scholar]
- 47. Musolino A, Bella MA, Bortesi B et al BRCA mutations, molecular markers, and clinical variables in early‐onset breast cancer: a population‐based study. Breast 2007; 16; 280–292. [DOI] [PubMed] [Google Scholar]
- 48. Ricketts C, Woodward ER, Killick P et al Germline SDHB mutations and familial renal cell carcinoma. J. Natl Cancer Inst. 2008; 100; 1260–1262. [DOI] [PubMed] [Google Scholar]
- 49. Gill AJ, Hes O, Papathomas T, Sedivcova M et al Succinate dehydrogenase (SDH)‐deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 2014; 38; 1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomlinson IP, Alam NA, Rowan AJ et al Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002; 30; 406–410. [DOI] [PubMed] [Google Scholar]
- 51. Winship IM, Dudding TE. Lessons from the skin – cutaneous features of familial cancer. Lancet Oncol. 2008; 9; 462–472. [DOI] [PubMed] [Google Scholar]
- 52. Ponti G, Losi L, Di Gregorio C et al Identification of Muir‐Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer 2005; 103; 1018–1025. [DOI] [PubMed] [Google Scholar]
- 53. Chan JK, Ip YT, Cheuk W. The utility of immunohistochemistry for providing genetic information on tumors. Int. J. Surg. Pathol. 2013; 21; 455–475. [DOI] [PubMed] [Google Scholar]
- 54. Argani P, Fritsch M, Kadkol SS, Schuster A, Beckwith JB, Perlman EJ. Detection of the ETV6–NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin‐embedded tissue: application to challenging pediatric renal stromal tumors. Mod. Pathol. 2000; 13; 29–36. [DOI] [PubMed] [Google Scholar]
- 55. Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017; 14; 735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cantile M, Marra L, Franco R et al Molecular detection and targeting of EWSR1 fusion transcripts in soft tissue tumors. Med. Oncol. 2013; 30; 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hall RD, Kudchadkar RR. BRAF mutations: signaling, epidemiology, and clinical experience in multiple malignancies. Cancer Control 2014; 21; 221–230. [DOI] [PubMed] [Google Scholar]
- 58. Valachis A, Ullenhag GJ. Discrepancy in BRAF status among patients with metastatic malignant melanoma: a meta‐analysis. Eur. J. Cancer 2017; 81; 106–115. [DOI] [PubMed] [Google Scholar]
- 59. Chen ZY, Zhong WZ, Zhang XC et al EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist 2012; 17; 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cai W, Lin D, Wu C et al Intratumoral heterogeneity of ALK‐rearranged and ALK/EGFR coaltered lung adenocarcinoma. J. Clin. Oncol. 2015; 33; 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Myers MB, Banda M, McKim KL, Wang Y, Powell MJ, Parsons BL. Breast cancer heterogeneity examined by high‐sensitivity quantification of PIK3CA, KRAS, HRAS, and BRAF mutations in normal breast and ductal carcinomas. Neoplasia 2016; 18; 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coenegrachts L, Garcia‐Dios DA, Depreeuw J et al Mutation profile and clinical outcome of mixed endometrioid‐serous endometrial carcinomas are different from that of pure endometrioid or serous carcinomas. Virchows Arch. 2015; 466; 415–422. [DOI] [PubMed] [Google Scholar]
- 63. Prahallad A, Sun C, Huang S et al Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483; 100–103. [DOI] [PubMed] [Google Scholar]
- 64. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017; 17; 676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Claeys S, Denecker G, Cannoodt R et al Early and late effects of pharmacological ALK inhibition on the neuroblastoma transcriptome. Oncotarget 2017; 8; 106820–106832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hyman DM, Piha‐Paul SA, Won H et al HER kinase inhibition in patients with HER2‐ and HER3‐mutant cancers. Nature 2018; 554; 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eroles P, Bosch A, Perez‐Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012; 38; 698–707. [DOI] [PubMed] [Google Scholar]
- 68. Heng YJ, Lester SC, Tse GM et al The molecular basis of breast cancer pathological phenotypes. J. Pathol. 2017; 241; 375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Califano A, Alvarez MJ. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 2017; 17; 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lusa L, McShane LM, Reid JF et al Challenges in projecting clustering results across gene expression‐profiling datasets. J. Natl Cancer Inst. 2007; 99; 1715–1723. [DOI] [PubMed] [Google Scholar]
- 71. Geyer FC, Weigelt B, Natrajan R et al Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J. Pathol. 2010; 220; 562–573. [DOI] [PubMed] [Google Scholar]
- 72. Potts SJ, Krueger JS, Landis ND et al Evaluating tumor heterogeneity in immunohistochemistry‐stained breast cancer tissue. Lab. Invest. 2012; 92; 1342–1357. [DOI] [PubMed] [Google Scholar]
- 73. Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 2014; 15; 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Goldhirsch A, Winer EP, Coates AS et al Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013; 24; 2206–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tang P, Tse GM. Immunohistochemical surrogates for molecular classification of breast carcinoma: a 2015 update. Arch. Pathol. Lab. Med. 2016; 140; 806–814. [DOI] [PubMed] [Google Scholar]
- 76. Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple‐negative breast cancer ‐ the road to new treatment strategies. Lancet 2017; 389; 2430–2442. [DOI] [PubMed] [Google Scholar]
- 77. de Beca FF, Caetano P, Gerhard R et al Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J. Clin. Pathol. 2013; 66; 187–191. [DOI] [PubMed] [Google Scholar]
- 78. Hagemann IS. Molecular testing in breast cancer: a guide to current practices. Arch. Pathol. Lab. Med. 2016; 140; 815–824. [DOI] [PubMed] [Google Scholar]
- 79. Guinney J, Dienstmann R, Wang X et al The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015; 21; 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017; 17; 79–92. [DOI] [PubMed] [Google Scholar]
- 81. Marisa L, de Reynies A, Duval A et al Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013; 10; 1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Trinh A, Trumpi K, De Sousa EMF et al Practical and robust identification of molecular subtypes in colorectal cancer by immunohistochemistry. Clin. Cancer Res. 2017; 23; 387–398. [DOI] [PubMed] [Google Scholar]
- 83. Ringner M, Jonsson G, Staaf J. Prognostic and chemotherapy predictive value of gene‐expression phenotypes in primary lung adenocarcinoma. Clin. Cancer Res. 2016; 22; 218–229. [DOI] [PubMed] [Google Scholar]
- 84. Chen F, Zhang Y, Parra E et al Multiplatform‐based molecular subtypes of non‐small‐cell lung cancer. Oncogene 2017; 36; 1384–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verhaak RG, Hoadley KA, Purdom E et al Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17; 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015; 129; 829–848. [DOI] [PubMed] [Google Scholar]
- 87. Korshunov A, Schrimpf D, Ryzhova M et al H3‐/IDH‐wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017; 134; 507–516. [DOI] [PubMed] [Google Scholar]
- 88. Reifenberger G, Wirsching HG, Knobbe‐Thomsen CB, Weller M. Advances in the molecular genetics of gliomas – implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017; 14; 434–452. [DOI] [PubMed] [Google Scholar]
- 89. Sturm D, Witt H, Hovestadt V et al Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22; 425–437. [DOI] [PubMed] [Google Scholar]
- 90. Louis DN, Perry A, Reifenberger G et al The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016; 131; 803–820. [DOI] [PubMed] [Google Scholar]
- 91. Ramaswamy V, Remke M, Bouffet E et al Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016; 131; 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ellison DW, Dalton J, Kocak M et al Medulloblastoma: clinicopathological correlates of SHH, WNT, and non‐SHH/WNT molecular subgroups. Acta Neuropathol. 2011; 121; 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kaur K, Kakkar A, Kumar A et al Integrating molecular subclassification of medulloblastomas into routine clinical practice: a simplified approach. Brain Pathol. 2016; 26; 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Clifford SC, Lusher ME, Lindsey JC et al Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub‐group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006; 5; 2666–2670. [DOI] [PubMed] [Google Scholar]
- 95. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983; 15; 10–17. [DOI] [PubMed] [Google Scholar]
- 96. Cancer Genome Atlas Research Network , Kandoth C, Schultz N, Cherniack AD et al Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497; 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Seiler R, Ashab HAD, Erho N et al Impact of molecular subtypes in muscle‐invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur. Urol. 2017; 72; 544–554. [DOI] [PubMed] [Google Scholar]
- 98. Dyrskjot L, Reinert T, Algaba F et al Prognostic impact of a 12‐gene progression score in non‐muscle‐invasive bladder cancer: a prospective multicentre validation study. Eur. Urol. 2017; 72; 461–469. [DOI] [PubMed] [Google Scholar]
- 99. Tomlinson DC, Baldo O, Harnden P, Knowles MA. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J. Pathol. 2007; 213; 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Christensen E, Birkenkamp‐Demtroder K, Nordentoft I et al Liquid biopsy analysis of FGFR3 and PIK3CA hotspot mutations for disease surveillance in bladder cancer. Eur. Urol. 2017; 71; 961–969. [DOI] [PubMed] [Google Scholar]
- 101. van Rhijn BW, van der Kwast TH, Vis AN et al FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004; 64; 1911–1914. [DOI] [PubMed] [Google Scholar]
- 102. Shariat SF, Karakiewicz PI, Palapattu GS et al Outcomes of radical cystectomy for transitional cell carcinoma of the bladder: a contemporary series from the Bladder Cancer Research Consortium. J. Urol. 2006; 176; 2414–2422; discussion 22. [DOI] [PubMed] [Google Scholar]
- 103. Uhlen M, Zhang C, Lee S et al A pathology atlas of the human cancer transcriptome. Science 2017; 357; pii: eaan2507. [DOI] [PubMed] [Google Scholar]
- 104. Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat. Rev. Clin. Oncol. 2015; 12; 511–526. [DOI] [PubMed] [Google Scholar]
- 105. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017; 17; 637–658. [DOI] [PubMed] [Google Scholar]
- 106. Lindstrom LS, Yau C, Czene K et al Intratumor heterogeneity of the estrogen receptor and the long‐term risk of fatal breast cancer. J. Natl Cancer Inst. 2018; 110; 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J. Clin. Invest. 2012; 122; 3416–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Apicella M, Corso S, Giordano S. Targeted therapies for gastric cancer: failures and hopes from clinical trials. Oncotarget 2017; 8; 57654–57669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ramon YCS, Castellvi J, Hummer S, Peg V, Pelletier J, Sonenberg N. Beyond molecular tumor heterogeneity: protein synthesis takes control. Oncogene 2018; 37; 2490–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ramon YCS, De Mattos‐Arruda L, Sonenberg N, Cortes J, Peg V. The intra‐tumor heterogeneity of cell signaling factors in breast cancer: p4E‐BP1 and peIF4E are diffusely expressed and are real potential targets. Clin. Transl. Oncol. 2014; 16; 937–941. [DOI] [PubMed] [Google Scholar]
- 111. Martinez A, Sese M, Losa JH et al Phosphorylation of eIF4E confers resistance to cellular stress and DNA‐damaging agents through an interaction with 4E‐T: a rationale for novel therapeutic approaches. PLoS One 2015; 10; e0123352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015; 14; 561–584. [DOI] [PubMed] [Google Scholar]
- 113. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 2013; 39; 1–10. [DOI] [PubMed] [Google Scholar]
- 114. Madore J, Vilain RE, Menzies AM et al PD‐L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti‐PD‐1/PD‐L1 clinical trials. Pigment Cell Melanoma Res. 2015; 28; 245–253. [DOI] [PubMed] [Google Scholar]
- 115. Hellmann MD, Ciuleanu TE, Pluzanski A et al Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018; 378; 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chalmers ZR, Connelly CF, Fabrizio D et al Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9; 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD‐1 inhibition. N. Engl. J. Med. 2017; 377; 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Faumont N, Durand‐Panteix S, Schlee M et al c‐Myc and Rel/NF‐kappaB are the two master transcriptional systems activated in the latency III program of Epstein‐Barr virus‐immortalized B cells. J. Virol. 2009; 83; 5014–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014; 4; pii: a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Guo X, Koff JL, Moffitt AB et al Molecular impact of selective NFKB1 and NFKB2 signaling on DLBCL phenotype. Oncogene 2017; 29; 4224–4232. [DOI] [PubMed] [Google Scholar]
- 121. Letovanec I, Finn S, Zygoura P et al Evaluation of NGS and RT‐PCR methods for ALK rearrangement in European NSCLC patients: results from the European Thoracic Oncology Platform Lungscape Project. J. Thorac. Oncol. 2018; 13; 413–425. [DOI] [PubMed] [Google Scholar]
- 122. Oliveira DM, Mirante T, Mignogna C et al Simultaneous identification of clinically relevant single nucleotide variants, copy number alterations and gene fusions in solid tumors by targeted next‐generation sequencing. Oncotarget 2018; 9; 22749–22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Han Z, Wei B, Zheng Y, Yin Y, Li K, Li S. Breast cancer multi‐classification from histopathological images with structured deep learning model. Sci. Rep. 2017; 7; 4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Vandenberghe ME, Scott ML, Scorer PW, Soderberg M, Balcerzak D, Barker C. Relevance of deep learning to facilitate the diagnosis of HER2 status in breast cancer. Sci. Rep. 2017; 7; 45938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yu KH, Zhang C, Berry GJ et al Predicting non‐small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016; 7; 12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rathore S, Akbari H, Rozycki M et al Radiomic MRI signature reveals three distinct subtypes of glioblastoma with different clinical and molecular characteristics, offering prognostic value beyond IDH1. Sci. Rep. 2018; 8; 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Werner HM, Mills GB, Ram PT. Cancer systems biology: a peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014; 11; 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kiel C, Serrano L. Structural data in synthetic biology approaches for studying general design principles of cellular signaling networks. Structure 2012; 20; 1806–1813. [DOI] [PubMed] [Google Scholar]
- 129. Russo M, Siravegna G, Blaszkowsky LS et al Tumor heterogeneity and lesion‐specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016; 6; 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gatenby RA, Cunningham JJ, Brown JS. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nat. Commun. 2014; 5; 5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T. The cancer cell map initiative: defining the hallmark networks of cancer. Mol. Cell 2015; 58; 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Paul JM, Templeton SD, Baharani A, Freywald A, Vizeacoumar FJ. Building high‐resolution synthetic lethal networks: a ‘Google map’ of the cancer cell. Trends Mol. Med. 2014; 20; 704–715. [DOI] [PubMed] [Google Scholar]
- 133. Alix‐Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016; 6; 479–491. [DOI] [PubMed] [Google Scholar]
- 134. Buder A, Tomuta C, Filipits M. The potential of liquid biopsies. Curr. Opin. Oncol. 2016; 28; 130–134. [DOI] [PubMed] [Google Scholar]
- 135. De Mattos‐Arruda L, Cortes J, Santarpia L et al Circulating tumour cells and cell‐free DNA as tools for managing breast cancer. Nat. Rev. Clin. Oncol. 2013; 10; 377–389. [DOI] [PubMed] [Google Scholar]
- 136. Murtaza M, Dawson SJ, Tsui DW et al Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497; 108–112. [DOI] [PubMed] [Google Scholar]
- 137. Murtaza M, Dawson SJ, Pogrebniak K et al Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015; 6; 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jamal‐Hanjani M, Wilson GA, Horswell S et al Detection of ubiquitous and heterogeneous mutations in cell‐free DNA from patients with early‐stage non‐small‐cell lung cancer. Ann. Oncol. 2016; 27; 862–867. [DOI] [PubMed] [Google Scholar]