

Abstract
Somatic mutation signatures may represent footprints of genetic and environmental exposures that cause different cancer. Few studies have comprehensively examined their association with germline variants, and none in an indigenous African population. SomaticSignatures was employed to extract mutation signatures based on whole‐genome or whole‐exome sequencing data from female patients with breast cancer (TCGA, training set, n = 1,011; Nigerian samples, validation set, n = 170), and to estimate contributions of signatures in each sample. Association between somatic signatures and common single nucleotide polymorphisms (SNPs) or rare deleterious variants were examined using linear regression. Nine stable signatures were inferred, and four signatures (APOBEC C>T, APOBEC C>G, aging and homologous recombination deficiency) were highly similar to known COSMIC signatures and explained the majority (60–85%) of signature contributions. There were significant heritable components associated with APOBEC C>T signature (h 2 = 0.575, p = 0.010) and the combined APOBEC signatures (h 2 = 0.432, p = 0.042). In TCGA dataset, seven common SNPs within or near GNB5 were significantly associated with an increased proportion (beta = 0.33, 95% CI = 0.21–0.45) of APOBEC signature contribution at genome‐wide significance, while rare germline mutations in MTCL1 was also significantly associated with a higher contribution of this signature (p = 6.1 × 10−6). This is the first study to identify associations between germline variants and mutational patterns in breast cancer across diverse populations and geography. The findings provide evidence to substantiate causal links between germline genetic risk variants and carcinogenesis.
Keywords: somatic mutation signatures, breast cancer, single‐nucleotide polymorphisms, rare deleterious variants
Short abstract
What's new?
Women of African ancestry are more likely to be diagnosed with clinically aggressive breast cancer than women of European or Asian ancestry. Here, the authors examined associations between germline variants and mutational signatures in breast cancers across different ethnicities, especially in a unique sample set of indigenous African women. They identified four stable mutational signatures that explained the majority of tumor mutations, leading to a better understanding of the complex interplay between germline genetics and somatic mutations across different ethnicities.
Abbreviations
- CADD
combined annotation dependent depletion
- CCDS
consensus coding domain sequence
- COSMIC
Catalog of Somatic Mutation in Cancer
- ER
estrogen receptor
- ExAC
Exome Aggregation Consortium
- GRM
genetic relationship matrix
- GWAS
genome‐wide association study
- HER2
human epidermal growth factor receptor 2
- HR
hormone receptor
- HRD
homologous recombination deficiency
- HWE
Hardy–Weinberg equilibrium
- indel
insertion and deletion
- MAF
minor allele frequency
- NER
nucleotide excision repair
- NMF
nonnegative matrix factorization
- PCA
principal component analysis
- PR
progesterone receptor
- SKAT‐O
optimal sequence kernel association test
- SNPs
single nucleotide polymorphisms
- SNVs
single nucleotide variants
- TCGA
The Cancer Genome Atlas
- VEP
variant effect predictor
- WES
whole‐exome sequencing
- WGS
whole‐genome sequencing
Introduction
Somatic mutation signatures act as the physiological readout of the biological history of a cancer, and provide a new bridge to connect cancer mutation to both exogenous and endogenous risk factors.1, 2, 3 Many signatures have been reported to be attributable to specific environmental or lifestyle mutagens.4 However, somatic mutations could also be influenced by heritable factors,5 and endogenous factors that influence cancer far more than previously considered.6, 7 A few studies have examined the interplay between germline genetics and somatic mutations in carcinogenesis with signals identified for germline variants in RAD51B,8 MC1R,9 APOBEC3 regions10, 11, 12 and DNA mismatch repair genes in different types of cancers.13 In addition, a few genes driven by rare deleterious variants were reported to be significant, which include: polymerase epsilon, catalytic subunit (POLE) gene associated with the Catalog of Somatic Mutation in Cancer (COSMIC) signature 10 in colorectal and endometrial carcinomas;1 homologous recombination deficiency (HRD) genes PALB2 and BRCA1/2 associated with HRD signature in various cancers;14, 15 and nucleotide excision repair (NER) genes associated with COSMiC signature 5 in urothelial tumors.16 The existing studies offered some evidences to the largely unexplored relationships between germline and somatic mutations.10, 17, 18, 19
The burden and severity of breast cancer vary widely across populations. Women of African ancestry are more likely to be diagnosed with clinically aggressive disease and have a higher mortality rate than age‐matched women of European or Asian ancestry.20 Emerging data from The Cancer Genome Atlas (TCGA) has identified mutation signatures that show differences across ancestry groups.21, 22 These studies suggest modest differences between African and non‐African ancestry groups but these datasets have relatively small number of women of African ancestry.23 To examine the association between common genetic variants, rare deleterious mutations and the contribution of mutational signatures across diverse populations, we used TCGA as the training set, and examined the whole‐genome sequencing (WGS) and whole‐exome sequencing (WES) from Nigeria as the validation set. We tested the hypothesis that biologically relevant germline variants associated with somatic mutation signatures in women of non‐African ancestry would be validated in women of African ancestry as well.
Methods
Ethics statement
Our study was embedded within the Nigerian Breast Cancer Study (NBCS) and approved by the Institutional Review Board of all participating institutions. Informed consent has been obtained from the participants.
Training dataset
A total of 1,037 breast cancer patients from TCGA were used as the training set. Demographics of participants, including clinical characteristics across ethnicity are summarized in Supporting Information Table S1. Somatic mutation data (single nucleotide variants [SNVs]) were generated as previously described.22 We included 1,035 WES and 84 WGS, and 82 patients owned both WGS and WES data in this analysis.
Common germline variants from genotyping array
Affymetrix Genome‐Wide Human SNP 6.0 Array data for 2,270 breast cancer samples were downloaded from TCGA on November 6, 2015. A total of 1,134 samples were excluded for different reasons (12: missing ID; 1,110: solid tumor tissue; 12: male patients). Among the remaining 1,136 female samples (1,087 unique patients), we excluded samples for 33 more patients due to genotyping missing rate ≥5%, and worked with samples for 1,054 patients. Of the 906,600 single nucleotide polymorphisms (SNPs) included in the microarray data, 1,466 SNPs were excluded for lacking chromosome information or mapping to Y chromosome and 47,140 SNPs were removed due to genotyping missing rate ≥5% or being monomorphic, and 857,994 SNPs remained.
Genetic ancestry of TCGA patients was estimated using principal component analysis (PCA).21 According to the estimated proportion of ancestry, patients were grouped into genomic Black (≥50% African ancestry), genomic White (≥90% European ancestry) and genomic Asian (≥90% Asian ancestry) as previously described.21
Genotype imputation was done using IMPUTE2 software with the 1,000 Genomes Project phase 3 integrated variant set as the reference panel.24 A total of 81,232,799 SNPs within autosomes were imputed, and 24,570,114 SNPs were kept after excluding singletons or SNPs with an imputation information score <0.7.
Rare deleterious variants from germline exomes
Using the Platypus (https://www.well.ox.ac.uk/research/research-groups/lunter-group/softwares/platypus-a-haplotype-based-variant-caller-for-next-generation-sequence-data) in single‐sample mode, variants within exonic regions were called as described previously.22 Briefly, variants were considered rare and deleterious if (i) they had an allele frequency <0.05 in The Exome Aggregation Consortium (ExAC)25 and across TCGA blood germline exomes from various cancer types,22 and (ii) they were predicted by variant effect predictor (VEP) to have “HIGH” impact, cause protein loss‐of‐function (stop‐gain, frameshift insertion and deletion [indel], etc.), or were missense mutations with a combined annotation dependent depletion (CADD)26 score >25. Only blood germline exomes were included. Additionally, variants were required to fall within consensus coding sequence (CCDS, version 17) exonic regions to be carried downstream. In total, 132,186 rare deleterious variants were found within 11,718 genes in 968 samples, of which 2,933 genes had only one mutation and 6,560 genes (56.0%) had less than five counts of rare coding deleterious variants among these samples. We therefore only focused on genes with at least five mutations (n = 5,158) in the downstream analyses. The Bonferroni corrected alpha levels for significance was 9.69 × 10−6.
Validation dataset
A total of 172 women with breast cancer from Nigeria were evaluated. All Nigerian patients were assumed to be 100% African with little to no admixture with other populations.27 These patients were younger and had more aggressive subtypes of breast cancer with higher proportion of estrogen receptor negative (ER−), progesterone receptor negative (PR−), human epidermal growth factor receptor 2 negative (HER2−; triple‐negative) subtypes. Characteristics of the Nigerian dataset were previously described.22
Common and rare genetic variants
Hundred breast tumor samples for WGS and 129 breast tumor samples for WES from 172 unique patients were processed for this analysis. Two WGS samples were excluded as outliers because their mutation counts were <100. Fifty‐seven patients had both WGS and WES. For the common SNP analysis, 98 WGS samples were involved, while 170 samples (129 WES, 41 WGS) were included for rare variants analysis. Rare coding deleterious variants were called as described above. In total, 22,035 rare coding deleterious variants were found within 5,439 genes in 170 samples, of which 2,196 genes had only one mutation among these samples and 3,876 genes (71.3%) had less than five rare coding deleterious counts. A total of 1,563 genes were included in the final analysis.
Mutation signature calling and comparison with reported signatures
Mutational signature calling
Somatic SNVs were called as described previously.22 Briefly, in order to be utilized in downstream analyses, SNVs needed to be called by both MuTect (https://software.broadinstitute.org/cancer/cga/mutect) and Strelka (https://github.com/Illumina/strelka) as well as be absent within a normal panel of exomes (n = 1,088) and genomes (n = 124) derived from blood. We employed a nonnegative matrix factorization (NMF) approach using SomaticSignatures28 to extract somatic mutational signatures and estimate their contributions to each sample.
The ability to reliably call mutation signatures depends on having sufficient number of mutations per sample. To this point, we used all high‐quality somatic exome SNVs, regardless of their position in the coding or noncoding region. A pilot study was conducted to compare the stability of signatures among different number of minimum SNVs (24, 48, 72 and 96, data not shown), and found that a cut point of 24 is sufficient. This is consistent with recent finding that 20 mutations gave an average classification accuracy of 80% across signatures.29 Any sample containing at least 24 SNVs was included for downstream assessment in order to enlarge the sample size as much as possible.
In order to obtain more stable signature estimates and ensure comparability between the training and validation sets, we combined both datasets to call the signatures, including 84 WGS samples and 1,008 WES samples from TCGA as well as 98 WGS samples and 129 WES samples from Nigeria. For samples with both WGS and WES, both data types were included.
We first used the nonnegative matrix factorization without normalization to obtain the mutation matrix as described by Alexandrov et al.,4 then calculated the percentage contributions of signatures for each sample by normalizing to the number of mutations of each sample. For subsequent analyses, we used the percentage of mutations assigned to each signature (contribution) rather than the total number of mutations attributed to a signature.30 The sum of mutation signatures’ contribution is 1. This distinction is important as high APOBEC contribution, for example, do not necessarily imply APOBEC hypermutation.
Nine signatures were retained at the end for further downstream analyses because nine signatures explained approximately 99% of variance in the current sample. For all nine signatures, we examined the correlation of contributions between exomes and genomes using 138 individuals who had both WES and WGS data using intraclass correlation coefficient (rho). Each signature's contribution was simultaneously called for WGS samples and WES samples for those 138 individuals. Signatures A1 (rho = 0.938), A2 (rho = 0.912) and A9 (rho = 0.866) all exhibited strong correlation). APOBEC C>T (Signature A1) and C>G (Signature A2) contributions were correlated (r = 0.43). We only focused on stable signatures between WGS and WES in order to reliably leverage all available WES samples.
Comparison with reported signatures
We compared our mutation signature matrices with the 30 previously reported signatures operative across a variety of cancer types downloaded from the COSMIC. Mean Kullback–Leibler Divergence was applied to evaluate the similarity between our signatures and those from COSMIC:31
P and Q are the distributions matrixes for our signatures and those from COSMIC, respectively. The smaller Kullback–Leibler Divergence is, the better the similarity. A similarity of 0.00 is an exact match. With this approach, we determined the best representative known COSMIC signature for each of the signature we identified, and named with the name from canonical COSMIC signature they most closely resemble. Two signatures A5 and A7 did not match any canonical COSMIC signature.
Statistical analysis
Signature contributions by demographic and clinical characteristics
We compared contribution of the selected signatures among categorical variables, including ancestry (Black, White, Nigerian) and hormone receptor (HR)/HER2 status (HER2+, HR+/HER2− and HR−/HER2−), using Mann–Whitney U test.
Heritability estimation of mutation signatures using GCTA
We used GCTA to calculate the pairwise genetic relationship between individuals and created the genetic relationship matrix (GRM).32 We used a GRM cut‐off value of 0.05,33 which excludes relationships that are approximately closer than second cousins. This removed around 77 samples in the TCGA dataset. All the heritability estimation analyses control for first 10 eigenvectors from PCA. We then estimated PCA‐adjusted heritability of each phenotype (signature contribution) by the restricted maximum likelihood method in GCTA. Power analysis (http://cnsgenomics.com/shiny/gctaPower/) indicated a 99% chance of detecting a SNP‐based heritability estimate (h 2) of 0.15 in the TCGA cohort.34
The heritability was not calculated for Nigeria samples due to limited sample size. In the training dataset, among 24,570,114 SNPs, 1,573,566 were removed due to missing genotype data (missing proportion >5%), 5,301,200 variants were removed due to minor allele frequency (MAF) <0.001, and 188,864 variants were removed due to deviation from the Hardy–Weinberg equilibrium (HWE). The remaining 17,506,484 SNPs were used for downstream analyses. Among 1,054 samples with SNP chip data, 178 samples were removed due to missing genotype data (cut‐off point as 1%), 77 were removed when pruning the GRM (relatedness <0.05) and 66 samples were excluded due to missing mutation signature. In the end, 733 samples were included in the model.
Association of common SNPs with signature contribution
For TCGA, we analyzed 7,177,790 common SNPs with MAF ≥0.05. The single‐SNP association tests with signature contribution were conducted using linear regression as implemented in SNPtest,35 with adjustment for age and the eigenvectors from PCA. For the validation data, on the other hand, we only adjusted for age given that we do not anticipate any admixture with other ethnic. We applied genome‐wide association study (GWAS) threshold (p = 5 × 10−8) as our cut‐off point in the discovery stage. We also filtered the association results of the GWAS‐identified risk SNPs by its location within the autosomal regions to check their role in the signature contributions. All SNPs included were previously reported to be associated with breast cancer risk (https://www.ebi.ac.uk/gwas/). In Nigerians, we only assessed SNPs that were nominally significant in the TCGA cohort.
Association of rare deleterious variants with signature contribution
Gene‐based association analyses for mutation signatures were conducted using the unified optimal sequence kernel association test (SKAT‐O),36 as implemented in SKAT.37 As a linear combination of the burden and SKAT tests,38 SKAT‐O achieves robust power whether a given gene has a high proportion of causal variants exerting effects in the same direction, or instead has many noncausal variants or variants exerting effects in opposite directions.39 We considered weighting parameter θ = 1 (burden) and θ = 0 (SKAT) tests and reported the optimal of the two tests. We included putatively deleterious variants with MAF ≤5%, and used the beta distribution weights proposed by Wu et al.,38 which upweights rarer variants, for both tests. Gene‐based analyses were adjusted for age and the eigenvectors from PCA for TCGA data. For validation with Nigerian dataset, we only adjusted for age. Subgroup analyses by subtype were also evaluated for all significant results. We used a Bonferroni adjustment for multiple testing to assess the significance of the gene‐based test results. We also calculate Benjamini–Hochberg's false positive rate in the discovery dataset. For genes with evidences of association in the discovery phase, we pooled data from TCGA and Nigeria and conducted SKAT‐O analysis after adjusting for age and proportion of African and Asian ancestry.
Two combined gene sets were examined to test their association with the contribution of HRD signature. One is a set of known breast cancer predisposition genes (BROCA gene panel: http://tests.labmed.washington.edu/BROCA), another one contained all homologous recombination related genes from Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, including atm, rad51c, bard1, brca1, brca2, brip1, fam175a, mre11a, nbn, palb2, xrcc2, pold1, rad51b, babam1, blm, bre, eme1, rad50, rad52, rad54b, rad54l, rbbp8, rpa1, sycp3, top3a, top3b, topbp1, uimc1 and xrcc3. All statistical calculations were completed in R or Stata version 15.0 (College Station, TX). All p values are two‐sided.
Results
Basic information
A total of 1,011 samples with somatic mutation count ≥24 (1,008 WES, 84 WGS) from TCGA were included as the training dataset, and 170 samples that satisfied the same criteria from Nigeria (129 WES, 98 WGS) were involved as the validation set (Fig. 1). The median (25th quartile and 75th quartile) for somatic mutation count per WES sample were 73 (50–144) for TCGA Caucasians, 100 (63–194) for TCGA African Americans and 131 (64–241) for Nigerians (Supporting Information Table S1 and Fig. S1). The number of mutation from WES was highly correlated with that from WGS (p < 0.001, Supporting Information Fig. S1C).
Figure 1.
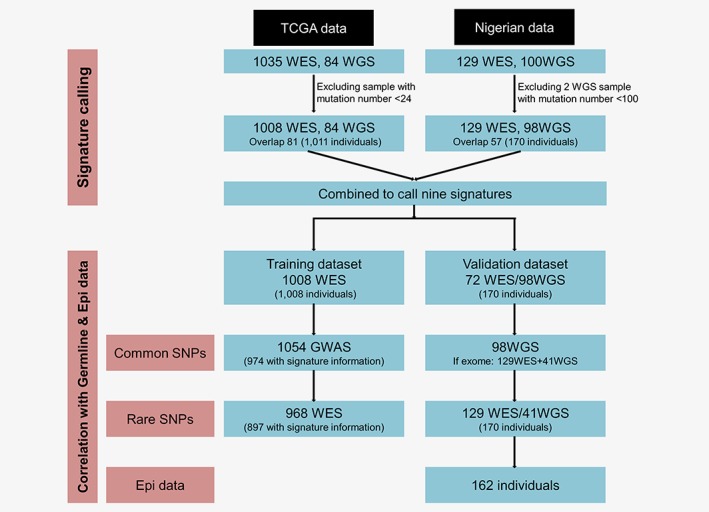
Flow chart of this study. [Color figure can be viewed at http://wileyonlinelibrary.com]
Nine mutational signatures were extracted (Supporting Information Fig. S2). Our signatures closely resembled breast cancer signatures from COSMiC: signatures A1 (S2: APOBEC C>T), A2 (S13: APOBEC C>G), A3 (S17: Unknown, breast cancer related), A4 (S10: Altered activity of polymerase POLE), A6 (S1: Aging), A8 (S8: Unknown etiology) and A9 (S9: HRD; Supporting Information Fig. S3). The common signatures including S2 (APOBEC C>T), S13 (APOBEC C>G), S1 (Aging), S8 (Unknown etiology) and S9 (HRD), explained the vast majority of mutations regardless of ethnicity or subtype (Supporting Information Figs. S4). The correlation between exomes and genomes were presented in Supporting Information Figures S5—there was high correlation for S2, S13, S1 and S9. Given the robustness of signature calls between these two data types, we focused on these four signatures for subsequent analyses.
Heritability of selected signatures’ contribution
The estimates of array‐based heritability, h 2, ranged from 0.040 to 0.575 in TCGA samples across four selected signatures (Table 1), with APOBEC C>T signature (h 2 = 0.575, p = 0.010) and combined APOBEC signature (h 2 = 0.432, p = 0.042) displaying the strongest heritable components. Of note, the heritability estimates of mutation signatures are unreliable due to limited sample size, although we showed that some of them were statistically significantly different from zero.
Table 1.
GWAS estimates of signature heritability for signature contributions in TCGA dataset
| Adjusted PCA, 733 samples (excluding relatedness) | ||||||||
|---|---|---|---|---|---|---|---|---|
| V(G) | V(E) | h 2 | ||||||
| Signatures | Value | SE | Value | SE | Value | SE | p | |
| 1 | APOBEC of cytidine deaminases (C>T) | 0.011 | 0.006 | 0.008 | 0.004 | 0.575 | 0.254 | 0.010 |
| 2 | APOBEC (C>G) | 0.001 | 0.004 | 0.010 | 0.002 | 0.108 | 0.307 | 0.350 |
| 1 + 2 | APOBEC | 0.022 | 0.016 | 0.028 | 0.010 | 0.432 | 0.272 | 0.042 |
| 6 | Spontaneous deamination of 5‐methylcytosine | 0.001 | 0.011 | 0.034 | 0.007 | 0.040 | 0.313 | 0.445 |
| 9 | Failure of DNA double‐strand break‐repair by homologous recombination | 0.004 | 0.012 | 0.026 | 0.007 | 0.154 | 0.358 | 0.349 |
Note: In TCGA, among 24,570,114 SNPs, 1,573,566 variants were removed due to missing genotype data (geno = 0.05), 5,301,200 variants were removed due to minor allele threshold (MAF, 0.001) and 188,864 variants were removed due to Hardy–Weinberg exact test. A total of 17,506,484 SNPs were kept in the end. Among 1,054 samples with GWAS information, 178 samples were removed due to missing genotype data (mind = 0.01), 77 were removed when pruning the GRM (relatedness = 0.05) and 66 samples were excluded due to missing of mutation signature. In the end, we have 733 samples in the model. Bold p values denote statistical significance at the p < 0.05.
Association of common SNPs with signature contribution
Next, we analyzed the association of common SNPs with mutation signatures. Nine hundred seventy‐four samples from TCGA and 170 samples from Nigeria with mutation signature results were further analyzed. No SNPs in TCGA showed significant association beyond genome‐wide significance level contributing to any of the four selected signatures (Supporting Information Fig. S6). When contributions from the two APOBEC signatures were combined together, seven SNPs on or near GNB5 showed evidence of association surpassing p < 5 × 10−8 (Fig. 2 a, Table 2). rs66866642/rs578194564 (MAF = 0.169) in the intronic region of the GNB5 gene (Fig. 2 b) had the strongest association, and this signal was also consistently observed for both APOBEC C>T and APOBEC C>G signatures (Supporting Information Fig. S7). The CT allele of rs66866642/rs578194564 was significantly associated with a 35.3% absolute increase in the contribution to APOBEC signature (95% confidence interval [CI]: 23.1–47.5, p = 1.03 × 10−8; Table 2, Fig. 2 c). The top seven SNPs in TCGA tagged a couple of SNPs having LD r 2 ≥ 0.85, and they are located in the region with several regulatory elements (Supporting Information Table S2, Fig. 2 d). Using the Nigerian cohort as the validation set, 13 of 30 top SNPs were detected with nonsignificant p values, but with consistent magnitude and direction in beta coefficients to those in TCGA (Table 2).
Figure 2.
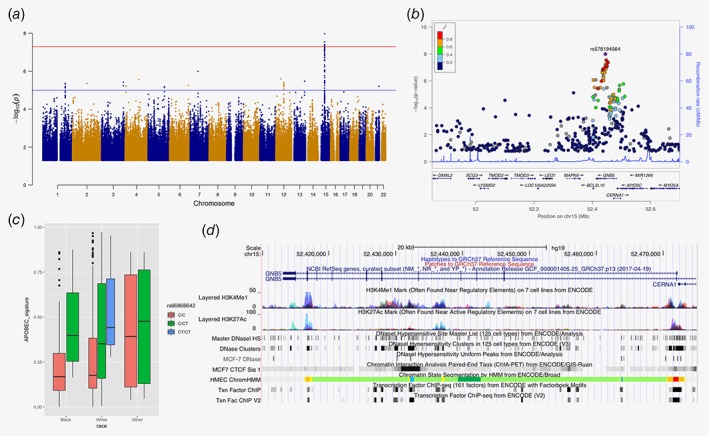
Common SNP rs66866642/rs578194564 in GNB5 is associated with APOBEC signatures. (a) Manhattan plot for associations between common SNPs and APOBEC signatures. (b) Plot of log‐transformed p values from single marker analysis for the GNB5 gene. The labeled marker, a purple diamond (rs66866642/rs578194564), is the most significant SNP (index SNP). The LD between the index SNP and other markers in the region was color coded, with red color indicating strong LD (r 2 > 0.8) and blue color indicating weak LD (r 2 < 0.2). (c) Genotype of rs66866642/rs578194564 among different ethnic groups. (d) Functional elements at the GNB5 gene region. Data is from ENCODE through UCSC Genome Browser, including histone modification marks for H3K4Me1 and H3K27Ac, transcription factor binding sites and DNase hypersensitivity sites of human mammary epithelial cells (HMEC), breast cancer cell (MCF7). [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 2.
The top 30 signal for common SNPs in association tests with combined APOBEC signature contributions
| ID | SNP | Chromosome | Position (base) | Ref allele | ALT allele | TCGA | Nigeria1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Information score | MAF | p value | Beta | Standard error | MAF | p value | Beta | Standard error | ||||||
| 1 | rs66866642/rs578194564 | 15 | 52,444,832 | C | CT | 0.948 | 0.169 | 1.03E‐08 | 0.353 | 0.062 | – | – | – | – |
| 2 | rs12902073 | 15 | 52,447,508 | G | A | 0.986 | 0.170 | 2.84E‐08 | 0.334 | 0.060 | – | – | – | – |
| 3 | rs12440354 | 15 | 52,452,434 | T | C | 0.983 | 0.175 | 3.90E‐08 | 0.328 | 0.060 | – | – | – | – |
| 4 | rs12901730 | 15 | 52,452,697 | T | A | 0.983 | 0.171 | 4.31E‐08 | 0.330 | 0.060 | – | – | – | – |
| 5 | rs12901891 | 15 | 52,452,834 | T | C | 0.983 | 0.171 | 4.33E‐08 | 0.330 | 0.060 | – | – | – | – |
| 6 | rs12438743 | 15 | 52,454,098 | C | T | 0.982 | 0.171 | 4.38E‐08 | 0.330 | 0.060 | – | – | – | – |
| 7 | rs373292393 | 15 | 52,440,257 | C | CAA | 0.983 | 0.155 | 4.98E‐08 | 0.340 | 0.062 | – | – | – | – |
| 8 | rs12910398 | 15 | 52,445,477 | C | T | 0.987 | 0.176 | 5.57E‐08 | 0.322 | 0.059 | 0.051 | 0.239 | 0.390 | 0.331 |
| 9 | rs12905698 | 15 | 52,445,090 | A | G | 0.986 | 0.176 | 6.16E‐08 | 0.321 | 0.059 | 0.051 | 0.239 | 0.390 | 0.331 |
| 10 | rs12909880 | 15 | 52,449,293 | T | C | 0.982 | 0.173 | 6.76E‐08 | 0.323 | 0.060 | – | – | – | – |
| 11 | rs12902522 | 15 | 52,448,036 | A | G | 0.986 | 0.177 | 7.59E‐08 | 0.319 | 0.059 | 0.051 | 0.239 | 0.390 | 0.331 |
| 12 | rs12909184 | 15 | 52,450,617 | G | T | 0.983 | 0.178 | 7.91E‐08 | 0.319 | 0.059 | 0.051 | 0.239 | 0.390 | 0.331 |
| 13 | rs12910052 | 15 | 52,449,360 | T | C | 0.983 | 0.178 | 7.98E‐08 | 0.318 | 0.059 | 0.051 | 0.239 | 0.390 | 0.331 |
| 14 | rs12903769 | 15 | 52,449,733 | G | A | 0.982 | 0.167 | 8.20E‐08 | 0.327 | 0.061 | – | – | – | – |
| 15 | rs12908833 | 15 | 52,449,195 | G | C | 0.982 | 0.174 | 8.80E‐08 | 0.320 | 0.060 | – | – | – | – |
| 16 | rs12908742 | 15 | 52,449,379 | A | C | 0.982 | 0.174 | 8.82E‐08 | 0.320 | 0.060 | 0.051 | 0.239 | 0.390 | 0.331 |
| 17 | rs3794543 | 15 | 52,441,524 | C | T | 0.993 | 0.158 | 1.07E‐07 | 0.327 | 0.061 | – | – | – | – |
| 18 | rs4776007 | 15 | 52,447,299 | A | G | 0.987 | 0.185 | 1.29E‐07 | 0.308 | 0.058 | 0.097 | 0.203 | 0.323 | 0.254 |
| 19 | rs8034097 | 15 | 52,437,278 | A | G | 0.995 | 0.151 | 1.32E‐07 | 0.330 | 0.063 | – | – | – | – |
| 20 | rs12438274 | 15 | 52,439,368 | T | G | 0.971 | 0.188 | 1.33E‐07 | 0.311 | 0.059 | – | – | – | – |
| 21 | rs12438194 | 15 | 52,439,096 | T | A | 0.993 | 0.152 | 1.35E‐07 | 0.330 | 0.063 | – | – | – | – |
| 22 | rs4636859 | 15 | 52,435,696 | T | C | 0.996 | 0.151 | 1.45E‐07 | 0.329 | 0.063 | – | – | – | – |
| 23 | rs12438937 | 15 | 52,441,686 | C | T | 0.992 | 0.170 | 1.79E‐07 | 0.313 | 0.060 | 0.092 | 0.216 | 0.320 | 0.259 |
| 24 | rs35709612 | 15 | 52,439,940 | A | C | 0.994 | 0.169 | 1.80E‐07 | 0.313 | 0.060 | 0.092 | 0.208 | 0.326 | 0.259 |
| 25 | rs35448038 | 15 | 52,434,758 | GT | G | 0.981 | 0.163 | 1.89E‐07 | 0.317 | 0.061 | – | – | – | – |
| 26 | rs12593041 | 15 | 52,443,107 | C | T | 0.988 | 0.171 | 2.49E‐07 | 0.310 | 0.060 | 0.097 | 0.203 | 0.323 | 0.254 |
| 27 | rs62014722 | 15 | 52,434,338 | C | G | 0.995 | 0.163 | 2.52E‐07 | 0.313 | 0.061 | 0.056 | 0.130 | 0.481 | 0.318 |
| 28 | rs62014721 | 15 | 52,434,284 | C | G | 0.994 | 0.163 | 2.53E‐07 | 0.313 | 0.061 | 0.056 | 0.130 | 0.481 | 0.318 |
| 29 | rs4238384 | 15 | 52,436,334 | C | T | 1.000 | 0.222 | 2.75E‐07 | 0.281 | 0.055 | 0.276 | 0.227 | 0.212 | 0.175 |
| 30 | rs71130134 | 15 | 52,437,673 | CA | C | 0.980 | 0.157 | 4.73E‐07 | 0.313 | 0.062 | – | – | – | – |
Bold p values denote statistical significance at the p < 5 × 10−8 (genome‐wide association study threshold).
A total of 17 SNPs were not covered in WES samples, so we filled the corresponding cell with “–”.
Abbreviation: MAF, minor allele frequency.
In addition, we evaluated the association between index SNPs identified in previous GWAS of breast cancer risk, and signatures contribution. We found a dozen SNPs associated at nominal significance level with either APOBEC signature or HRD signature (Supporting Information Table S3). The genes tagged by SNPs associated with APOBEC signature included ANKRD16, ZNF365, CHST9, ARRDC3, FGFR2 and ESR1. Most of the genes associated with HRD signature function to decrease homologous recombination repair frequency, including RNF115, ANXA13, DNAJC1, DNAH11 and TFAP2A.
Associations between rare deleterious variants and signature contribution
Manhattan plots for the gene‐based association analyses are shown in Figure 3a and Supporting Information Figure S8 for selected mutation signatures in TCGA cohort. Five genes showed evidence of association with contribution of APOBEC C>T signature or APOBEC C>G signature surpassing defined significance level (threshold as 9.69 × 10−6, Table 3). MTCL1 is the top signal for combined APOBEC signature (pooled p = 6.11 × 10−6), and women with MTCL1 mutations had higher APOBEC signature contribution. Two additional genes (HIVEP1 and TMEM104) had marginal nominal significant association with contribution of combined APOBEC signature in both datasets. As the direction of the association for HIVEP1 was consistent across the TCGA and Nigerian datasets (pooled p = 0.0009), while the TMEM104 association was positive in the TCGA cohort but negative in the Nigerian cohort (pooled p = 0.079) (Supporting Information Fig. S9), suggesting HIVEP1 is likely a true association but TMEM104 may be false negative. Mutations in three genes, including MTCL1, ERC1 and HIP1, were associated with higher contribution of APOBEC C>G signature alone.
Figure 3.
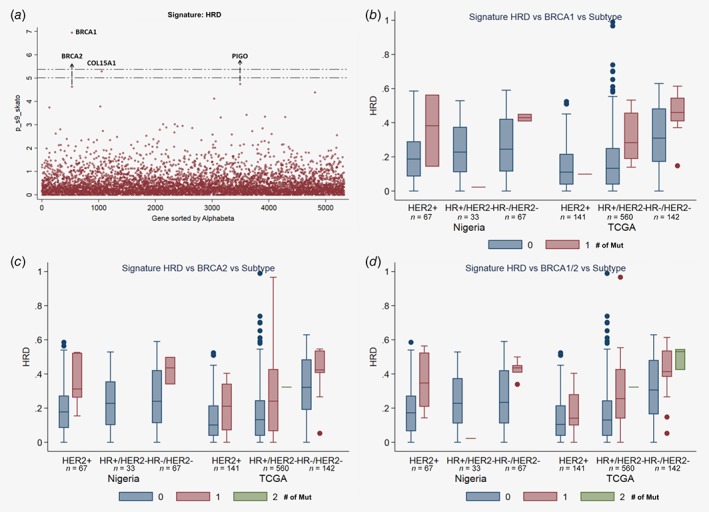
Associations between rare deleterious variants and HRD signature. (a) Plot of log‐transformed p values from gene‐based analysis for rare deleterious variants. (b–d) Comparison of contribution of HRD signature, grouped by breast cancer tissue subgroups, datasets, and the number of BRCA1 (b), BRCA2 (c) and BRCA1/BRCA2 mutations (d). [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 3.
Significant results of gene‐based analysis in the training and validation datasets
| Signatures | Gene | TCGA data | Nigeria data | Pooled data | ||||
|---|---|---|---|---|---|---|---|---|
| Nominal p value | False discovery rate | Theta1 | # of variants in the test | p value | # of variants in the test | p value2 | ||
| APOBEC C>T | ATP13A1 | 9.04 × 10−6 | 0.006 | 1 | 5 | 0.580 | 5 | 0.0089 |
| ERC1 | 0.186 | 1.000 | 1 | 9 | 0.442 | 1 | 0.282 | |
| HIP1 | 0.773 | 1.000 | 1 | 13 | 0.494 | 2 | 1.000 | |
| QARS | 0.0023 | 1.154 | 0 | 6 | 0.434 | 1 | 0.0036 | |
| MTCL1 | 5.21 × 10−5 | 0.053 | 0 | 16 | 0.538 | 2 | 0.0005 | |
| HIVEP1 | 0.149 | 1.000 | 1 | 20 | 0.0657 | 9 | 0.0798 | |
| TMEM104 | 0.0002 | 0.061 | 0 | 15 | 0.156 | 7 | 0.0609 | |
| APOBEC C>G | ATP13A1 | 0.356 | 1.000 | 1 | 5 | 0.737 | 5 | 0.580 |
| ERC1 | 4.45 × 10−7 | 0.0005 | 0 | 9 | 0.444 | 1 | 1.63 × 10 −7 | |
| HIP1 | 3.46 × 10−6 | 0.005 | 0 | 13 | 0.219 | 2 | 9.18 × 10 −7 | |
| QARS | 6.46 × 10−7 | 0.001 | 0 | 6 | 0.740 | 1 | 2.86 × 10−5 | |
| MTCL1 | 1.35 × 10−6 | 0.001 | 0 | 16 | 0.360 | 2 | 4.21 × 10 −7 | |
| HIVEP1 | 2.23 × 10−5 | 0.015 | 0 | 20 | 0.119 | 9 | 2.57 × 10−5 | |
| TMEM104 | 0.0095 | 0.417 | 1 | 15 | 0.0238 | 7 | 0.0445 | |
| APOBEC | ATP13A1 | 0.1274 | 0.883 | 1 | 5 | 0.611 | 5 | 0.507 |
| ERC1 | 0.0006 | 0.165 | 0 | 9 | 0.957 | 1 | 0.0014 | |
| HIP1 | 0.0154 | 0.452 | 0 | 13 | 0.808 | 2 | 0.0075 | |
| QARS | 1.50 × 10−5 | 0.056 | 0 | 6 | 0.520 | 1 | 0.0002 | |
| MTCL1 | 1.91 × 10−6 | 0.012 | 1 | 16 | 0.393 | 2 | 6.11 × 10 −6 | |
| HIVEP1 | 0.0024 | 0.253 | 1 | 20 | 0.054 | 9 | 0.0009 | |
| TMEM104 | 0.0004 | 0.189 | 1 | 15 | 0.041 | 7 | 0.079 | |
| HRD | BRCA1 | 1.03 × 10−8 | 0.0001 | 1 | 25 | 0.139 | 6 | 7.02 × 10 −9 |
| BRCA2 | 1.05 × 10−5 | 0.008 | 1 | 50 | 0.014 | 8 | 7.93 × 10 −7 | |
| COL15A1 | 5.15 × 10−6 | 0.005 | 0 | 10 | 0.391 | 1 | 5.50 × 10 −6 | |
| PIGO | 1.80 × 10−5 | 0.009 | 0 | 5 | – | 0 | 8.68 × 10 −6 | |
Bold p values denote statistical significance at the p < 9.69 × 10−6 (Bonferroni corrected alpha level).
The weighting parameter theta indicates whether the SKAT test (theta = 0) or burden test (theta = 1) gave the smallest p value.
p value from optimal sequence kernel association test (SKAT‐O).
Germline mutations in four genes, BRCA1, BRCA2, COL15A1 and PIGO, were significantly associated with contributions from the HRD signature (all p ≤ 5.5 × 10−6). The contribution of HRD signature was consistently increasing along with the number of rare deleterious variants in both BRCA1 and BRCA2 across different tumor subtypes (HER2+, HR+/HER2− and HR−/HER2+) and different datasets (Figs. 3 b and 3 c). We combined all mutations in BRCA1 and BRCA2 together. The combined gene set was significantly associated with the contribution to HRD signature in both TCGA (p = 5.0 × 10−12) and Nigeria samples (p = 0.0036). This association was also highly consistent across different tumor subtypes and datasets (Fig. 3 d). Despite genome‐wide significance, mutations in COL15A1 and PIGO are rare and further validation of their associations is desirable.
In addition, when we combined all mutations occurring in a set of known breast cancer predisposition genes (BROCA gene panel; 17 genes), or when we merged all homologous recombination related genes (29 genes), we found that both compounded gene sets were significantly associated with the contribution of HRD signature (p = 2.6 × 10−7 and 2.8 × 10−5, respectively). The significance level remained in the validation set (p = 0.0007 and 0.014, respectively). However, after excluding BRCA1 and BRCA2, neither of these gene sets remained significantly associated with HRD signature contribution. Two of HR related genes, BRIP1 and RAD50, probably contributed to the association signal since many Nigerian samples had HRD contributions present yet no germline mutation in BROCA or homologous recombination genes (Supporting Information Table S4).
Discussion
Using breast cancer data from 1,011 patients from TCGA (the training set) and 170 Nigerian women (the validation set), we extracted four stable mutational signatures including APOBEC C>T, APOBEC C>G, aging and HRD, which is consistent with previous studies.1, 4, 40, 41 These signatures alone were able to explain the majority of the tumor mutations across both cohorts. Heritability analysis suggests that germline genetic factors could explain some of the heterogeneity in mutation signatures across patients with breast cancer. We confirmed the association between rare deleterious variants in BRCA1/2 and increased HRD signature activity. We found that common variants proximal to GNB5 as well as rare exonic mutations within MTCL1 were associated with increased APOBEC C>T/G activity, acting as plausible contributors to the observed heritability. Rare variants in HIVEP1 were also marginally associated with the signature although this did not reach genome‐wide significance, probably because of the small sample size.
APOBEC signatures, the most widespread mutational signatures in human cancers, have been reported in 16 of 30 tumor types from TCGA, implicating APOBEC enzymes as some of the most pervasive mutators in human cancers.10 We found seven common SNPs in/near GNB5 to be significantly associated with APOBEC signature contribution in TCGA. However, this signal could not be replicated in Nigerians, possibly due to limited sample size. Previous studies suggested a potential role for GNB5 in cancer. First, GNB5 encodes guanine nucleotide‐binding protein beta‐5, and this protein is expressed in breast tissue.42 Evidence from the Human Protein Atlas at the protein and mRNA levels also suggested the favorable prognostic role of GNB5 in renal cancer and endometrial cancer. Second, overexpression of GNB5 promoted a migratory phenotype in lung adenocarcinoma cells.43 However, it remains unclear whether GNB5 directly affects the transcription, translation or activity of APOBEC enzymes. Further molecular analyses are required to functionally characterize the observed relationship and its generalizability to other tumor types.
The association between HIVEP1 and APOBEC signature could be explained by their functional roles in viral defense.44, 45 The presence of viral element can stimulate APOBEC expression through a complex network of innate immunity signaling, including components like Toll‐like receptors, interferons, interleukins and even P53.46 It is possible that HIVEP1 proteins play a more direct role in APOBEC regulation as it binds enhancer elements containing the GGGACTTTCC motif.47 In the context of disease, downregulation of another family member, HIVEP2 gene, has already been shown as one of the genetic events responsible for breast cancer.48 We did not find prior evidence directly implicating MTCL1, also known as suppressor of glucose autophagy associated 2 (SOGA2), in APOBEC activity. Suggestive association between MTCL1 and breast cancer risk has been reported in women of Indonesian ancestry, further indicating the potential importance of germline variation proximal to this gene.49 To further understand the biological basis of our observations, it will be necessary to conduct functional studies to uncover the exact causal mechanisms that may ultimately inform innovative therapeutic approaches.
We found that the combined effects of rare, deleterious alleles in BRCA1 and BRCA2 were consistently associated with higher HRD signature contributions in both TCGA and Nigerian data. This finding was in accordance with other studies done with prostate and gastric cancers,1, 14, 15 and the mechanisms promoting HRD activity have been explored in several experimental studies.30, 41, 50, 51, 52 However, to the best of our knowledge, this is the first report of association in an indigenous African population in Nigeria, and our findings could have significant implications for cancer control in underserved and underrepresented population with higher burden of aggressive young onset breast cancer.20
In previous work, Zhu and colleagues observed a significant inverse association (p = 8.75 × 10−6) between the risk allele in rs2588809 of the gene RAD51B and total somatic mutation count across 638 breast cancer patients of European ancestry from TCGA.8 In our study, rs2588809 was not associated with the contribution of any signatures, but rare deleterious variants within RAD51B were found to be nominal significantly correlated with the contributions of aging and HRD signatures. It has been shown that loss‐of‐function mutations in homologous recombination genes other than BRCA1/2 can facilitate HRD signature activity.41 It is possible that RAD51B deficiency increases the mutational burden of tumors through HRD, potentially making these patients good candidates for immunotherapy due to their increased likelihood of harboring neoantigens.53 Taken together, these findings suggest that genetic ancestry may play an important role in mutational development and cancer progression. This highlights the need to accumulate more genetic data from diverse populations in order to better understand the heightened aggressive breast cancer risk observed in individuals of African ancestry.54
There are multiple advantages in our study. First, samples from Nigeria added a substantial value in terms of validation, which expanded the findings from a primarily US European ancestry population to an indigenous African population.55 Second, we harmonized data from a number of sources using the same pipeline, which avoided batch effect due to bioinformatics software. Third, we inferred mutations signatures across all samples using nonnegative matrix factorization and used the estimated signature contributions directly for analysis. This offered advantages over previous approaches which used the burden of mutations found in specific trinucleotide contexts as a proxy for APOBEC signature activity.3, 8, 56, 57 Fourth, we comprehensively explored the association between common genetic variants, rare deleterious variants and the contribution of mutational signatures across different ethnicities, and provided insights to the relationship between germline variants and somatic mutational processes.10 Several study limitations should also be noted. First, we only focused on the substitutions, and did not consider other types of somatic mutation, such as indels and structural variants.3, 41 Second, while WES allowed us to acquire a larger number of data, our analyses were confined to somatic mutations within protein coding regions. WGS could uncover microscale and macroscale somatic alterations58 and provide us more stable estimates of mutation signature contributions. As such, we only focused on the four signatures with strong concordance between WGS data and WES data. Although common practice,1, 22 estimating mutation signatures from exome sequencing could cause potential biases due to sequence motif representation as compared to whole genome. APOBEC enzymes have been shown to preferentially target genic regions, which is likely due to the availability of ssDNA substrates during transcription.59, 60, 61 Additional work has demonstrated that APOBEC mutations are also enriched within early‐replicating regions regardless of transcriptional activity.60 It is unclear if identical mutagenic processes affect all early‐replicating and transcriptionally active regions. Even though we observed high mutation signature correlations between exomes and genomes, we cannot rule out the possibility that different molecular mechanisms generated similar mutational signatures in coding and noncoding regions. It is also possible that a single mutational process is responsible for generating somewhat different signatures in these regions. As such, it is important to remember that signature contribution estimates only serve as a phenotypic marker and may not fully capture the nuances of underlying mechanistic processes. Third, causal links between germline and somatic mutational processes are one explanation for the associations presented here, but other explanations cannot be ruled out.30
Conclusions
In summary, our study identified associations between germline variants and mutational patterns in breast cancer across different ethnicities, especially in African women for the first time. This finding may advance our understanding of breast cancer etiology with potential implications for prevention and treatment. However, further replications in larger and diverse populations are needed to validate our findings. Meanwhile, future work focused on understanding the biological basis of cancer susceptibility alleles will be instrumental in better understanding the complex interplay between germline genetics and somatic mutations.
Author contributions
Experimental design: White KP, Huo D and Olopade OI. Patient recruitment and clinical annotation of samples supervision: Olopade OI. Genomic materials preparation at UCH: Odetunde A. Operation of sample preparation supervision at UCH: Falusi A. Patient's recruitment and harvesting specimens from patients in Nigeria: Popoola A, Ogundiran T and Ibrahim N. Pathological assessment of patient specimens at UCH: Oluwasola O and Ajani M. Study supervision at UCH: Ojengbede O. Pathological assessment at LASUTH: Sanni A and Obafunwa J. Pathological assessment at the University of Chicago: Khramtsova G. Study operation at the University of Chicago: Yoshimatsu TF and Hurley I. Quality assessment of sequence data: Fitzgerald D and Pitt JJ. Data analysis: Wang S, Pitt JJ, Zheng Y, Yoshimatsu TF, Fitzgerald D, Gao G and Huo D. Statistical analysis supervision: Huo D. WES data production and analysis supervision: Barretina J. Data analysis and result interpretation supervision: White KP, Huo D and Olopade OI. Article writing: Wang S, Pitt JJ, Zheng Y, Yoshimatsu TF, White KP, Huo D and Olopade OI. Final approval of the article: all authors.
Data availability
Raw TCGA data used in this analysis were downloaded from TCGA Data Portal or Cancer Genomics Hub. Access to the harmonized variant calls that support the findings of our study are available on request from the corresponding author (O.I.O.). The raw sequencing data from Nigerian cases is available through dbGaP under Study Accession phs001687.v1.p1.
Supporting information
Table S1. Summary of basic information for WES and WGS samples
Table S2. Potential regulatory function of the top seven significant SNPs and their tagged SNPs in GNB5
Table S3. Associations of GWAS index SNPs for breast cancer with contributions of selected mutation signatures
Table S4. Associations of selected genes for breast cancer with contributions of selected mutation signatures
Figure S1. Distribution of the somatic mutation counts across ethnicity and testing methods (WES vs. WGS)
Figure S2. Nine mutational signatures found in breast cancer created from both TCGA and Nigeria samples
Figure S3. The mean Kullback–Leiber divergence for mutation signatures between COSMIC and ours
Figure S4. The proportions explained by each signature for TCGA samples and Nigeria samples
Figure S5. Concordance between signature contribution in WGS and WES in the 138 patients
Figure S6. Manhattan plot showing genome‐wide p values of SNP's association for four selected signature (with signals)
Figure S7. Plots of 10‐based log‐transformed p values in association with contribution of APOBEC signatures
Figure S8. Manhattan plot showing genome‐wide p values of gene's association for four selected signature (with signals)
Figure S9. Associations between contribution of APOBEC signature and number of rare deleterious variants in selected genes
Acknowledgements
We are greatly indebted to all the patients who agreed to participate in our study and graciously donated their biological materials. Our study was supported by U01 CA161032 awarded to D.H., K.W., and O.I.O.; R01 CA228198 to D.H.; Susan G. Komen for the Cure (SAC110026) to O.I.O; Breast Cancer Research Foundation grants awarded to O.I.O. and D.H.; as well as by funding from the Novartis Institutes for Biomedical Research granted to O.I.O. Computational resources were provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory, under grant 1S10OD018495‐01. We want to thank the New York Genome Center for the quality of the sequencing and analysis services they offered. We also want to thank QIAGEN for their generous donation of PAXgene Tissue Containers and DNA Extraction Kits for our study. We also want to thank Yoo‐Jeong Han, Jing Zhang and Vineet Dhiman for their discussion of this project. S.W. was supported by a University of Chicago Global Health Fellowship and the National Natural Science Foundation of China (Grant No. 81502884). Y.Z. is supported by Paul Calabresi Career Development Award for Clinical Oncology (K12 CA139160, PI O.I.O.). This article is dedicated to the memory of the late Professor Abideen Olayiwola Oluwasola who was supported by D43 TW009112.
Conflict of interest: K.P.W. serves as President, and is a shareholder of Tempus Labs Inc. O.I.O. serves as a board member of Healthy Life for all Foundation, is a co‐founder, Chief Scientific Advisor and equity stockholder of CancerIQ, serves as Scientific Adviser and is a stockholder of Tempus. J.B. was a former Novartis employee and stock holder.
Data Availability: Raw TCGA data used in this analysis were downloaded from TCGA Data Portal or Cancer Genomics Hub. Access to the harmonized variant calls that support the findings of our study are available on request from the corresponding author (O.I.O.). The raw sequencing data from Nigerian cases is available through dbGaP under Study Accession phs001687.v1.p1.
Contributor Information
Kevin P. White, Email: kevin@tempus.com.
Dezheng Huo, Email: dhuo@health.bsd.uchicago.edu.
Olufunmilayo I. Olopade, Email: folopade@medicine.bsd.uchicago.edu.
References
- 1. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Segovia R, Tam AS, Stirling PC. Dissecting genetic and environmental mutation signatures with model organisms. Trends Genet 2015;31:465–74. [DOI] [PubMed] [Google Scholar]
- 3. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science (New York, NY) 2015;349:1483–9. [DOI] [PubMed] [Google Scholar]
- 4. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013;3:246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu S, Powers S, Zhu W, et al. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016;529:43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brucher BL, Jamall IS. Somatic mutation theory—why it's wrong for Most cancers. Cell Physiol Biochem 2016;38:1663–80. [DOI] [PubMed] [Google Scholar]
- 7. Alderton GK. Cancer risk: debating the odds. Nat Rev Cancer 2016;16:68. [DOI] [PubMed] [Google Scholar]
- 8. Zhu B, Mukherjee A, Machiela MJ, et al. An investigation of the association of genetic susceptibility risk with somatic mutation burden in breast cancer. Br J Cancer 2016;115:752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Robles‐Espinoza CD, Roberts ND, Chen S, et al. Germline MC1R status influences somatic mutation burden in melanoma. Nat Commun 2016;7:12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Middlebrooks CD, Banday AR, Matsuda K, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC‐signature mutations in tumors. Nat Genet 2016;48:1330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nik‐Zainal S, Wedge DC, Alexandrov LB, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC‐dependent mutations in breast cancer. Nat Genet 2014;46:487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starrett GJ, Luengas EM, McCann JL, et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun 2016;7:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phipps AI, Ahnen DJ, Cheng I, et al. PIK3CA somatic mutation status in relation to patient and tumor factors in racial/ethnic minorities with colorectal cancer. Cancer Epidemiol Biomarkers Prev 2015;24:1046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Decker B, Karyadi DM, Davis BW, et al. Biallelic BRCA2 mutations shape the somatic mutational landscape of aggressive prostate Tumors. Am J Hum Genet 2016;98:818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sahasrabudhe R, Lott P, Bohorquez M, et al. Germline mutations in PALB2, BRCA1, and RAD51C, which regulate DNA recombination repair, in patients with gastric cancer. Gastroenterology 2017;152:983–6.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J, Mouw KW, Polak P, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016;48:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rahman N. Realizing the promise of cancer predisposition genes. Nature 2014;505:302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Machiela MJ, Ho BM, Fisher VA, et al. Limited evidence that cancer susceptibility regions are preferential targets for somatic mutation. Genome Biol 2015;16:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ding L, Bailey MH, Porta‐Pardo E, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell 2018;173:305–320.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huo D, Ikpatt F, Khramtsov A, et al. Population differences in breast cancer: survey in indigenous African women reveals over‐representation of triple‐negative breast cancer. J Clin Oncol 2009;27:4515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huo D, Hu H, Rhie SK, et al. Comparison of breast cancer molecular features and survival by African and European ancestry in the cancer genome atlas. JAMA Oncol 2017;3:1654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pitt JJ, Riester M, Zheng Y, et al. Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun 2018;9:4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mcclellan JM, Lehner T, King MC. Gene discovery for complex traits: lessons from Africa. Cell 2017;171:261–4. [DOI] [PubMed] [Google Scholar]
- 24. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huo D, Zheng Y, Ogundiran TO, et al. Evaluation of 19 susceptibility loci of breast cancer in women of African ancestry. Carcinogenesis 2012;33:835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gehring JS, Fischer B, Lawrence M, et al. SomaticSignatures: inferring mutational signatures from single‐nucleotide variants. Bioinformatics (Oxford, England) 2015;31:3673–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Temko D, Tomlinson I, Severini S, et al. The effects of mutational process and selection on driver mutations across cancer types. Nat Commun 2018;9:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Helleday T, Eshtad S, Nik‐Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014;15:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katainen R, Dave K, Pitkanen E, et al. CTCF/cohesin‐binding sites are frequently mutated in cancer. Nat Genet 2015;47:818–21. [DOI] [PubMed] [Google Scholar]
- 32. Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010;42:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaitlen N, Kraft P, Patterson N, et al. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 2013;9:e1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou K, Donnelly L, Yang J, et al. Heritability of variation in glycaemic response to metformin: a genome‐wide complex trait analysis. Lancet Diab Endocrinol 2014;2:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchini J, Howie B. Genotype imputation for genome‐wide association studies. Nat Rev Genet 2010;11:499–511. [DOI] [PubMed] [Google Scholar]
- 36. Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare‐variant association testing with application to small‐sample case–control whole‐exome sequencing studies. Am J Hum Genet 2012;91:224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 2012;13:762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu MC, Lee S, Cai T, et al. Rare‐variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011;89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee S, Abecasis GR, Boehnke M, et al. Rare‐variant association analysis: study designs and statistical tests. Am J Hum Genet 2014;95:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nik‐Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149:979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature 2016;534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong H, Claffey KP, Brocke S, et al. Expression of phosphodiesterase 6 (PDE6) in human breast cancer cells. SpringerPlus 2013;2:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang SH, Li CF, Chu PY, et al. Overexpression of regulator of G protein signaling 11 promotes cell migration and associates with advanced stages and aggressiveness of lung adenocarcinoma. Oncotarget 2016;7:31122–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ejima T, Hirota M, Mizukami T, et al. An anti‐HIV‐1 compound that increases steady‐state expression of apoplipoprotein B mRNA‐editing enzyme‐catalytic polypeptide‐like 3G. Int J Mol Med 2011;28:613–6. [DOI] [PubMed] [Google Scholar]
- 45. Radwan MO, Sonoda S, Ejima T, et al. Zinc‐mediated binding of a low‐molecular‐weight stabilizer of the host anti‐viral factor apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like 3G. Bioorg Med Chem 2016;24:4398–405. [DOI] [PubMed] [Google Scholar]
- 46. Roberts SA, Lawrence MS, Klimczak LJ, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45:970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baldwin AS Jr, LeClair KP, Singh H, et al. A large protein containing zinc finger domains binds to related sequence elements in the enhancers of the class I major histocompatibility complex and kappa immunoglobulin genes. Mol Cell Biol 1990;10:1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fujii H, Gabrielson E, Takagaki T, et al. Frequent down‐regulation of HIVEP2 in human breast cancer. Breast Cancer Res Treat 2005;91:103–12. [DOI] [PubMed] [Google Scholar]
- 49. Haryono SJ, Datasena IG, Santosa WB, et al. A pilot genome‐wide association study of breast cancer susceptibility loci in Indonesia. Asian Pac J Cancer Prev 2015;16:2231–5. [DOI] [PubMed] [Google Scholar]
- 50. Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003;22:5784–91. [DOI] [PubMed] [Google Scholar]
- 51. Ngeow J, Nizialek E, Eng C. Into the eye of the storm: breast cancer's somatic mutation landscape points to DNA damage and repair. Transl Cancer Res 2013;2:59–61. [PMC free article] [PubMed] [Google Scholar]
- 52. Zamborszky J, Szikriszt B, Gervai JZ, et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017;36:746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rieke DT, Ochsenreither S, Klinghammer K, et al. Methylation of RAD51B, XRCC3 and other homologous recombination genes is associated with expression of immune checkpoints and an inflammatory signature in squamous cell carcinoma of the head and neck, lung and cervix. Oncotarget 2016;7:75379–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramakodi MP, Kulathinal RJ, Chung Y, et al. Ancestral‐derived effects on the mutational landscape of laryngeal cancer. Genomics 2016;107:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Amar D, Izraeli S, Shamir R. Utilizing somatic mutation data from numerous studies for cancer research: proof of concept and applications. Oncogene 2017;36:3375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morganella S, Alexandrov LB, Glodzik D, et al. The topography of mutational processes in breast cancer genomes. Nat Commun 2016;7:11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Van den Eynden J, Larsson E. Mutational signatures are critical for proper estimation of purifying selection pressures in cancer somatic mutation data when using the dN/dS metric. Front Genet 2017;8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagarajan N, Bertrand D, Hillmer AM, et al. Whole‐genome reconstruction and mutational signatures in gastric cancer. Genome Biol 2012;13:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Swanton C, McGranahan N, Starrett GJ, et al. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov 2015;5:704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kazanov MD, Roberts SA, Polak P, et al. APOBEC‐induced cancer mutations are uniquely enriched in early‐replicating, gene‐dense, and active chromatin regions. Cell Rep 2015;13:1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seplyarskiy VB, Andrianova MA, Bazykin GA. APOBEC3A/B‐induced mutagenesis is responsible for 20% of heritable mutations in the TpCpW context. Genome Res 2017;27:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of basic information for WES and WGS samples
Table S2. Potential regulatory function of the top seven significant SNPs and their tagged SNPs in GNB5
Table S3. Associations of GWAS index SNPs for breast cancer with contributions of selected mutation signatures
Table S4. Associations of selected genes for breast cancer with contributions of selected mutation signatures
Figure S1. Distribution of the somatic mutation counts across ethnicity and testing methods (WES vs. WGS)
Figure S2. Nine mutational signatures found in breast cancer created from both TCGA and Nigeria samples
Figure S3. The mean Kullback–Leiber divergence for mutation signatures between COSMIC and ours
Figure S4. The proportions explained by each signature for TCGA samples and Nigeria samples
Figure S5. Concordance between signature contribution in WGS and WES in the 138 patients
Figure S6. Manhattan plot showing genome‐wide p values of SNP's association for four selected signature (with signals)
Figure S7. Plots of 10‐based log‐transformed p values in association with contribution of APOBEC signatures
Figure S8. Manhattan plot showing genome‐wide p values of gene's association for four selected signature (with signals)
Figure S9. Associations between contribution of APOBEC signature and number of rare deleterious variants in selected genes
Data Availability Statement
Raw TCGA data used in this analysis were downloaded from TCGA Data Portal or Cancer Genomics Hub. Access to the harmonized variant calls that support the findings of our study are available on request from the corresponding author (O.I.O.). The raw sequencing data from Nigerian cases is available through dbGaP under Study Accession phs001687.v1.p1.