

Summary
Aims
The classifications of occluding vasculopathies may present some difficulties. Firstly, classifications may follow different principles, e.g. clinicopathological findings, etiology or pathomechanism. Secondly, authors sometimes do not distinguish between vasculitis and vasculopathy. Thirdly, vasculopathies are often systemic diseases. Organ‐specific variations make morphologic findings difficult to compare. Moreover, subtle changes may be recognized in the skin, but be invisible in other organs. Our aim was to use the skin and subcutis as tools and clinicopathological correlation as the basic process for classification.
Methods and results
In the first step, we differentiate between small and medium vessel occluding vasculopathies in the skin, and focus in this part on small vessel occluding vasculopathies. In the second step, we differentiate among subtypes of small vessels. In the final step, we differentiate according to the time point of the coagulation/reorganization process and the involved inflammatory cells/stromal features. Applying the same procedure to the various entities and visualizing the findings with bar codes makes the similarities and differences more apparent, both clinically and with histopathology.
Conclusion
Occluding vasculopathies are often not separate entities, but reaction patterns and epiphenomena. Distinguishing them from vasculitides is crucial because of differences in pathogenesis, therapeutic approach and prognosis.
Introduction
Vasculopathies are diseases evolving in and around vessels. There are two large groups: vasculitides and occluding vasculopathies. With vasculitis (discussed previously in an algorithmic context 1), the pathogenetic process starts in the vessel wall. We find increased numbers of inflammatory cells in and/or around the vessel wall accompanied by vascular damage 2, 3, 4, 5, 6, 7, 8, 9, 10. Damage can be recognized by leukocytoclasia, endothelial and smooth muscle necrosis, as well as fibrin and connective tissue degeneration. Thrombi can also be observed, but they are a secondary phenomenon caused by damage to the vessel wall. Occluding vasculopathies 11 can be caused by proliferations such as malignancies, by embolization of different materials (cholesterol, oxalate, microorganisms) or most commonly by coagulopathies with occlusion by thrombi. Coagulopathies differ from vasculitides in their initiation by abnormal blood coagulation. We find partial or complete occlusion of one or more vessels due to hypercoagulability, in particular by thrombi and emboli. Inflammatory cells in and/or around the vessel wall appear as a secondary phenomenon.
Distinguishing between primary vasculitis and primary vasculopathy can be difficult, because the two pathological processes are often intertwined. On the one hand, vasculitis may not be recognized because it may trigger vaso‐occlusive events that dominate the clinical and histopathological picture, e.g. in septic vasculitis (primary vasculitis due to activation of endothelial cells is often subtle, while vascular occlusion due to disseminated intravasal coagulation is dominating). On the other hand, vasculopathies may be misinterpreted as vasculitis because they may show a secondary inflammatory infiltrate as the dominant feature in due course.
We have already reported our view on vasculitides (and their classification based on clinicopathological correlation) as illustrated by the “vasculitic wheel” 1. We now aim to apply similar criteria to occluding vasculopathies.
Comparison of previous classifications 2, 3, 4, 5, 6, 8, 9, 10, 12, 13, 14, 15, of vasculopathies is difficult. Firstly, classifications may follow different principles, including clinicopathological findings, etiology, pathomechanism, prognosis or therapeutic options. Unfortunately, all widely used current and past classifications confuse different features such as size of the vessels, etiological factors and pathogenetic considerations. Secondly, colleagues often fail to distinguish clearly between the primary event as vasculitis or vasculopathy, which has important therapeutic consequences 16, 17; classical vasculitides benefit from immunosuppressive and anti‐inflammatory therapies; microorganism‐associated vasculitides (septicemia) need specific antimicrobial agents; and coagulopathies require anticoagulant therapy (acetylsalicylic acid, heparin, coumarin, new oral anticoagulants). Thirdly, a vasculopathy may be a relatively benign, single‐organ (e.g. cutaneous) disease or a systemic multiorgan disease (CNS, kidney, lungs) with a poor prognosis 15, 18, 19, 20. The differing conditions from organ to organ modify the pathological process (for example, a proclivity to hemorrhage in loose lung tissue or accumulation of capillary loops make glomeruli a predisposed focus of vascular processes) and make morphological findings difficult to compare. Moreover, subtle changes are easily recognized in the skin and may be reflected in CNS symptoms, but changes are sometimes asymptomatic or invisible in other organs, as in patients with Sneddon syndrome.
Our approach focuses on clinical signs and histopathology. Etiological and pathogenetic data are not primarily used in our approach. However, pathogenetic evaluation is crucial. Laboratory tests (Table 1a) 21, 22, and imaging (especially in medium vessel vasculopathies; Table 1b), are necessary and used to verify or falsify diagnoses, based primarily on clinical and histopathological data. In addition, other specialties, in particular cardiology, pneumology, nephrology, rheumatology, neurology and ophthalmology, must frequently be included in a multidisciplinary approach to optimal patient management.
Table 1a.
Diagnostic laboratory procedure
|
Table 1b.
Diagnostic imaging procedures
| 1. Abdominal ultrasound | SLE, APS |
| 2. Cerebral MRI | Sneddon syndrome, SLE, APS |
| 3. PET‐CT | PAN, GP, EGP, MPA, Takayasu syndrome |
Abbr.: SLE, systemic lupus erythematosus; APS, anti‐phosholipid syndrome; PAN, polyarteritis nodosa; GP, granulomatosis with polyangiitis; EGP, eosinophilic granulomatosis with polyangiitis; MPA, microscopic polyangiitis.
We use the skin and subcutis as a tool and clinicopathological correlation as the basic process for classification. We apply an algorithmic approach with pattern analysis, which allows consistent and reliable reporting of microscopic findings. With an approach similar to that of the International Chapel Hill Consensus Conference on the nomenclature of vasculitides from 1994 13, 2012 14 and 2018 23, we first differentiate between small and medium vessel coagulopathies (there are no large vessels in the skin). In part I of our review, we focus on small vessel vasculopathies (Figure 1). In the second step, we differentiate the subtypes of small vessels (capillaries versus postcapillary venules). Capillaries are found in dermal papillae, in the perifollicular and periglandular connective tissue, between collagen fibers in reticular dermis and within the lobules of the panniculus; in contrast, postcapillary venules contribute to the superficial and deep perivascular plexus – the former at the border between the papillary and reticular dermis, the latter at the border between the reticular dermis and subcutis. Other venules include the interconnecting venules between superficial and deep plexus and the septal venules. In the final step, we differentiate according to the life cycle of the event. All vasculo/coagulopathies have a characteristic life cycle of histopathological events. Early stages are dominated by fibrin thrombi without significant inflammation. This process can favor capillaries (e.g. livedo vasculopathy, coumarin/heparin necrosis, septic vasculitis), postcapillary venules (e.g. systemic lupus erythematosus, anti‐phospholipid syndrome) or larger vessels (e.g. Sneddon syndrome, calciphylaxis, cholesterol emboli), especially when the intensity of the process increases (Figure 1). At this early stage, there is frequently prominent hemorrhage, usually presenting as non‐inflammatory retiform purpura, probably due to ischemia with hemorrhage prior to complete occlusion. According to the extent of disease, one will find necrotic lesions with erosion to ulceration, cellular debris with crust formation, and granulation tissue with a mixed infiltrate of neutrophils, lymphocytes and macrophages. In due course, degradation of these thrombi leads to “lymphocytic vascular reorganization”, a process dominated more or less by a dense perivascular lymphocytic infiltrate that invades the walls of thrombosed or damaged vessels to a variable extent. This is a histopathological reaction pattern that is referred to as lymphocytic vasculitis by some dermatopathologists. The term is misleading as it is not vasculitis and is not consistently used, so it should be avoided. Finally, there is healing with complete reconstitution of vessels with lumina, and/or partial to complete occlusion of vessels by fibroblasts and collagen. Deep erosions (in particular ulcers) heal with scars, as seen for example in atrophie blanche.
Figure 1.
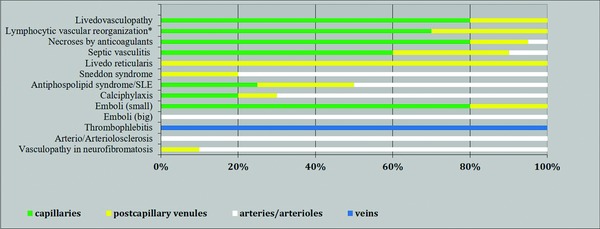
Association of entities/reaction patterns with vessel size.
Abbr.: SLE, systemic lupus erythematosus
Emboli (small): fat, air, gas; emboli (large): cholesterol, oxalate, embolia cutis medicamentosa.
*All inflammatory and even proliferative/neoplastic processes may be associated with coagulation disorders and thus with fibrin thrombi that can lead to lymphocytic vascular reorganization.
This life cycle of histopathological events in occluding vasculopathies is seen in a variety of instances. Any erosion, ulceration or necrotic lesion (Figure 2) causes hypercoagulability with fibrin thrombi in the surrounding tissue. The clue to differentiation from vasculitis is the distribution around capillaries and accentuation around postcapillary venules. The process is accentuated near and/or closer to erosions/ulcers in the case of occluding vasculopathies and decreases gradually with increasing distance. Nuclear dust is present in both instances and does not help with differentiation. However, the accentuation of the process helps: vasculopathy will focus the process towards the epidermis while vasculitis will focus on dermal vessels, usually postcapillary venules – another important clue. This histopathological process caused by occluding vasculopathies has been referred to as “secondary vasculitis”. As it is not vasculitis, we recommend that this term also be avoided. The histopathological presentation does not always allow one to differentiate between the various causes of occluding vasculopathies, which must be done by clinicopathological correlation.
Figure 2.
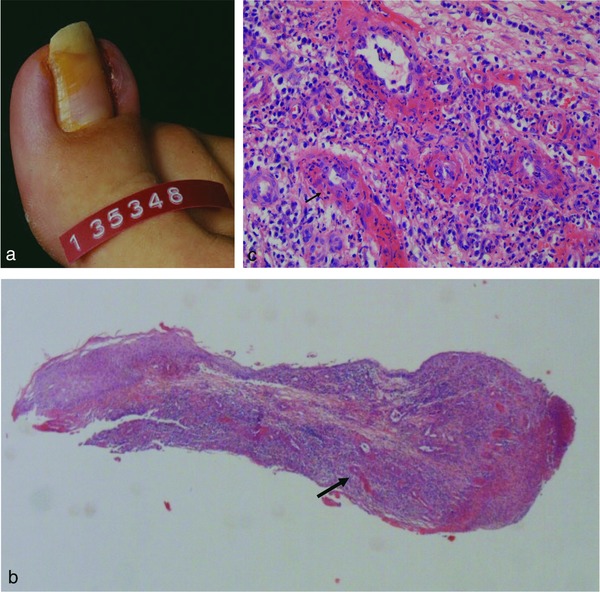
Occluding vasculopathy secondary to infection. Acute paronychia on the right big toe with erythema, swelling, erosion and crusting (a). Histology of Nicoladoni surgery shows eroded epidermis with dense mixed inflammatory infiltrate that is perivascular and diffuse (b). Fibrin thrombi surrounded by lymphocytes, neutrophils and nuclear dust. Arrows indicate corresponding areas of Figure 2b and 2c (c).
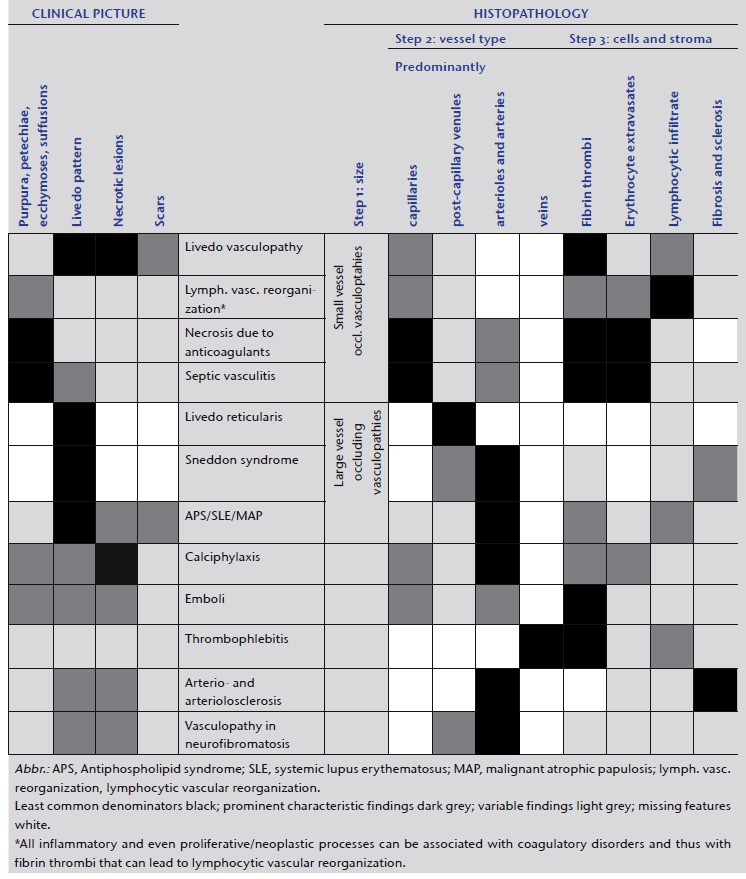
In our algorithmic approach, we use tables and shading to highlight the different features, in order to facilitate comparison between the different manifestations and to grade the importance of certain features (Tables 2–5). We mark the least common denominators in black, prominent characteristic findings in dark gray, variable findings in light gray and missing features in white. By applying the same procedure to the various entities, the overlaps and differences, based on the clinical picture as well as histopathology, become more apparent. We try to visualize these in the form of bar codes; this helps to simplify the findings and render them comparable. However, we are aware that this method of classifying vasculopathies has its limits until clear molecular tools are discovered.
Table 2.
Livedo vasculopathy
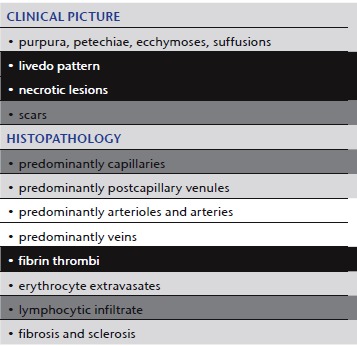
|
Table 5.
Septic vasculitis
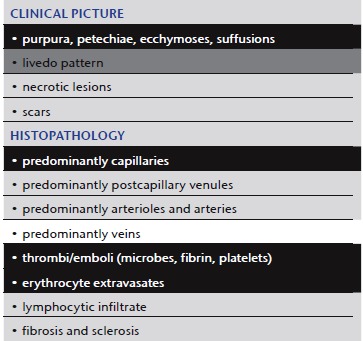
|
Livedo vasculopathy
Although many aspects of its pathogenesis have not been clearly delineated, we regard livedo vasculopathy as a characteristic clinically and histopathologically defined process 24, 25, caused by a variety of coagulation disorders 11, 22, (such as AT‐III deficiency, protein‐C and protein‐S deficiency, APC resistance, cryoglobulinemia, cold agglutinin disease, cryofibrinogenemia), even though one cannot always detect or define such a defect. Clinical appearance and histopathological findings (Table 2) are characteristically independent of etiology and pathomechanism. However, when the cause is identified, the therapeutic approach can be correlated and be more successful. Locoregional factors such as stasis, high blood pressure in the veins of the lower leg, as well as perforator veins and thus blood flow problems are also important. Some features of livedo vasculopathy can also be observed complicating other diseases (for example chronic venous insufficiency, anti‐phospholipid syndrome, lupus erythematosus, Sneddon syndrome, hepatitis B or C infections, multiple myeloma, paroxysmal nocturnal hemoglobulinemia, polycythemia, thrombocytosis, leukemia, lymphoma) or can be caused by drugs such as hydroxyurea.
Clinic: The triad of livedo racemosa, very painful ulcers (acute phase) and atrophie blanche (scarring regeneration) is highly characteristic of this process (Figure 3a, b) 26.
Figure 3.
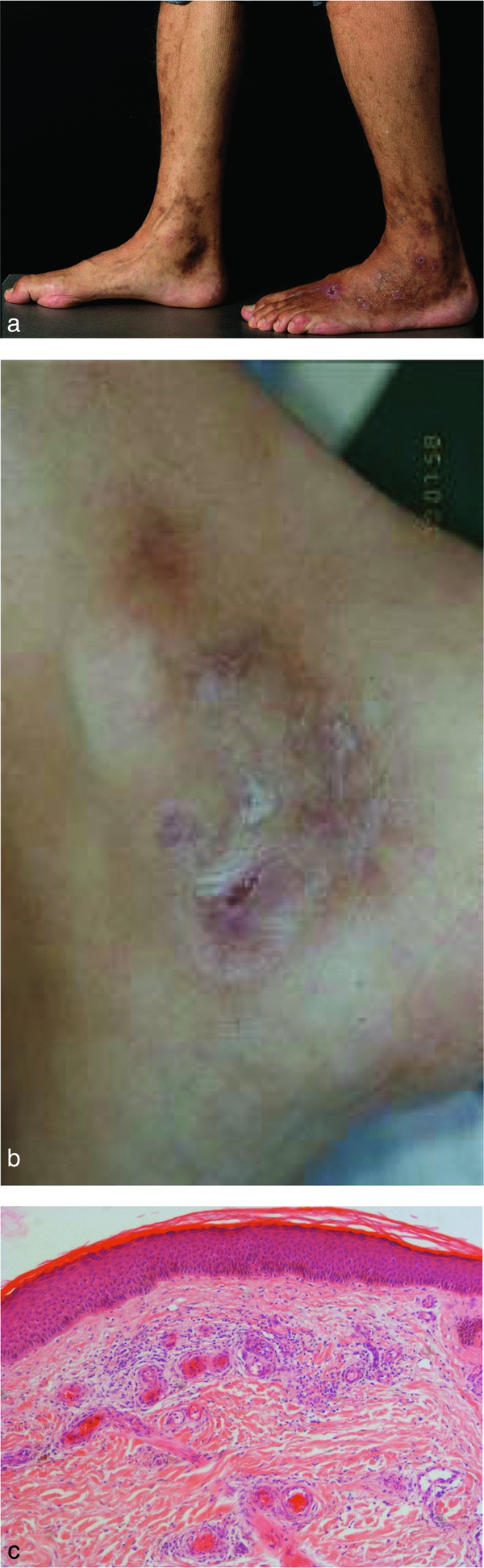
Livedo vasculopathy. Painful bizarre ulcers with hemorrhagic to livid‐brown livedo racemosa in a characteristic location around the ankle (a). Late scarring regeneration, “atrophie blanche” (b). Occluding thrombi of capillaries and postcapillary venules surrounded by lymphocytes (the histology of septic vasculitis is mostly identical, except for the presence of neutrophils in the area around primary damage due to septicemia/bacteremia) (c).
Synonyms: Livedo vasculitis, livedoid vasculitis, (idiopathic) atrophie blanche, atrophie blanche vasculitis, livedo reticularis with winter ulcerations, livedo reticularis with summer ulcerations, segmental hyalinizing vasculitis, PPURPLE (painful purpuric ulcers with reticulated patterning of lower extremities).
Histology: Occluding thrombi of capillaries and postcapillary venules are sparsely surrounded by lymphocytes. We suggest the term “lymphocytic vascular reorganization”, previously known as “lymphocytic vasculitis” (Figure 3c). So‐called “hyalinized” vessels in the superficial dermis showing thrombi as well as fibrin in/around the vessel walls (mostly without involvement of muscular vessels) are typical.
Comment: Livedo vasculitis suggests a vasculitic process. This is due in part to the histopathological presentation with “lymphocytic vasculitis”. However, this is mostly due to the report of Richard Winkelmann and others in the 1970s 27, 28, 29, who described immunofluorescence findings of IgG, C3, C4 and fibrinogen in these disorders and equated these with vasculitis; thus, he introduced the term livedo vasculitis for this disease, which was previously known as atrophie blanche. In our interpretation, this process is primarily based on coagulopathies mimicking vasculitis. Thus, the term livedo vasculopathy seems more appropriate.
“Lymphocytic vascular reorganization” (previously known as lymphocytic vasculitis)
In lymphocytic vascular reorganization (referred to by some histopathologists as lymphocytic vasculitis) (Table 3) we see a characteristic histological reaction pattern in a variety of instances 30 (Figure 2). The least common denominator is a coagulopathy with fibrin thrombi. The coagulation cascade must keep a balance between coagulation and fibrinolysis. Slight dysregulation can cause fibrin thrombi on the one hand or varying degrees of blood extravasation on the other. Thus, clinical pictures can be broad and may vary between purpura (i.e. pigmented purpura [Schamberg disease], previously also known as capillaritis) 31 and livedo pattern (i.e. malignant atrophic papulosis, Degos disease). In our view, every inflammatory and proliferative neoplastic disease that shows imbalance of coagulation resulting in the formation of fibrin thrombi can be followed by lymphocytic vascular reorganization.
Table 3.
“Lymphocytic vascular reorganization”
|
Comment: Although some authors see “lymphocytic vasculitis” as an authentic form of vasculitis 3, 4, 12, 32, in our experience it is mostly a consequence of a basic pathological process and not itself a fundamental pathological process 19, 30. Thus, we suggest that it be referred to as “lymphocytic vascular reorganization”. We find lymphocytes indicative of regenerative “secondary vasculitis”, i.e. due to coagulopathies. In a T‐cell mediated process, lymphocytic vasculitis would be an appropriate term, but to the best of our knowledge this has not been described so far.
Necrosis induced by anticoagulants (heparin, coumarin)
Clinical picture: These forms of disease/coagulopathy (Table 4) are usually associated with prominent hemorrhage. Patients develop ecchymoses, suffusions and large hematomas (Figure 4). With heparin they occur primarily in areas of subcutaneous injection 33; with coumarin, they occur mainly in areas rich in subcutaneous adipose tissue, e.g. the gluteal region 34.
Table 4.
Necrosis induced by anticoagulants
|
Figure 4.
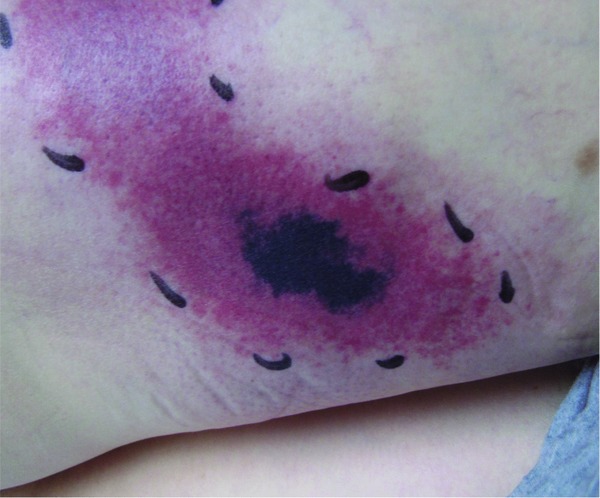
Coumarin necrosis. Hematoma, ecchymoses and necrosis after initiation of anticoagulation therapy with acenocoumarol (Sintrom ®).
Histology: In our experience, thrombi predominantly occlude capillaries, but can also affect all vessels of the superficial and deep dermal plexus. Heparin tends to induce platelet thrombi, while coumarin (as a vitamin K antagonist) tends to induce fibrin thrombi.
Comment: It is a paradox that therapies initiated to prevent coagulation cause hypercoagulation with hemorrhage and necrotic tissue 35.
Septic vasculitis
Septic vasculitis (Table 5) presents another paradox 3, 36, 37. The pathomechanism starts at the vessel wall as classic vasculitis. However, coagulopathy often dominates the histopathological picture. The process appears clinically as vasculitis, while histopathologically mimicking a coagulopathy. Although the coagulopathy is not the primary event, it is dominant. Septic vasculitis is therefore included in this section.
Bacteremia, septicemia or sepsis are by definition the presence of microorganisms within the vascular system. This causes an intravascular reaction via coagulatory proteins such as von Willebrand factor, with inflammation, activation of endothelial cells and fibrin, which can be followed by thrombus clot formation 38, 39, 40. Consequently, septic emboli mainly occlude capillaries, and with time the progressing coagulation cascade will carry the process far beyond the focus of disease and affect larger vessels with fibrin thrombi. While the initial septic emboli will include microorganisms (Figure 5c), the following events will result in a histological picture identical to that of other coagulopathies, with fibrin thrombi, erythrocyte extravasates and necrotic tissue (Figure 5b). Some microorganisms may be more prominent, such as bacteria in ecthyma gangrenosum in Pseudomonas septicemia 37, or fungi in systemic aspergillosis or Aspergillus septicemia. In many instances, the septic core of the process can be difficult to find, for example in meningococcemia, gonococcemia or subacute or smoldering sepsis due to staphylococci associated with subacute bacterial endocarditis. For histopathologists, it is often like looking for needles in a haystack, with the needles representing the microorganisms surrounded by exaggerated hypercoagulation. In other cases, not a single bacterium can be found in sections or cultures, as endotoxins such as lipopolysaccharides (LPSs) (at least in murine models of vasculitis) may destroy vessels and cause vasculitis by activating endothelial cells, followed by adhering neutrophils that release toxic products in immediate proximity to the vessel wall while initiating an uncontrolled coagulation cascade; this is usually the scenario in meningococcal meningoencephalitis. With or without bacteria in the blood, this process is known as disseminated intravascular coagulopathy (DIC) or one of its synonyms such as consumption coagulopathy, Schwartzman‐Sanarelli phenomenon, purpura fulminans or Waterhouse‐Friderichsen syndrome, although strictly speaking it is not correct to list the latter processes as septic vasculitis without histological evidence of bacteria or positive blood cultures.
Figure 5.
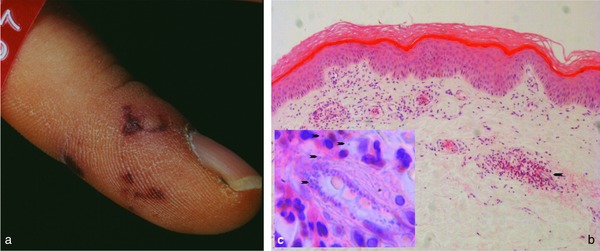
Septic vasculitis. Hemorrhagic, partially livedo‐like macules with acral predilection (retiform purpura) (a). Occluding thrombi of capillaries and postcapillary venules surrounded by lymphocytes (b). Inset indicated by arrow in 5b: neutrophils accentuated and clustered around area with intravascular cocci, also seen in surrounding tissue (arrows) (c).
Clinical picture: There is a wide variety of presentations. According to the size of the involved vessels and the severity of the process, the clinical picture may be asymptomatic or show (frequently with acral accentuation) hemorrhagic macules to nodules, splinter hemorrhages of nails, necrotic tissue or a livedo pattern (retiform purpura) (Figure 5) as well as concomitant migratory arthritis and tendovaginitis. The mechanism and coagulatory outcome depend on the causative toxins, bacteria or fungi. There may be intravascular plasmatic coagulation, platelet activation 41 or endothelial cell damage or endothelial cell activation with the release of coagulatory proteins. Although the clinical appearance can differ, histopathology can only distinguish between bacteria and fungi if the responsible agents can be identified (Gram or fungal staining).
Distinguishing entities in the spectrum of small vessel coagulopathies/vasculopathies can benefit from an algorithmic approach. Applying the same questions makes the clinical and histopathological findings clearer. The same method of bar codes can also be applied to medium vessel coagulopathies (Table 6), which is presented in detail in Part II of our review. There is no sharp border between small and medium vessel coagulopathies, but a continuum. Small vessel coagulopathies can also involve medium vessels and medium vessel coagulopathies may affect small vessels.
Table 6.
Vasculo/coagulopathies – systematic approach and comparison reminiscent of a bar code reader
|
The use of bar codes simplifies the results and may lead to inaccuracy, because it is rarely possible to describe biology in the form of tables. However, it can help in making these entities more understandable and comparable.
Conflict of interest
None.
This manuscript was presented in part at the National Specialist Dermatopathology EQA Scheme in Warwick, United Kingdom, November 8, 2013.
Acknowledgement
We remember our deceased mentor and friend Prof. Dr. Walter Burgdorf and thank him for his valuable input in the conceptual design of this paper.
We thank Reviewer 2 for his effort and suggestions that led to improvement of the manuscript.
References
- 1. Ratzinger G, Zelger BG, Carlson JA et al. Vasculitic wheel – an algorithmic approach to cutaneous vasculitides. J Dtsch Dermatol Ges 2015; 13: 1092–117. [DOI] [PubMed] [Google Scholar]
- 2. Carlson JA, Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopathol 2006; 28: 486–506. [DOI] [PubMed] [Google Scholar]
- 3. Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol 2008; 9: 71–92. [DOI] [PubMed] [Google Scholar]
- 4. Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology 2010; 56: 3–23. [DOI] [PubMed] [Google Scholar]
- 5. Crowson AN, Mihm MC, Jr. , Magro CM. Cutaneous vasculitis: a review. J Cutan Pathol 2003; 30: 161–73. [DOI] [PubMed] [Google Scholar]
- 6. Jorizzo JL. Classification of vasculitis. J Invest Dermatol 1993; 100: 106S–10S. [DOI] [PubMed] [Google Scholar]
- 7. Daoud MS, Gibson LE, De Remee RA et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol 1994; 31: 605–12. [DOI] [PubMed] [Google Scholar]
- 8. Csernok E, Gross WL. Primary vasculitides and vasculitis confined to skin: clinical features and new pathogenic aspects. Arch Dermatol Res 2000; 292: 427–36. [DOI] [PubMed] [Google Scholar]
- 9. Blanco R, Martinez‐Taboada VM, Rodriguez‐Valverde V, Garcia‐Fuentes M. Cutaneous vasculitis in children and adults. Associated diseases and etiologic factors in 303 patients. Medicine (Baltimore) 1998; 77: 403–18. [DOI] [PubMed] [Google Scholar]
- 10. Luqmani RA, Bacon PA, Moots RJ et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994; 87: 671–8. [PubMed] [Google Scholar]
- 11. Llamas‐Velasco M, Alegria V, Santos‐Briz A et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol 2017; 39: 637–62. [DOI] [PubMed] [Google Scholar]
- 12. Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol 2005; 27: 504–28. [DOI] [PubMed] [Google Scholar]
- 13. Jennette JC, Falk RJ, Andrassy K et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187–92. [DOI] [PubMed] [Google Scholar]
- 14. Jennette JC, Falk RJ, Bacon PA et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013; 65: 1–11. [DOI] [PubMed] [Google Scholar]
- 15. Yus ES, Simon RS, Requena L. Vein, artery, or arteriole? A decisive question in hypodermal pathology. Am J Dermatopathol 2012; 34: 229–32. [DOI] [PubMed] [Google Scholar]
- 16. Chen KR. The misdiagnosis of superficial thrombophlebitis as cutaneous polyarteritis nodosa: features of the internal elastic lamina and the compact concentric muscular layer as diagnostic pitfalls. Am J Dermatopathol 2010; 32: 688–93. [DOI] [PubMed] [Google Scholar]
- 17. Dalton SR, Fillman EP, Ferringer T et al. Smooth muscle pattern is more reliable than the presence or absence of an internal elastic lamina in distinguishing an artery from a vein. J Cutan Pathol 2006; 33: 216–9. [DOI] [PubMed] [Google Scholar]
- 18. Ackermann AB, Chongchitnant N, Sanchez J. Histologic Diagnosis of Inflammatory Skin Diseases. Williams & Wilkins, Baltimore, 1997. [Google Scholar]
- 19. Zelger BG, Zelger B. Fibrin in vasculitis: criterion for or mere clue to diagnosis? Dermatopathology: Practical & Conceptual, 2002. [Google Scholar]
- 20. Zelger B. Vasculitis or vasculopathy? What comes first? Chicken or egg? Dermatopathology: Practical & Conceptual, 2004. [Google Scholar]
- 21. Zelger B, Plorer A, Sepp N, Fritsch PO. [Differential livedo syndrome diagnosis]. Hautarzt 1995; 46: 369–79; quiz 77–8. [DOI] [PubMed] [Google Scholar]
- 22. Dabiri G, Damstetter E, Chang Y et al. Coagulation disorders and their cutaneous presentations: Diagnostic work‐up and treatment. J Am Acad Dermatol 2016; 74: 795–804; quiz 05–6. [DOI] [PubMed] [Google Scholar]
- 23. Sunderkotter CH, Zelger B, Chen KR et al. Nomenclature of Cutaneous Vasculitis: Dermatologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol 2018; 70: 171–84. [DOI] [PubMed] [Google Scholar]
- 24. Kerk N, Goerge T. Livedoid vasculopathy – current aspects of diagnosis and treatment of cutaneous infarction. J Dtsch Dermatol Ges 2013; 11: 407–10. [DOI] [PubMed] [Google Scholar]
- 25. Fritsch P, Zelger B. [Livedo vasculitis]. Hautarzt 1995; 46: 215–24; quiz 22–3. [DOI] [PubMed] [Google Scholar]
- 26. Monshi B, Posch C, Vujic I et al. Efficacy of intravenous immunoglobulins in livedoid vasculopathy: long‐term follow‐up of 11 patients. J Am Acad Dermatol 2014; 71: 738–44. [DOI] [PubMed] [Google Scholar]
- 27. Winkelmann RK. Livedoid vasculitis (segmental hyalinizing vasculitis). Jpn J Dermatol B 1972; 82: 84–9. [PubMed] [Google Scholar]
- 28. Winkelmann RK, Schroeter AL, Kierland RR, Ryan TM. Clinical studies of livedoid vasculitis: (segmental hyalinizing vasculitis). Mayo Clin Proc 1974; 49: 746–50. [PubMed] [Google Scholar]
- 29. Bard JW, Winkelmann RK. Livedo vasculitis. Segmental hyalinizing vasculitis of the dermis. Arch Dermatol 1967; 96: 489–99. [DOI] [PubMed] [Google Scholar]
- 30. Ackermann AB, Mones J. Ackerman's Resolving Quandaries in Dermatology, Pathology & Dermatopathology. NY Ardor Scribendi, New York, 2001. [Google Scholar]
- 31. Piette WW. The differential diagnosis of purpura from a morphologic perspective. Adv Dermatol 1994; 9: 3–23; discussion 24. [PubMed] [Google Scholar]
- 32. Carlson JA, Cavaliere LF, Grant‐Kels JM. Cutaneous vasculitis: diagnosis and management. Clin Dermatol 2006; 24: 414–29. [DOI] [PubMed] [Google Scholar]
- 33. Prechel M, Walenga JM. Heparin‐induced thrombocytopenia: an update. Semin Thromb Hemost 2012; 38: 483–96. [DOI] [PubMed] [Google Scholar]
- 34. Magnolo N, Jeskowiak A, Goerge T. Extensive coumarin‐induced necrosis – a vasculopathy following oral anticoagulation. J Dtsch Dermatol Ges 2014; 12: 263–4. [DOI] [PubMed] [Google Scholar]
- 35. Carlson JA, Chen KR. Cutaneous pseudovasculitis. Am J Dermatopathol 2007; 29: 44–55. [DOI] [PubMed] [Google Scholar]
- 36. Shapiro L, Teisch JA, Brownstein MH. Dermatohistopathology of chronic gonococcal sepsis. Arch Dermatol 1973; 107: 403–6. [PubMed] [Google Scholar]
- 37. Brunsting LA, Goeckerman WM, O'Leary PA. Pyoderma (ecthyma) gangraenosum, clinial and experimental observations in 5 cases occurring in adults. Arch Dermatol Syphilol 1932; 12: 655. [Google Scholar]
- 38. Argenbright LW, Barton RW. Interactions of leukocyte integrins with intercellular adhesion molecule 1 in the production of inflammatory vascular injury in vivo. The Shwartzman reaction revisited. J Clin Invest 1992; 89: 259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sunderkotter C, Seeliger S, Schonlau F et al. Different pathways leading to cutaneous leukocytoclastic vasculitis in mice. Exp Dermatol 2001; 10: 391–404. [DOI] [PubMed] [Google Scholar]
- 40. Scholzen TE, Sunderkotter C, Kalden DH et al. Alpha‐melanocyte stimulating hormone prevents lipopolysaccharide‐induced vasculitis by down‐regulating endothelial cell adhesion molecule expression. Endocrinology 2003; 144: 360–70. [DOI] [PubMed] [Google Scholar]
- 41. Ghosh SA, Polanowska‐Grabowska RK et al. Shiga toxin binds to activated platelets. J Thromb Haemost 2004; 2: 499–506. [DOI] [PubMed] [Google Scholar]