

Abstract
The bottom‐up construction of cell mimics has produced a range of membrane‐bound protocells that have been endowed with functionality and biochemical processes reminiscent of living systems. The contents of these compartments, however, experience semidilute conditions, whereas macromolecules in the cytosol exist in protein‐rich, crowded environments that affect their physicochemical properties, such as diffusion and catalytic activity. Recently, complex coacervates have emerged as attractive protocellular models because their condensed interiors would be expected to mimic this crowding better. Here we explore some relevant physicochemical properties of a recently developed polymer‐stabilized coacervate system, such as the diffusion of macromolecules in the condensed coacervate phase, relative to in dilute solutions, the buffering capacity of the core, the molecular organization of the polymer membrane, the permeability characteristics of this membrane towards a wide range of compounds, and the behavior of a simple enzymatic reaction. In addition, either the coacervate charge or the cargo charge is engineered to allow the selective loading of protein cargo into the coacervate protocells. Our in‐depth characterization has revealed that these polymer‐stabilized coacervate protocells have many desirable properties, thus making them attractive candidates for the investigation of biochemical processes in stable, controlled, tunable, and increasingly cell‐like environments.
Keywords: block copolymers, complex coacervates, macromolecular crowding, self-assembly, synthetic cells
It′s a lot like life: The physicochemical characterization of polymer‐stabilized coacervate protocells (such as macromolecular diffusion and buffering capacity within the core, structure and permeability of the polymer membrane) were explored, thus establishing this system as an attractive platform for investigating biochemical processes and enzymatic reactions in stable, tunable, and increasingly cell‐like environments.
Introduction
The question of what constitutes life and how it came about is an ancient one. In bottom‐up synthetic biology, the search for an answer lies in mimicking the complexity of contemporary life forms by combining biological, organic, and inorganic elements of varying origins to create simple cell mimics with life‐like properties.1 Certain structural facets of living systems—such as compartmentalization and the molecular crowding in the cytosol—have been successfully, but separately, mimicked in different approaches to building synthetic life. In order to replicate such complex and finely tuned living cells, these structural facets must be combined, along with other hallmarks, in a step‐by‐step approach to produce functional life‐like systems: so‐called protocells.
One such facet of living systems is the crowded internal environments within cells, where up to 30 % of the intracellular volume is occupied by biomolecules.2 Crowdedness has been reported to induce the organization of codependent molecules and can be a protection mechanism used by cells under stress conditions.3 In recent years, these crowded environments have been mimicked in bottom‐up platforms through the use of condensed polymer phases,4 in which oppositely charged polyelectrolytes condense into a polymer‐rich phase due to their mutual electrostatic interactions. This process is called complex coacervation. The formation of a coacervate phase has been demonstrated with a variety of polyionic molecules, most notably with use of cationic peptides or unnatural polymers in combination with nucleotides,5 RNA,4b, 6 or fatty acids,7 or with use of elastin‐like polypeptides.8 In addition, complex coacervates can readily be loaded with a wide range of cargos, including proteins, nucleic acids, and metabolites, and have been shown to support various cell‐like processes such as RNA folding and ribosomal activity,9 transcription and translation,10 and dissipative self‐assembly of cytoskeletal proteins.11 This makes complex coacervates ideal mimics both for the liquid organelles that perform specialized functions in the cell, such as nucleoli,3b and for the cell cytosol.4
Despite these important benefits that coacervates offer as cell mimics, their use as protocells is severely hindered by the absence of physical stabilization imparted by a membrane. In contrast, the commonly used membrane‐bound cell mimics, such as liposomes and polymersomes, do profit from the mechanical stability offered by their membranes. However, they are internally unstructured and do not offer the crowded environments that are present inside cells and that coacervates mimic more closely. Combining the internal structure offered by coacervates and the stabilization provided by membrane‐bound structures would yield compartments superbly suited to mimicking cellular physicochemical properties. For complex coacervates, only a few existing strategies have been reported for membranization and subsequent stabilization of coacervate microdroplets to allow their use in longer‐term, more physically demanding experiments.4b, 6b, 12
We recently described a solution to this problem, utilizing a block terpolymer to stabilize complex coacervate microdroplets.13 In this process, coacervation is started by mixing positively charged amylose (modified with a quaternary amine, Q‐Am) with negatively charged amylose (modified with a carboxymethyl group, CM‐Am). A carefully designed block terpolymer—poly(ethylene glycol)‐b‐poly(caprolactone‐gradient‐trimethylene carbonate)‐block‐poly(glutamic acid) (PEG‐PCLgTMC‐PGA)—is subsequently introduced; because of electrostatic interactions this self‐assembles on the surfaces of nascent coacervate droplets and stabilizes them against coalescence.
Our platform combines both structural facets found in living systems: a crowded cytosol capable of encapsulating bio‐macromolecules, as well as a semipermeable membrane that permits chemical communication between cargo and the outside world. However, some fundamental physicochemical parameters of our coacervate system remain unexplored: the extent to which cytosolic mimicry is imparted by the coacervates, the selective uptake of cargo into the polymer‐dense phase, and the membrane structure and permeability, for example, are yet to be fully characterized.
Here we first explored the tunable uptake of protein cargos into the coacervates. This is based on electrostatic interactions and so provides an insight into the uptake mechanism. Next, the structure of the stabilizing terpolymer membrane was clarified, and the functional consequences of this structure for its permeability towards a range of molecules with different charges and molecular weights were determined. We then characterized the ability of the coacervate interior to mimic the crowded and buffered cytosol by probing both the diffusion of fluorescent cargos, with the aid of fluorescence recovery after photobleaching (FRAP), and the pH response to externally added acid/base. Finally, our characterization of this protocell platform enabled the study of an enzymatic reaction within the coacervate phase; this revealed Michaelis–Menten parameters consistent with enzymatic reactions occurring in crowded environments. We thus describe a coacervate protocell that incorporates multiple facets of living systems, thus making it a unique platform on which additional cell‐like functionalities can readily be incorporated and bringing us one step closer to mimicking life itself.
Results and Discussion
Programmable protein uptake through tuning of electrostatic interactions
Coacervate systems have been reported to sequester a wide range of biomolecules spontaneously, with reported partitioning coefficients of up to ∼10 000.14 This strong uptake of molecules into complex coacervates is largely driven by electrostatic interactions,4a, 14 although other factors, such as protein conformation or persistence length, also influence the partitioning. Thanks to its negatively charged backbone, for example, RNA is generally taken up effectively.9, 14, 15
Uptake of proteins is less clear‐cut, however, because their charges at neutral pH differ, and both hydrophobic and conformational effects play larger roles than in the case of polynucleotides. To investigate the effect of charge on protein uptake into our coacervates, three variants of GFP with increasing isoelectric points—GFP(−30), GFP(−8), and GFP(+36)—were recombinantly expressed and purified (Figures S2–S4 in the Supporting Information).16 Protein uptake was studied by mixing the Q‐Am and the CM‐Am with each of the GFP mutants, followed by the addition of terpolymer to stabilize the coacervate protocells for analysis by confocal laser‐scanning microscopy. Charge‐dependent partitioning of GFP into the coacervate phase was observed, with strong uptake of GFP(−30) into the coacervate interior, as opposed to the exclusion of GFP(−8) (Figure 1 A). The heterogeneous distribution of GFP(+36) is likely due to this protein aggregating, because the salt concentrations of the phosphate‐buffered saline (PBS) used in these experiments were too low (<600 mm salt) to maintain protein stability.16b Taken together, these observations support the hypothesis that electrostatic interactions constitute an important factor in the uptake of folded proteins into the coacervate phase, and that these can to an extent be controlled by the total charge on the protein.
Figure 1.

The uptake of proteins into the coacervates is charge‐driven. A) The net charges on three GFP mutants dictate their uptake into coacervates formed from 2:1 Q‐Am/CM‐Am (DS Q=DS CM=0.8). The contrast for GFP(−8) has been increased to compensate for very low fluorescence from the polymer‐poor phase. B) The uptake of negatively charged GFP(−30) is dependent on the ratio of Q‐Am to CM‐Am. The normalized GFP fluorescence of the coacervate phase is indicated for each set of conditions. Scale bar: 5 μm.
To study whether protein partitioning can also be controlled by tuning the net charge of the coacervate phase, the uptake of GFP(−30) was studied for several charge balances. The charge balance in the coacervates can be tuned by varying the ratio of the positively charged Q‐Am and negatively charged CM‐Am. Our standard protocol for coacervate formation uses an excess of positive charge on the amylose chains (Q‐Am/CM‐Am=2:1, with DS Q=2, DS CM=1; see the Experimental Section), but here we used a Q‐Am and a CM‐Am with a similar number of ionizable groups per chain (DS=0.8). This was done to exclude any effects attributable to charge density on the amylose and to focus on the effect of charge ratio.
Coacervate formation was successful for Q/CM ratios in the range from 1:1 to 3:1. At ratios below 1:1, coacervation did not occur, indicating a preference for an excess of positively charged polymer (Table S1). At a neutral charge balance (Q/CM 1:1), GFP(−30) uptake was negligible, whereas an excess of positively charged amylose clearly increased the partitioning of the GFP into the coacervate phase (Figure 1 B). However, GFP(−8) was excluded from the coacervate at all charge balances (Figure S5), thus indicating that some proteins are not sufficiently negatively charged to be taken up at all. As demonstrated previously, however, proteins can be endowed with sufficient negative charge by converting basic amino acids on the surface of the protein into negative hemisuccinate groups.13
In summary, these experiments demonstrate that cargo loading into the coacervate protocells can be programmed both by tuning the charge balance of the coacervate phase and by considering the charge density of the cargo.
Terpolymer membrane structure and permeability
One of the most important aspects of a protocell is compartmentalization—a physical interface separating internal environments from the outside world.1a, 13, 17 Although our previous initial report on this system demonstrated the unique ability of the PEG‐PCLgTMC‐PGA terpolymer to form stable and discrete populations of protocells,13 little was known about the precise molecular arrangement of molecules at the interface, nor about its permeability characteristics. It is hypothesized that the molecular structure is a monolayer, with the negatively charged glutamic acid block facing inwards, interacting electrostatically with the predominantly positively charged coacervate core (Figure 2 A). This is due to the fact that, without the PGA block, self‐assembly of the polymer on the membrane and subsequent stabilization did not occur (Figure S6).
Figure 2.
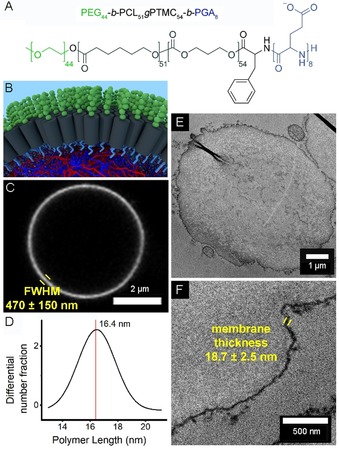
Detailed characterization of the stabilizing terpolymer membrane. A) Chemical structure of the terpolymer. B) A 3D representation of the hypothesized structural organization at the protocell interface, with terpolymer molecules aligned in a monolayer with the glutamic acid block (light blue) interacting with the coacervate core and the PEG block (green) oriented into the solution. C) Confocal micrograph of a single terpolymer‐stabilized coacervate protocell, with Nile Red indicating the membrane. D) Gaussian fit of polymer lengths dissolved in THF as determined by multi‐angle light scattering measurements. E) TEM analysis of a single coacervate protocell prepared by microtomy. F) Zoomed region of a different protocell.
The terpolymer membrane was rapidly and easily visualized in solution by using the fluorophore Nile Red, which fluoresces in hydrophobic environments. However, although this analysis indicated that the membrane was uniform over the entire protocell surface (Figure S7), the measured thickness of (470±150) nm was orders of magnitude too large to correspond to a monolayer (Figure 2 C); this overestimation is due to the diffraction‐limited nature of traditional light microscopy.
To investigate the membrane structure further, a multi‐angle light scattering (MALS) experiment of the terpolymer was performed, providing information on the polymer length in tetrahydrofuran (THF), a plasticizing solvent. This analysis indicated that the modal polymer length was (16.4±2.4) nm (Figure 2 D).
In order to confirm the monolayered membrane structure, and to rule out multilayered, aggregated, or pickering emulsion conformations of the terpolymer, ultrathin sections of chemically fixed coacervate protocells were prepared by microtomy and imaged under a transmission electron microscope after negative staining. These images (Figure 2 E) clearly show a continuous region of high contrast at the interface of each protocell, corresponding to the terpolymer membrane. The aspherical and folded nature of the protocells in the transmission electron micrographs is an artifact of sample preparation. The measured thickness of this interfacial region is (18.7±2.5) nm; this correlates closely with the MALS data, and confirms the hypothesized monolayered arrangement of terpolymer molecules on the surfaces of the coacervate microdroplets. These structural investigations frame the following permeability experiments, because the monolayered structure of the membrane should be more permeable to molecules than other, bilayer membranes.
To function as a catalytically active cell mimic, the coacervates need to be able to exchange molecular cargo, such as substrates and cofactors, with their environment. Previously, certain uncharged or weakly charged substrates, such as hydrogen peroxide, glucose, and Amplex Red (AR), had been indirectly observed to diffuse through the terpolymer membrane.13 To study this permeability towards a range of small molecules with varying net charges at physiological pH (see Figures S8 and S9 for full structural formulas), stabilized protocells with no cargo were first formed, followed by the addition of a small fluorescent molecule (molecular weight <1000 g mol−1). A fourfold excess of positive charge on the amylose was used, so a strong partitioning of polyanionic fluorophores was expected. It was indeed observed (Figure 3), with uptake showing a charge dependency in which higher anionic charge density promoted partitioning into the coacervate. Other physicochemical properties, such as hydrophobicity or hydrogen bonding, might account for the small differences in partitioning for molecules of similar net charge.
Figure 3.
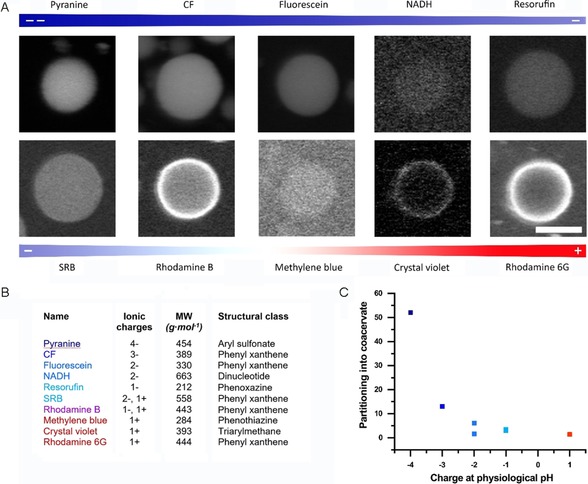
The uptake of small molecules into the coacervates is dominated by electrostatic interactions. A) Confocal micrographs of the partitioning of ionic low‐molecular‐weight fluorophores of different net charges into coacervates. Scale bar: 5 μm. B) Molecular properties of the fluorophores in A). The charge states of the molecules are estimated for physiological pH. CF: carboxyfluorescein. SRB: sulforhodamine B. C) The uptake of low‐molecular‐weight fluorophores into the coacervates as a function of the net charge at physiological pH. Averaged partitioning coefficients for the fluorophores in A), excluding those that preferentially partition into the membrane (rhodamine B, Crystal Violet, and rhodamine 6G). Exact values can be found in Figure S8.
Interestingly, positively charged or zwitterionic molecules showed two types of behavior: they either partitioned weakly into the coacervate or were localized to the periphery of the coacervate phase. We attribute this localization to a combination of electrostatic interactions with the polyglutamate block of the terpolymer, which is anchored in the coacervate phase, and also, possibly, hydrophobic interactions with the PCLgTMC block. The possible association of cationic species with the terpolymer is supported by the observation that cationic molecules, when mixed with unstabilized coacervates, were not sequestered at the interface but partitioned inside the coacervate phase (Figure S10).
These findings show that the partitioning and localization of small molecules is dependent on the charge and other chemical properties of the molecule. This has important implications for implementing biochemical reactions inside coacervates.
In addition to permeability towards small molecules, we were interested in investigating the molecular weight cut‐off for our semipermeable terpolymer membrane assembled on the coacervate surface, because the controlled translocation of functional macromolecules such as DNA, polypeptides, and fully folded enzymes across membranes is an important characteristic for future mimicking of protocellular applications. In order to determine the molecular weight at which a molecule is too large readily to permeate the membrane, stabilized protocells with no cargo were first formed, followed by the addition of a fluorescently labeled macromolecule. Confocal microscopy was then used to determine the fates of these externally added macromolecules (Figure 4).
Figure 4.
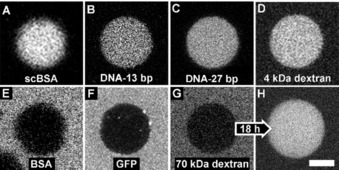
The permeability of the terpolymer membrane to macromolecules is predominantly dependent on their charge densities. Macromolecules with high negative charge—A) succinylated BSA, B) single stranded DNA with 13 base pairs, and C) single stranded DNA with 27 base pairs—are sequestered immediately despite their large size, similarly to small, neutral compounds such as D) 4 kDa dextran. Macromolecules with lower negative charge density, such as E) non‐succinylated BSA, and F) sfGFP, are excluded. Large, neutral molecules such as the 70‐kDa dextran are G) initially excluded, but H) slowly permeate the membrane. The protocells imaged in G) and H) are not the same particle; hence the small size difference. Scale bar: 5 μm.
Interestingly, the permeability of the membrane is independent of size, and depends more strongly on the charge on the macromolecule, thus supporting our observations made in the case of small molecules. For negatively and neutrally charged species, the rate of membrane translocation is influenced by the density of charge, because even a large macromolecule, such as 70 kDa dextran, is attracted to the coacervate interior through the terpolymer membrane. This is likely due to the fact that the PCLgTMC hydrophobic block is relatively thin for a polymeric membrane, and in addition is highly disordered, with a glass transition temperature below 0 °C. This results in a membrane that is highly mobile and easily penetrated by species that experience an attractive force to the coacervate core. Such high permeability, even to macromolecules, is likely due to this explanation rather than to a patchy or incomplete membrane, because experiments show that, even with a large excess of added terpolymer, succinylated BSA can still cross the membrane (Figure S11). In addition, when the terpolymer is added in excess, increased amounts of unstructured hydrophobic aggregates are observed, thus suggesting that the interface is saturated under normal formation conditions. These findings demonstrate the strong sequestration potential of the coacervate, and that the terpolymer membrane, in its current form, is vital only for the structural integrity of the coacervate protocell itself and has a limited role in permeability. Together with the small‐molecule permeability data, we observe that the terpolymer membrane is both non‐selective and readily permeable. It also imparts spatial organization on small positively charged or zwitterionic species, which can become localized at the coacervate periphery. Importantly, the terpolymer membrane enables our system to maintain an open environment for the easy exchange of molecules—which is a feature that limits many liposomal systems. As the coacervate protocell is a modular and modifiable system, these findings should enable us to design next‐generation membranes with programmed permeability. For example, it should be feasible to reliably functionalize the internal or external face of the membrane, or to take advantage of the monolayered structure to insert membrane‐bound proteins to add biomimetic functionality to this protocellular platform.
Coacervate interior as crowded and buffered cytosol mimic
Although coacervates have been touted as mimics of the crowdedness in the cellular cytosol,1b the specific characteristics of macromolecular diffusion and pH are rarely interrogated for each particular system. Here we have studied the diffusion of biomolecules inside the terpolymer‐stabilized coacervates by using fluorescence recovery after photobleaching (FRAP) experiments. The coacervates were loaded with three different biomolecules: fluorescein isothiocyanate (FITC)‐labeled dextran, a negatively charged green fluorescent protein (GFP) variant [GFP(−30)], or succinylated bovine serum albumin (BSA) labeled with Alexa488 dye. These negatively charged biomolecules have molecular weights of 4, ≈29, and ≈66 kDa, respectively.
Clear differences in fluorescence recovery between the different protocell populations containing each of the biomolecules were observed (Figure 5 A). Fitting the time‐dependent fluorescence recovery in the bleached area (Figure 5 B) enabled the apparent diffusion constants (D app) to be calculated; which were (0.46±0.04), (0.39±0.03), and (0.10±0.01) μm2 s−1 for the dextran, GFP(−30), and succinylated BSA, respectively. The D app values for these biomolecules are one order of magnitude lower than those measured for similar molecules in bacterial cells.18 However, the decrease in D app with increasing molecular weight that we observed here was also reported for proteins of similar molecular weights inside Escherichia coli cells;18b this is to be expected because larger macromolecules will inevitably diffuse more slowly. The apparent diffusion of bio‐macromolecules in free solution19 is at least two orders of magnitude higher than our measured values, thereby establishing our system as a closer approximation to the cellular cytosol than traditional protocell models such as liposomes or polymersomes, in which encapsulated bio‐macromolecules essentially experience a solvent environment unchanged in relation to the bulk.
Figure 5.
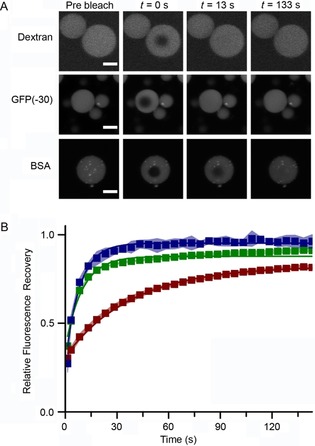
FRAP experiments undertaken to determine the diffusion characteristics of macromolecular cargos in coacervate protocells. A) Representative confocal images indicating fluorescence recovery due to diffusion of cargo [variously fluorescein‐labeled 4 kDa dextran, GFP(−30) (a negatively charged variant of GFP), or an Alexa488‐labeled succinylated BSA] within the protocell core over time. B) The relative fluorescence recovery of dextran (▪), GFP(−30) (▪), and BSA (▪), over time as determined by FRAP analysis. The errors are estimated from three separate measurements.
Biochemical interactions are often dictated and influenced by the pH of a solution, so pH within the protocell is a vital parameter to monitor. In nature, the pH of the cytosol is tightly controlled and is typically 7.2 in mammalian cells20 and from 7.2 to 7.8 in E. coli cells,21 depending on the external environment of the cell and whether or not it is proliferating.
A micro pH probe was used to measure the pH of a 2:1 Q‐Am/CM‐Am coacervate phase, which is a representative environment as found in coacervate microdroplets. The pH of the polymer‐rich phase after formation in PBS was 7.3. This is slightly more acidic than the value measured for neat PBS, a variation likely attributable to the change in hydrogen ion activity in the highly charged and viscous coacervate phase.
Another important characteristic of the cytosol is its buffering capacity and resistance to external changes in pH.22 This was investigated by measuring the coacervate pH in response to added acid or base in comparison with neat PBS (Figure 6). The internal coacervate pH largely follows the behavior of neat PBS, with a slightly worse buffering capacity under acidic conditions and a significantly better buffering capacity under alkaline conditions. This can be explained in terms of the chemical structures of the functional groups within the complex coacervate droplets. The quaternary amine group of Q‐Am is non‐ionizable in the pH range examined and thus has no capacity to react with any added H+ ions. This is in contrast to the carboxylic acid moiety on CM‐Am, which is able to react with added OH− ions. It should be noted that the pH values of the polymer‐rich and ‐poor phases were roughly the same throughout the titration experiments, within 0.05 pH units. Furthermore, the buffering capacity of this system makes it attractive for the incorporation of chemical reactions that are sensitive to changes in pH. However, it should be noted that the buffering capacity of this system is finite, in contrast to those of living cells, which employ an array of both active and passive processes to maintain cytosolic pH.
Figure 6.
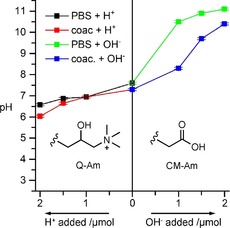
The buffering capacity of the complex coacervate phase utilized in this protocellular system. The pH was determined by using a micro pH meter. The x‐axis represents the amount of acid or base added to the system from a 1 m stock solution (1 μL of 1 m stock is 1 μmol).
Coacervates as platforms for study of enzyme kinetics in cell‐like environs
Living systems are characterized by extraordinary numbers of concurrent enzymatic reactions occurring in compartmentalized and crowded microenvironments, a characteristic that bottom‐up protocells struggle to recreate. We have shown that our coacervate protocells are permeable to most small molecules and can readily sequester negatively charged macromolecules into their interiors, thus allowing us to study the effects of their crowded microenvironments on enzyme activity. The rate of enzyme activity is highly system‐dependent, because different phase‐separated systems can variably partition not only enzymes, but also their substrates, intermediates, and products.4a, 23 In addition, macromolecular crowding has been observed to have a variable effect on enzymatic activity, depending on the nature of the system.2b, 23, 24 Therefore, it is crucial to investigate the effect of coacervate encapsulation on enzyme activity for this system, because this can have downstream effects on the application of the protocells.
Negatively charged proteins were shown to be readily taken up into the positively charged coacervate interior (Figure 1). To encourage the uptake of relatively neutral proteins, succinylation can be employed to increase the number of negatively charged surface residues.25 However, this strategy might not be applicable to all proteins, because this modification is known to affect protein oligomerization26 and to alter enzyme activity.27 Indeed, we have also observed this for several proteins, such as catalase, alcohol dehydrogenase, and esterase, with which either aggregation occurred, or key active‐site residues were modified, thereby destroying enzyme activity (data not shown).
Horseradish peroxidase (HRP, EC 1.11.1.7), however, was amenable to succinylation and, after this modification, was readily taken up into the positively charged coacervate interior (Figure S12).13 HRP was chosen here as a model enzyme because it has been extensively used to study the effects of molecular crowding.24b, 24e, 28 To study the effects of the coacervate microenvironment on enzymes, the activity of succinylated HRP encapsulated in the coacervate phase was compared with that of the same amount of protein in bulk solution (in the absence of coacervates). HRP consumes profluorescent substrate AR and hydrogen peroxide, to produce the fluorescent product resorufin (Figure 7 A). The enzyme activity was studied over a range of substrate concentrations, to afford the Michaelis–Menten curve fits for both substrates (Figure 7 B, C).
Figure 7.
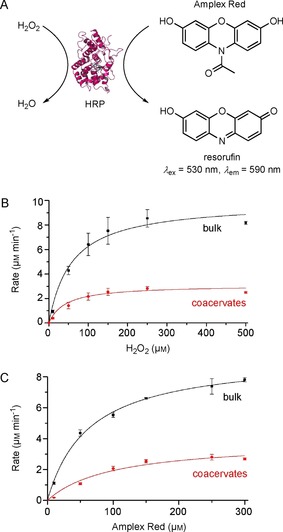
A) HRP consumes H2O2 and AR to produce a fluorescent product, resorufin. B) The Michaelis–Menten curves for succinylated HRP (30 pm final concentration) in a bulk solution (black) or within coacervates (red) containing 250 μm AR and varied concentrations of H2O2. C) The Michaelis–Menten curves for succinylated HRP (30 pm final concentration) in a bulk solution (black) or within coacervates (red) containing 250 μm H2O2 and varied concentrations of AR. The kinetic constants derived from the Michaelis–Menten fits are given in Table 1. The error bars are standard deviations of the rates estimated from duplicates of at least two separate experiments.
The Michaelis–Menten reaction parameters revealed interesting enzymatic behavior inside the coacervates, in comparison with the free enzyme in solution. The K M(H2O2) values were comparable for HRP inside and outside the coacervate. This suggests similar substrate binding capacity; this is unsurprising because H2O2 is a small substrate unlikely to be hindered within the coacervate environment. However, a threefold decrease in V max(H2O2) was observed for HRP localized inside coacervates, in relation to HRP in bulk solution.
Similar decreases in V max, but similar K M values, have also been observed for other enzyme kinetics occurring in crowded milieux,24b, 24c, 29 and have been attributed both to the coacervate–protein interactions and to the excluded volume effect hindering the movement of the enzyme active site. This delayed exploration of conformational space can hinder substrate binding, decreasing the maximum velocity of catalysis for the enzyme.
Similar decreases in V max were observed for samples of HRP within coacervates, in relation to those in bulk solutions, when AR concentrations were varied. However, the K M(AR) for coacervate‐localized proteins is significantly higher, by about 57 %, than that for bulk proteins. This increase in K M(AR) indicates that the substrate could be interacting with the coacervate interior, thus limiting its interaction with the HRP active site. AR interacting with the coacervate interior would be expected, because we have observed that the resorufin product—which shares many structural characteristics with AR—also readily localizes within the coacervate interior (Figure 3).
Similar differences in reaction parameters (increased K M and decreased V max values) were also observed for HRP when substrates interacted with increasing concentrations of crowding agents.24b These results confirm that this particular enzyme exhibits kinetic parameter changes that align with the effect of placing enzymes in crowded milieus, thus further demonstrating that our polymer‐stabilized coacervate protocells constitute an ideal platform for the study of enzyme kinetics in a tailorable, controlled, and open environment that resembles the cellular cytosol.
Conclusion
The condensed cores of our polymer‐stabilized coacervate protocells create a crowded environment in which the diffusivity of bio‐macromolecules is decreased so as to resemble that found in the cytosol of cells more closely than it does that seen in the semidilute conditions of many other protocells. This molecularly dense coacervate core is stabilized by a terpolymer that forms a homogeneous monolayered membrane, the semipermeable nature of which allows the exchange of small molecules of various net charges and of macromolecules with sufficient charge density. We have demonstrated that the loading of proteins into the coacervate phase can be controlled both by protein charge and by the composition of the coacervate phase, whereas the sequestration of a model enzyme does not abolish its activity. These experiments show that these coacervate protocells are highly interesting mimics of the crowding in the cellular cytosol, a facet that has remained relatively unexplored in the bottom‐up construction of protocells. The control over protein loading and the semipermeable nature of the membrane excellently position this system to expand the scope of biochemical reaction networks that can be studied in the crowded protocell interior.
Experimental Section
Materials: All chemicals were used as received unless otherwise stated. For the synthesis of terpolymer, poly(ethylene glycol) monomethyl ether 2 kDa was purchased from Rapp Polymere, and trimethylene carbonate (1,3‐dioxan‐2‐one) was purchased from TCI Europe. For the preparation of modified amylose derivatives, amylose (12–16 kDa) was supplied by Carbosynth and (3‐chloro‐2‐hydroxypropyl)trimethylammonium chloride (65 wt % in water) by TCI Europe. 10‐Acetylphenoxazine‐3,7‐diol was purchased from Sanbio. All other chemicals and reagents were supplied by Sigma–Aldrich.
Methods
Terpolymer synthesis: This block terpolymer was synthesized as described previously.13 Briefly, monomethoxy poly(ethylene glycol) monomethyl ether 2 kDa was used to initiate the ring‐opening polymerization of ϵ‐caprolactone and trimethylene carbonate. The terminal alcohol of this polymer was subsequently modified by Steglich esterification with Boc‐l‐Phe‐OH to yield a primary amine after TFA deprotection. The final poly(l‐glutamic acid) block was introduced by the ring‐opening polymerization of N‐carboxyanhydride γ‐benzyl l‐glutamate followed by hydrogenolysis.
Amylose modification: Both the quaternized amylose (Q‐Am) and the carboxymethylated amylose (CM‐Am) were prepared in accordance with previously published procedures.13 Briefly, amylose was dissolved in aqueous NaOH. For Q‐Am, (3‐chloro‐2‐hydroxypropyl)trimethylammonium chloride solution (60 %, w/v, in water) was added dropwise to the stirring reaction mixture, which was subsequently left to react overnight at 35 °C. For CM‐Am, chloroacetic acid was added and the reaction mixture was left to stir for 2 h at 70 °C. Both were purified by precipitation into cold ethanol followed by extensive dialysis against ultrapure water before lyophilization.
FRAP experiments: Coacervate protocells were formed by mixing 300 μL of 1 mg mL−1 Q‐Am (DS Q=2) and CM‐Am (DS CM=1) in a 2:1 ratio (v/v), together with, variously, 4 kDa Dex‐FITC (1 mg mL−1), GFP(−30) (3.6 mg mL−1), or succinylated BSA‐Alexa488 (1 mg mL−1), with mixing at 1500 rpm in an Eppendorf shaker for 4.5 min, after which terpolymer (50 mg mL−1 in PEG350, 9 μL) was added for stabilization. For FRAP analysis, an aliquot (100 μL) of each sample was loaded in an eight‐well glass‐bottomed slide (Ibidi). The samples were analyzed with a Zeiss LSM510 META NLO instrument equipped with a C‐Apochromat, 63× 1.2 UV/VIS/IR water objective and a photomultiplier tube detector. For imaging, an argon laser set at 488 nm 25 % laser power was used, with pinhole set to 1 Airy Unit (110 μm). Transmission and detector gain were optimized for each different fluorophore to use the maximum amount of gray values of the detector. FRAP experiments were performed by use of the Zen 2009 software bleaching interface. Firstly, an image of 1024×1024 pixels with a pixel dwell time of 0.8 μs/pixel was obtained. A region of interest (ROI) of 7 μm in diameter was defined. The samples were measured three times pre bleaching, followed by bleaching with a Chameleon by Coherent two‐photon laser set at 810 nm, 50 % laser power for 10 iterations. Afterwards images were acquired every 5 s for a total of 33 images. Images were analyzed by using ImageJ to generate the recovery curves. The data were normalized as described by Jia et al.4c by using Equation (1).
| (1) |
The data were fitted with a first‐order exponential equation [Eq. (2), in which F(t) is the normalized fluorescence at time point t, A is the amplitude of the recovery, and C represents the y intercept].
| (2) |
As described by Poudayl et al.,4d the half‐life of the recovery was determined by using Equation (3), which was used to calculate D app by using Equation (4), in which ω is the radius of the ROI.
| (3) |
| (4) |
Determination of coacervate pH and buffer capacity: The pH of the polymer‐rich coacervate phase was measured with a micro pH meter. To obtain sufficient volume of this phase to cover the pH electrode (ca. 50 μL), Q‐Am (10 mg mL−1, 2 mL, DS Q=2) was mixed with CM‐Am (10 mg mL−1, 1 mL, DS CM=1), followed by centrifugation of the suspension of coacervate droplets at 4000 g for 10 min. The buffer capacity of the system was determined by adding either NaOH (1 m, 1 μL) or HCl (1 m, 1 μL), vortexing briefly, and centrifuging again before measuring the pH of the polymer‐rich phase.
Protein preparations: A full description of recombinant GFP (Figures 1, 4, and 5) expression and purification is provided in the Supporting Information. The succinylated and fluorescently labeled BSA (Figures 4 and 5) was synthesized by using a previously reported protocol.13 The succinylated HRP (Figure 7) was also made according to the same protocol13 and purified from excess succinic acid by extensive dialysis into PBS (pH 7.4).
GFP partitioning inside coacervates: All experiments were performed in × PBS [pH 7.4, Na2HPO4 (10 mm), KH2PO4 (1.8 mm), NaCl (137 mm), KCl (2.7 mm)]. Q‐Am (DS Q=0.80) was mixed with CM‐Am (DS CM=0.85) to a final concentration of 0.33 mg mL−1 in 375 μL and shaken at 1500 rpm. Immediately after mixing, GFP (2 μL) was added to a final concentration of 100 nm and the sample was shaken for 5 min at room temperature. Dissolved terpolymer (50 mg mL−1 in DMSO, 7.5 μL) was gently added, and samples were shaken for another 30 s. Imaging was performed within 2 h after coacervate formation with a Zeiss LSM 510 Meta NLO by using a 63× water immersion objective and excitation at 488 nm, with emission recorded at 500–550 nm.
Microtomy and transmission electron microscopy: For TEM analysis of coacervates, coacervate suspension (same method as FRAP experiments, 300 μL) was spun down for 3 min at 2700 g, and the pellet was washed three times for 5 min in sodium cacodylate buffer (0.1 m, centrifugation for 3 min at 2700 g between washes). Subsequently, the sample was stained with osmium tetroxide (1 %, w/v) in cacodylate (0.1 m), CaCl2 (5 mm) buffer for 1 h and washed twice for 5 min with cacodylate (0.1 m), CaCl2 (5 mm) buffer. The sample was then washed twice with deionized water, filtered through a 0.22 μm filter (Milli‐Q water) for 5 min, and subsequently stained with uranyl acetate (2 %, w/v) in Milli‐Q water for 1 h. After the sample had been washed twice with Milli‐Q water for 5 min, it was dehydrated [2×10 min acetone in Milli‐Q water (50 %, v/v), 2×10 min acetone (70 %), 2×10 min acetone (96 %), 3×10 min anhydrous acetone (100 %)]. For infiltration, the sample was incubated with increasing concentrations of Epon resin in anhydrous acetone [Epon resin in anhydrous acetone (30 %, v/v) for 2 h, Epon resin (50 %) for 16 h, Epon resin (70 %) for 8 h, Epon resin (100 %) for 16 h] and subsequently incubated in freshly prepared Epon resin for 3 h before transfer to an embedding mold filled with freshly prepared Epon resin. After polymerization at 60 °C for 24 h, ultrathin sections (70 nm) were cut from the sample with a diamond knife and transferred to TEM grids (FCF‐200‐Cu, EMS). The samples were then studied with a FEI Tecnai 20 TEM (type Sphera, operated at 200 kV, equipped with a LaB6 filament and a FEI BM‐Ceta CCD camera).
Light scattering analysis of terpolymer: The gel permeation chromatography (with a PL gel 5 μm mixed D column and THF as an eluent)—multi‐angle light scattering (GPC‐MALS) experiments were performed with a Wyatt DAWN HELEOS II light scattering detector (MALS), which is operated with a 664.5 nm laser. Detectors were installed at the following angles: 12.9, 20.6, 29.6, 37.4, 44.8, 53.0, 61.1, 70.1, 80.1, 90.0, 99.9, 109.9, 120.1, 130.5, 149.1 and 157.8°. Prior to the measurements, SLS detectors were normalized with BSA. The processing and analysis of the data were performed with Astra 7.1.2 (random coil model).
Small‐molecule partitioning: All experiments were performed in PBS (1×, pH 7.4). Q‐Am (DS Q=2, 133 μL, 5.0 mg mL−1 was mixed with CM‐Am (DS CM=1, 66 μL, 5.0 mg mL−1) and shaken at 1500 rpm for 4 min at room temperature. Terpolymer (6 μL, 50 mg mL−1 in PEG350) was gently added, and samples were shaken for another 30 s. The coacervate solution was mixed with the appropriate fluorophore (20 μm) and imaged over 30 min. Imaging was performed with a Zeiss LSM 510 Meta NLO and use of a 63× water immersion objective and the following laser and filter settings: λ ex=488 nm, λ em=500–550 nm (fluorescein); λ ex=543 nm, λ em=565–615 nm (rhodamine); λ ex=458 nm, λ em=500–530 nm (pyranine); multiphoton laser: λ ex=725 nm, λ em<650 nm (NADH); λ ex=633 nm, λ em=650–710 nm (Methylene Blue); λ ex=543 nm, λ ex=650–710 nm (Crystal Violet). Partitioning into the coacervate was determined by calculating the ratio of fluorescence intensity inside and outside the coacervate phase.
Macromolecule partitioning: Coacervate protocells were formed by mixing Q‐Am/CM‐Am in a 2:1 ratio (1 mg mL−1, 300 μL) at 1500 rpm for 4.5 min, after which terpolymer (50 mg mL−1 in PEG350, 9 μL) was added for stabilization. Protocells were transferred into an eight‐well glass‐bottomed slide (Ibidi), followed by the addition of the labeled macromolecules (1 μL). Protocells were measured by confocal microscopy under the above conditions.
Enzyme assays and derivation of kinetic parameters: Succinylated HRP was synthesized and purified as previously described by using commercially available lyophilized protein (77 332 from Sigma).13 The protein concentration was determined by using the Nanodrop method with the heme absorbance at 403 nm and the extinction coefficient ϵ 403=91 000 m −1 cm−1.30 For the bulk reaction samples, the protein was diluted in PBS (pH 7.4) prior to mixing with other assay reagents. For the coacervate samples, the succinylated protein was added to forming coacervates (Q‐Am/CM‐Am in a 2:1 ratio, 0.5 mg mL−1, 300 μL, with DS Q=2 and DS CM=1), and the samples were mixed at 1500 rpm for 4.5 min, after which terpolymer (50 mg mL−1 in PEG350, 10 μL) was added to stabilize the coacervate samples.
The enzymatic assays were performed in a total volume of 150 μL per reaction at 25 °C in PBS buffer, pH 7.4 (P3813 from Sigma). The substrates, H2O2 and AR, were both premixed in the wells to achieve the final total concentrations given in Figure 7 B and C. To initiate the reaction, a protein sample (130 μL, either in bulk or in coacervates) was added to the wells to give 30 pm final concentration of enzyme (whether as free protein or as proteins encapsulated inside coacervates) in each reaction mixture. The reaction was monitored over 10 min through resorufin fluorescence (λ ex=530 nm, λ em=590 nm) with a Spark 10 M microplate reader (TECAN), and the fluorescence values were background‐subtracted. A standard curve of resorufin was used to determine the concentration of product from the fluorescence values. To obtain the Michaelis–Menten plots, the initial rate was determined by measuring the linear increase in resorufin production over the first minute, and this rate was plotted against substrate concentration. The error bars on the plot are the standard deviations between the rates of duplicates from at least two different experiments. The Michaelis–Menten curves were fitted by using Origin 2015 software, with the kinetic parameters from this fit indicated in Table 1.
Table 1.
Michaelis–Menten reaction parameters for succinylated HRP in bulk solution or localized inside the coacervate. Reactions were performed in PBS (pH 7.4) at 25 °C. The values and errors are derived from curve fitting performed by using Origin software.
|
Location of |
H2O2 |
Amplex Red |
||
|---|---|---|---|---|
|
succinylated HRP |
K M [μm] |
V max [μm min−1] |
K M [μm] |
V max [μm min−1] |
|
bulk |
58.1±9.5 |
9.9±0.4 |
63.2±4.4 |
9.3±0.2 |
|
coacervates |
42.2±8.1 |
3.1±0.1 |
99.2±14.8 |
3.8±0.2 |
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035) and the ERC Advanced grant Artisym 694120 are acknowledged for funding.
N. A. Yewdall, B. C. Buddingh, W. J. Altenburg, S. B. P. E. Timmermans, D. F. M. Vervoort, L. K. E. A. Abdelmohsen, A. F. Mason, J. C. M. van Hest, ChemBioChem 2019, 20, 2643.
Contributor Information
Dr. Alexander F. Mason, Email: a.f.mason@tue.nl.
Prof. Jan C. M. van Hest, Email: j.c.m.v.hest@tue.nl.
References
- 1.
- 1a. Buddingh B. C., van Hest J. C. M., Acc. Chem. Res. 2017, 50, 769–777; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Yewdall N. A., Mason A. F., van Hest J. C. M., Interface Focus 2018, 8, 20180023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Ellis R. J., Trends Biochem. Sci. 2001, 26, 597–604; [DOI] [PubMed] [Google Scholar]
- 2b. Zhou H. X., Rivas G., Minton A. P., Annu. Rev. Biophys. 2008, 37, 375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Shin Y., Brangwynne C. P., Science 2017, 357, eaaf4382; [DOI] [PubMed] [Google Scholar]
- 3c. Woodruff J. B., Hyman A. A., Boke E., Trends Biochem. Sci. 2018, 43, 81–94. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Crowe C. D., Keating C. D., Interface Focus 2018, 8, 20180032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Aumiller W. M., Pir Cakmak F., Davis B. W., Keating C. D., Langmuir 2016, 32, 10042–10053; [DOI] [PubMed] [Google Scholar]
- 4c. Jia T. Z., Hentrich C., Szostak J. W., Origins Life Evol. Biospheres 2014, 44, 1–12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Poudyal R. R., Guth-Metzler R. M., Veenis A. J., Frankel E. A., Keating C. D., Bevilacqua P. C., Nat. Commun. 2019, 10, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koga S., Williams D. S., Perriman A. W., Mann S., Nat. Chem. 2011, 3, 720. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Aumiller W. M., Keating C. D., Nat. Chem. 2016, 8, 129–137; [DOI] [PubMed] [Google Scholar]
- 6b. Williams D. S., Patil A. J., Mann S., Small 2014, 10, 1830–1840. [DOI] [PubMed] [Google Scholar]
- 7. Garenne D., Beven L., Navailles L., Nallet F., Dufourc E. J., Douliez J. P., Angew. Chem. Int. Ed. 2016, 55, 13475–13479; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13673–13677. [Google Scholar]
- 8. Simon J. R., Carroll N. J., Rubinstein M., Chilkoti A., Lopez G. P., Nat. Chem. 2017, 9, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drobot B., Iglesias-Artola J. M., Le Vay K., Mayr V., Kar M., Kreysing M., Mutschler H., Tang T. D., Nat. Commun. 2018, 9, 3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dora Tang T. Y., van Swaay D., deMello A., Ross Anderson J. L., Mann S., Chem. Commun. 2015, 51, 11429–11432. [DOI] [PubMed] [Google Scholar]
- 11. Te Brinke E., Groen J., Herrmann A., Heus H. A., Rivas G., Spruijt E., Huck W. T. S., Nat. Nanotechnol. 2018, 13, 849–855. [DOI] [PubMed] [Google Scholar]
- 12. Dora Tang T. Y., Rohaida Che Hak C., Thompson A. J., Kuimova M. K., Williams D. S., Perriman A. W., Mann S., Nat. Chem. 2014, 6, 527. [DOI] [PubMed] [Google Scholar]
- 13. Mason A. F., Buddingh B. C., Williams D. S., van Hest J. C. M., J. Am. Chem. Soc. 2017, 139, 17309–17312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frankel E. A., Bevilacqua P. C., Keating C. D., Langmuir 2016, 32, 2041–2049. [DOI] [PubMed] [Google Scholar]
- 15. Vieregg J. R., Tang T. Y. D., Curr. Opin. Colloid In 2016, 26, 50–57. [Google Scholar]
- 16.
- 16a. Azuma Y., Zschoche R., Tinzl M., Hilvert D., Angew. Chem. Int. Ed. 2016, 55, 1531–1534; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1555–1558; [Google Scholar]
- 16b. Lawrence M. S., Phillips K. J., Liu D. R., J. Am. Chem. Soc. 2007, 129, 10110–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Brea R. J., Hardy M. D., Devaraj N. K., Chem. Eur. J. 2015, 21, 12564–12570; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Szostak J. W., Bartel D. P., Luisi P. L., Nature 2001, 409, 387–390. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Elowitz M. B., Surette M. G., Wolf P. E., Stock J., Leibler S., Curr. Biol. 1997, 7, 809–812; [DOI] [PubMed] [Google Scholar]
- 18b. Kumar M., Mommer M. S., Sourjik V., Biophys. J. 2010, 98, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Luby-Phelps K. in Microcompartmentation and Phase Separation in Cytoplasm, Vol. 192 (Eds.: H. Walter, D. E. Brooks, P. A. Srere), Academic Press, San Diego, 1999, 189–221; [Google Scholar]
- 19b. Nauman J. V., Campbell P. G., Lanni F., Anderson J. L., Biophys. J. 2007, 92, 4444–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Casey J. R., Grinstein S., Orlowski J., Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61; [DOI] [PubMed] [Google Scholar]
- 20b. Theillet F. X., Binolfi A., Frembgen-Kesner T., Hingorani K., Sarkar M., Kyne C., Li C., Crowley P. B., Gierasch L., Pielak G. J., Elcock A. H., Gershenson A., Selenko P., Chem. Rev. 2014, 114, 6661–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilks J. C., Slonczewski J. L., J. Bacteriol. 2007, 189, 5601–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldsmith D. J. A., Hilton P. J., Kidney Int. 1992, 41, 43–49. [DOI] [PubMed] [Google Scholar]
- 23. Davis B. W., Aumiller W. M., Hashemian N., An S., Armaou A., Keating C. D., Biophys. J. 2015, 109, 2182–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Dhar A., Samiotakis A., Ebbinghaus S., Nienhaus L., Homouz D., Gruebele M., Cheung M. S., Proc. Natl. Acad. Sci. USA 2010, 107, 17586–17591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Aumiller W. M., Davis B. W., Hatzakis E., Keating C. D., J. Phys. Chem. B 2014, 118, 10624–10632; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Balcells C., Pastor I., Vilaseca E., Madurga S., Cascante M., Mas F., J. Phys. Chem. B 2014, 118, 4062–4068; [DOI] [PubMed] [Google Scholar]
- 24d. Samanta N., Das Mahanta D., Patra A., Mitra R. K., Int. J. Biol. Macromol. 2018, 118, 209–215; [DOI] [PubMed] [Google Scholar]
- 24e. Gao Y., Liu X., Sun L., Xu Y., Yang S., Fan C., Li D., J. Phys. Chem. Lett. 2019, 10, 82–89. [DOI] [PubMed] [Google Scholar]
- 25. Aitken A., Learmonth M. in The Protein Protocols Handbook (Ed.: J. M. Walker), Humana, Totowa, 2002, pp. 459–460. [Google Scholar]
- 26. Klotz I. M., Keresztes-Nagy S., Nature 1962, 195, 900–901. [Google Scholar]
- 27. Riordan J. F., Vallee B. L., Biochemistry 1964, 3, 1768–1774. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Baumann P., Spulber M., Fischer O., Car A., Meier W., Small 2017, 13, 1603943; [DOI] [PubMed] [Google Scholar]
- 28b. Altikatoglu M., Basaran Y., Protein J. 2011, 30, 84–90. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Maximova K., Wojtczak J., Trylska J., Anal. Biochem. 2019, 567, 96–105; [DOI] [PubMed] [Google Scholar]
- 29b. Pastor I., Pitulice L., Balcells C., Vilaseca E., Madurga S., Isvoran A., Cascante M., Mas F., Biophys. Chem. 2014, 185, 8–13. [DOI] [PubMed] [Google Scholar]
- 30. Keilin D., Hartree E. F., Biochem. J. 1951, 49, 88–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary