

Abstract
The two most abundant molecules on synaptic vesicles (SVs) are synaptophysin and synaptobrevin‐II (sybII). SybII is essential for SV fusion, whereas synaptophysin is proposed to control the trafficking of sybII after SV fusion and its retrieval during endocytosis. Despite controlling key aspects of sybII packaging into SVs, the absence of synaptophysin results in negligible effects on neurotransmission. We hypothesised that this apparent absence of effect may be because of the abundance of sybII on SVs, with the impact of inefficient sybII retrieval only revealed during periods of repeated SV turnover. To test this hypothesis, we subjected primary cultures of synaptophysin knockout neurons to repeated trains of neuronal activity, while monitoring SV fusion events and levels of vesicular sybII. We identified a significant decrease in both the number of SV fusion events (monitored using the genetically encoded reporter vesicular glutamate transporter‐pHluorin) and vesicular sybII levels (via both immunofluorescence and Western blotting) using this protocol. This revealed that synaptophysin is essential to sustain both parameters during periods of repetitive SV turnover. This was confirmed by the rescue of presynaptic performance by the expression of exogenous synaptophysin. Importantly, the expression of exogenous sybII also fully restored SV fusion events in synaptophysin knockout neurons. The ability of additional copies of sybII to fully rescue presynaptic performance in these knockout neurons suggests that the principal role of synaptophysin is to mediate the efficient retrieval of sybII to sustain neurotransmitter release.
Keywords: endocytosis, neurotransmission, presynapse, synaptophysin, VAMP2, vesicle
Despite controlling the packaging of the essential fusogenic protein synaptobrevin II onto synaptic vesicles, the absence of synaptophysin results in surprisingly mild effects on neurotransmission. We hypothesised that the abundance of vesicular synaptobrevin may mean that effects on exocytosis only appear during periods of heightened activity. To test this, we subjected synaptophysin knockout neurons to repeated trains of activity, and revealed that synaptophysin is essential to sustain the number of fusion events and vesicular synaptobrevin levels during periods of repetitive vesicle turnover. Therefore, the principal role of synaptophysin is to mediate the efficient retrieval of synaptobrevin to sustain neurotransmitter release.
Abbreviations used
- anova
analysis of variance
- AP180
adaptor protein 180
- AP‐2
adaptor protein complex 2
- DIV
days in vitro
- KO
knockout
- mCer
mCerulean
- PBS
phosphate‐buffered saline
- RRID
research resource identifier
- SDS‐PAGE
sodium dodecyl sulphate polyacrylamide gel electrophoresis
- SNARE
soluble NSF attachment protein receptor
- STP
short‐term plasticity
- SV
synaptic vesicle
- sybII
synaptobrevin‐II
- SYP
synaptophysin
- vGLUT1
vesicular glutamate transporter
The maintenance of high fidelity neurotransmitter release requires tight regulation of synaptic vesicle (SV) reformation via endocytosis. SV endocytosis is integrated with the clustering and recruitment of cargo molecules at the presynaptic plasma membrane such that recycled SVs contain the appropriate protein cargo in the correct stoichiometry (Kelly and Owen 2011; Cousin 2017; Kaempf and Maritzen 2017). This combination of both cargo selection and vesicle retrieval ensures a constant supply of fusion‐competent SVs that sustain neurotransmitter release. One such cargo is synaptobrevin‐II (sybII, also known as vesicle‐associated membrane protein 2; VAMP2), which is the most abundant molecule on SVs (Takamori et al. 2006; Wilhelm et al. 2014) and is essential for neurotransmitter release (Schoch et al. 2001).
Multiple mechanisms exist at central nerve terminals to cluster SV cargo at the plasma membrane for retrieval by endocytosis. The majority of SV cargoes possess recognition motifs for classical adaptor protein complexes such as adaptor protein complex 2 (Kelly and Owen 2011; Rao et al. 2012). However, several key SV proteins, including sybII, lack an adaptor protein complex 2 recognition sequence, and thus their retrieval from the plasma membrane during endocytosis must be facilitated by alternative mechanisms. In agreement, sybII binds to the monomeric adaptor protein, adaptor protein 180 (AP180) (Miller et al. 2011; Koo et al. 2011). Organisms that lack the gene encoding for AP180 display inefficient sybII retrieval during endocytosis coupled to an increase in surface sybII with a concomitant decrease in vesicular sybII (Nonet et al. 1999; Bao et al. 2005; Koo et al. 2011; Vanlandingham et al. 2014; Koo et al. 2015). A similar phenotype has been observed in rodent neurons lacking the gene encoding the highly abundant integral SV protein synaptophysin (Gordon et al. 2011; Gordon and Cousin 2013; Harper et al. 2017), indicating that it is also required for efficient sybII retrieval during endocytosis. In addition to this role, synaptophysin is proposed to clear sybII from the active zone after SV fusion events (Rajappa et al. 2016).
Synaptophysin knockout neurons do not display overt defects in either evoked or spontaneous neurotransmission (McMahon et al. 1996; Janz et al. 1999) and only small perturbations in short‐term plasticity (Janz et al. 1999; Kwon and Chapman 2011; Rajappa et al. 2016). The absence of a stronger neurotransmitter release defect is puzzling, since it would be predicted that inefficient sybII retrieval would result in sybII depletion from SVs. However, the large reservoir of sybII on SVs (approximately 70 copies) (Takamori et al. 2006; Wilhelm et al. 2014) and the fact that only 1‐3 sybII molecules are required for SV fusion (van den Bogaart et al. 2010, Sinha et al. 2011, Mohrmann et al. 2010) may mean presynaptic defects only become apparent during periods of frequent SV turnover. To test this hypothesis, we subjected synaptophysin knockout neurons to repeated trains of neuronal activity while monitoring effects on SV fusion and levels of vesicular sybII. Using this protocol, we revealed a progressive decline in the amplitude of exocytic events, with a concomitant reduction in vesicular sybII, in the absence of synaptophysin. The expression of exogenous synaptophysin fully rescued exocytic capacity, and intriguingly, so did the expression of sybII. The ability of additional copies of sybII to fully recue SV fusion suggests that the principal role of synaptophysin at the presynaptic nerve terminals is to ensure the efficient retrieval of sybII.
Methods
Materials
Vesicular glutamate transporter 1 (vGLUT1)‐pHluorin (a pH‐sensitive variant of GFP) was kindly provided by Prof. R. Edwards (University of California). mCerulean (mCer) empty vector and synaptophysin‐mCer (syp‐mCer) were generated as described (Gordon et al. 2011). Flag empty vector was a gift from Dr Gareth Evans (University of York, (Dunning et al. 2016)). Synaptobrevin‐II (sybII‐flag) was cloned into this vector with BamHI and XhoI restriction sites using the primers CTCGAGATGTCGGCTACCGCTGCCACCGTCC and TGGATCCCGAGTGCTGAAGTAAACGATGATGATG (restriction sites underlined). Custom‐made vectors will be made available upon reasonable request. Neurobasal media (cat # 21103049), B‐27 supplement (cat # 17504044), penicillin/streptomycin (cat # 15140122), Minimum Essential Media (MEM; cat # 11095080), Lipofectamine 2000 (11668019), goat anti‐chicken IgY (H + L) secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific Cat# A‐11039, RRID:AB_2534096) and goat anti‐rabbit IgG (H + L) cross‐adsorbed secondary antibody conjugated to AlexaFluor 568 (Thermo Fisher Scientific Cat# A‐11011, RRID:AB_143157) were obtained from Fisher Scientific (Loughborough, UK). Chicken anti‐GFP (Abcam Cat# ab13970, RRID: AB_300798), and rabbit anti‐sybII (Abcam Cat# ab3347, RRID: AB_2212462) antibodies were from Abcam (Cambridge, UK). Rabbit anti‐synaptotagmin‐1 (Synaptic Systems Cat# 105 103, RRID: AB_11042457) was from Synaptic Systems (Gottingen, Germany). Odyssey blocking phosphate‐buffered saline (PBS) buffer (cat # 92740000), IRDye anti‐mouse and anti‐rabbit secondary antibodies were from LI‐COR Biosciences (Lincoln, Nebraska, USA). Papain (cat # LK003178) was obtained from Worthington (Lakewood, NJ, USA). Foetal bovine serum (cat # FB‐1001/500, lot # 013BS715) was from Biosera (Nuaille, France). All other reagents (including mouse anti‐ ß‐actin, (Sigma‐Aldrich Cat# A3854, RRID: AB_262011)) were from Sigma‐Aldrich (Poole, UK).
Hippocampal neuronal culture
All animal work was performed in accordance with the UK Animal (Scientific Procedures) Act 1986, under Project and Personal Licence authority and was approved by the Animal Welfare and Ethical Review Body at the University of Edinburgh (Home Office project licence – 70/8878). Specifically, all animals were killed by schedule 1 procedures in accordance with UK Home Office Guidelines; adults were killed by cervical dislocation followed by destruction of the brain, and embryos were killed by decapitation followed by destruction of the brain. Synaptophysin knockout mice (originally obtained from R. Leube (RWTH Aachen University, Germany) (Eshkind and Leube 1995) were maintained as heterozygous breeding pairs on a C57BL/6J background and genotyped as described (Gordon et al. 2011) using primers ACTTCCATCCCTATTTCCCACACC, TTCCACCCACCAGTTCAGTAGGA and TCGCCTTCTTGACGAGTTCTTCTG and cycle conditions of 95°C for 3 min, then 30 cycles of 95°C for 1 min, 58°C for 30 s and 72°C for 2 min, followed by 72°C for 10 min. Mice were timed mated as homozygous pairs. Wild‐type C57BL/6J mice were sourced from an in‐house colony at the University of Edinburgh. All mouse colonies were housed in standard open top caging on a 14 h light/ dark cycle (light 07:00–21:00). Breeders were fed RM1 chow, whereas stock mice were maintained on RM3 chow.
Dissociated primary hippocampal‐enriched neuronal cultures were prepared from E16.5‐E18.5 wild‐type or synaptophysin knockout mouse embryos of both sexes as outlined (Baker et al. 2015). In brief, isolated hippocampi (combined from at least four individual embryos for fluorescence imaging assays and from single embryos for cell fractionation assays) were digested in 10 U/mL papain in Dulbecco’s PBS, washed in MEM supplemented with 10 % foetal bovine serum and triturated to single‐cell suspension. Single‐cell suspensions of hippocampal neurons were then plated at a density of 3–5 × 104 (for fluorescence imaging assays) or 7 × 105 (cell fractionation) cells per coverslip on poly‐D‐lysine and laminin‐coated (50 μL laminin spot for fluorescence imaging, 200 μL for cell fractionation) 25 mm coverslips. Cultures were maintained in Neurobasal media supplemented with B‐27, 0.5 mM L‐glutamine and 1% v/v penicillin/streptomycin. After 72 h, cultures were further supplemented with 1 μM cytosine β‐d‐arabinofuranoside to inhibit glial proliferation. Cells were transfected after 7–8 days in culture with 2 µL Lipofectamine 2000 and 1 µg/DNA construct per well as described (Gordon et al. 2011). In all experiments, two constructs were co‐expressed: vGLUT1‐pHluorin was co‐transfected with either mCerulean empty vector (mCer) or syp‐mCer, or flag empty vector (flag) or sybII‐flag. Cells were fixed or imaged after 13–16 days in vitro (DIV).
Fixation and immunolabelling
Hippocampal cultures were mounted in a Warner imaging chamber with embedded parallel platinum wires (RC‐21BRFS) in the presence of saline buffer (in mM: 136 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgCl2, 10 glucose, 10 HEPES, pH 7.4 supplemented with 10 μM 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione and 50 μM DL‐2‐Amino‐5‐phosphonopentanoic acid). Cultures were fixed in 4% paraformaldehyde in PBS either at rest, or following 4 min 30 s recovery from stimulation with four trains of 300 action potentials delivered at 10 Hz (100 mA, 1 ms pulse width), interspersed at 5‐min intervals.
Fixed cells were incubated at room temperature (21 ‐ 23 °C) in 50 mM NH4Cl in PBS for 10 min, washed with PBS and permeabilized with 0.1 % v/v Triton X‐100, 1 % v/v bovine serum albumin (BSA) in PBS for 5 min. The cells were washed with PBS, blocked with 1 % BSA in PBS for 1 h before being incubated in a humidified chamber with antibodies in 1 % BSA in PBS for 1–2 h (chicken anti‐GFP 1 : 2000 with either rabbit anti‐synaptotagmin‐1, 1 : 100 or rabbit anti‐sybII, 1 : 1000) followed by extensive washing with PBS. Cells were then labelled with fluorescent secondary antibodies (goat anti‐chicken conjugated to Alexa Fluor 488 and goat anti‐rabbit conjugated to Alexa Fluor 568, both 1 : 500) in a dark humidified chamber and then washed extensively in PBS. Coverslips were rinsed with ddH2O, air‐dried in the dark, and mounted onto glass slides with Mowiol® 4‐88.
Fluorescence imaging
Fixed, immunolabelled cells or live neuronal cultures mounted in a Warner imaging chamber with embedded parallel platinum wires (RC‐21BRFS) were placed on the stage of Zeiss Axio Observer D1 epifluorescence microscope and imaged using a Zeiss Plan Apochromat x40 oil immersion objective (NA 1.3). Neurons transfected with mCer vectors were visualised at 430 nm excitation, whereas vGLUT1‐pHluorin was visualised at 500 nm excitation. The same emission collection was applied in both instances (using a 525 nm dichroic filter and long‐pass emission filter, > 535 nm). GFP immunolabelling was visualised at 480 nm excitation (with emission monitored using a 526/26 nm band‐pass filter and 495 nm dichroic filter), whereas sybII or synaptotagmin‐1 immunolabelling was visualised at 550 nm excitation (with emission monitored using a long‐pass > 565 nm filter and a 560 dichroic filter). Fluorescent images were captured using a Hamamatsu Orca‐ER digital camera and processed offline using Image J 1.43 software.
For live fluorescence imaging assays, cultures were stimulated with four trains of 300 action potential stimulation delivered at 10 Hz (100 mA, 1 ms pulse width) every 5 min during continuous perfusion with saline buffer (21 – 23°C). They were then challenged with alkaline buffer (saline buffer with 50 mM NH4Cl substituted for 50 mM NaCl) to reveal total vGLUT1‐pHluorin fluorescence. Images were captured at 4 s intervals. Regions of interest of identical size were placed over nerve terminals and the total fluorescence intensity was monitored over time. Only regions that responded to action potential stimulation were selected for analysis. The vGLUT1‐pHluorin fluorescence change was calculated as ΔF/F0, normalised to the peak height of first stimulus train, and n refers to the number of individual coverslips examined.
To quantify synaptic sybII or synaptotagmin‐1 expression in immunolabelled neurons, identically sized regions of interest were placed over transfected puncta and non‐transfected puncta in the same field of view, along with background regions and total fluorescence intensity measured. The level of protein expression in knockout versus rescued neurons was calculated by subtracting background autofluorescence and determining the ratio of fluorescence in non‐transfected or transfected puncta.
Cell fractionation assay
DIV 13‐16 cultured hippocampal neurons (plated at a density of 7 × 105 cells/ 25 mm coverslip) were washed in saline buffer, and then exposed four times to either saline or depolarisation buffer (saline buffer with 50 mM KCl substituted for 50 mM NaCl) for 10 s, interspersed with 5‐min recovery in saline buffer. At the completion of the 5‐min recovery from the fourth depolarisation, cells were hypotonically lysed for 10 s in 20 mM Tris, pH 7.4 supplemented with 2 μL/mL protease inhibitors and 1 mM PMSF. KCl was added to the cells to a final concentration of 150 mM to restore osmolarity and cells were harvested. Cells from three coverslips were combined for each condition. Cells were spun down at 19 700 g at 4°C for 2 min to separate heavy membranes from intracellular organelles. The membrane pellet, containing the plasma membrane, was separated from the supernatant, containing intracellular SVs. Both samples were directly lysed in sample buffer (67 mM Tris pH 6.8, 2 mM EDTA, 67 mM SDS, 9 % Glycerol, 0.0002 % Bromophenol blue, 0.04 % β‐mercaptoethanol).
Samples were resolved by SDS‐PAGE on 12.5% SDS gels and transferred to nitrocellulose membranes for Western blotting. Membranes were blocked in Odyssey blocking PBS buffer, and were then subjected to immunoblotting analysis using the following primary antibodies: synaptotagmin‐1 (1 : 3000), ß‐actin (1 : 50 000) and sybII (1 : 4000). Membranes were washed four times for 5 min with PBS 0.1 % v/v Tween 20 before antibodies were amplified using IRDye anti‐mouse and anti‐rabbit secondary antibodies (all 1 : 10 000). Membranes were again washed as described, and then imaged using an Odyssey 9120 Infrared Imaging System (LI‐COR Biosciences) and analysed using Image Studio Lite (LI‐COR Biosciences). The basal and stimulated intensity of the plasma membrane and SV samples from each experiment was calculated and expressed as a plasma membrane/SVs ratio for each protein, after normalisation to actin.
Statistical analysis
Independent fields of view from individual coverslips, repeated across at least two independent culture preparations, are indicated by n. The exception is the cell fractionation assay, where N indicates individual cultures. All statistical analyses were performed using Microsoft Excel (Excel 2016) and GraphPad Prism (version 8) software. All data passed a normality test (Shapiro–Wilk test). Data were analysed by one‐way analysis of variance (anova) with Sidak’s multiple comparison test, two‐way anova with Tukey’s multiple comparison post hoc test, or repeated measures two‐way anova with Sidak’s multiple comparison test, with p < 0.05 considered significantly different; individual p values are reported in figure legends. No blinding was performed in this study. Study groups were not randomised, but were performed interleaved to ensure any inter‐group variation in experimental conditions was minimised. Post hoc outlier analysis (ROUT (Q = 1%)) revealed no outliers in any of the data sets, with the exception of two data points in Figure S3. All data are reported as mean ± standard error of the mean (SEM) with individual data points shown.
Results
Loss of synaptophysin results in a progressive reduction of SV fusion events during repeated action potential trains
We have previously established that synaptophysin is required for the efficient retrieval of sybII during SV endocytosis (Gordon et al. 2011; Gordon and Cousin 2013). However, the physiological consequence of such a major defect appears to be relatively minor, with an absence of synaptophysin having no effect on either the size of the readily releasable pool or release probability (McMahon et al. 1996; Janz et al. 1999) and very small effects on short‐term plasticity (Janz et al. 1999; Kwon and Chapman 2011; Rajappa et al. 2016). Because of the large disparity of sybII molecules present on SVs (Takamori et al. 2006; Wilhelm et al. 2014) when compared to the copies required for fusion of a SV (Mohrmann et al. 2010, van den Bogaart et al. 2010, Sinha et al. 2011), we reasoned that the outcome of inefficient sybII retrieval would only become apparent when neurons were challenged with a physiological stressor, such as repeated patterns of neuronal activity. Therefore, we first determined whether the absence of synaptophysin impacted on the ability of neurons to sustain SV exocytosis during repeated trains of action potential stimuli. In these experiments, SV fusion was monitored using the genetically encoded fluorescent reporter vGLUT1‐pHluorin (Voglmaier et al. 2006). The fluorescence of this reporter is quenched at rest when pHluorin is internalised in the acidic lumen of SVs, but is unquenched when action potential stimulation induces the fusion of SVs with the plasma membrane. The fluorescence of vGLUT1‐pHluorin is then re‐quenched by the subsequent endocytosis and acidification of SVs (Kavalali and Jorgensen 2014), enabling it to report the extent of both activity‐dependent SV fusion and SV endocytosis.
To examine the requirement for synaptophysin in sustaining neurotransmitter release, synaptophysin knockout neurons were transfected with vGLUT1‐pHluorin and either a vector expressing the fluorescent protein mCerulean or exogenous synaptophysin tagged with mCerulean (synaptophysin‐mCerulean; syp‐mCer) to rescue normal function (Gordon and Cousin 2013). Neurons were challenged with four trains of action potential stimulation (300 action potentials at 10 Hz every 5 min) (Fig 1a) and the vGLUT1‐pHluorin fluorescence response was monitored over time. In synaptophysin knockout neurons expressing syp‐mCer, the first stimulus train evoked a classical pHluorin response, with a rapid increase in fluorescence occurring during stimulation and then a slower return to baseline post‐stimulation (Fig 1b). The first stimulus train in synaptophysin knockout neurons evoked a very similar vGLUT1‐pHluorin response. When the peak amplitude of vGLUT1‐pHluorin fluorescence was monitored over multiple stimulus trains, SV fusion was sustained in synaptophysin knockout neurons that were rescued with syp‐mCer (Fig 1b). However, synaptophysin knockout neurons displayed a progressive reduction in the peak amplitude of the vGLUT1‐pHluorin fluorescence response, suggesting that synaptophysin is required for maintaining the extent of SV fusion during periods of sustained activity (Fig 1b). A comparison of the amplitude of peak vGLUT1‐pHluorin fluorescence responses across the four stimulus trains confirmed that the peak vGLUT1‐pHluorin response at later stimuli was significantly lower in neurons lacking synaptophysin than those expressing syp‐mCer (Fig 1c; fourth stimulus train: syp‐mCer 94.81 ± 7.50 %; mCer 68.95 ± 2.41 %; p < 0.01). Therefore, synaptophysin is required to sustain neurotransmitter release during repeated patterns of neuronal activity.
Figure 1.
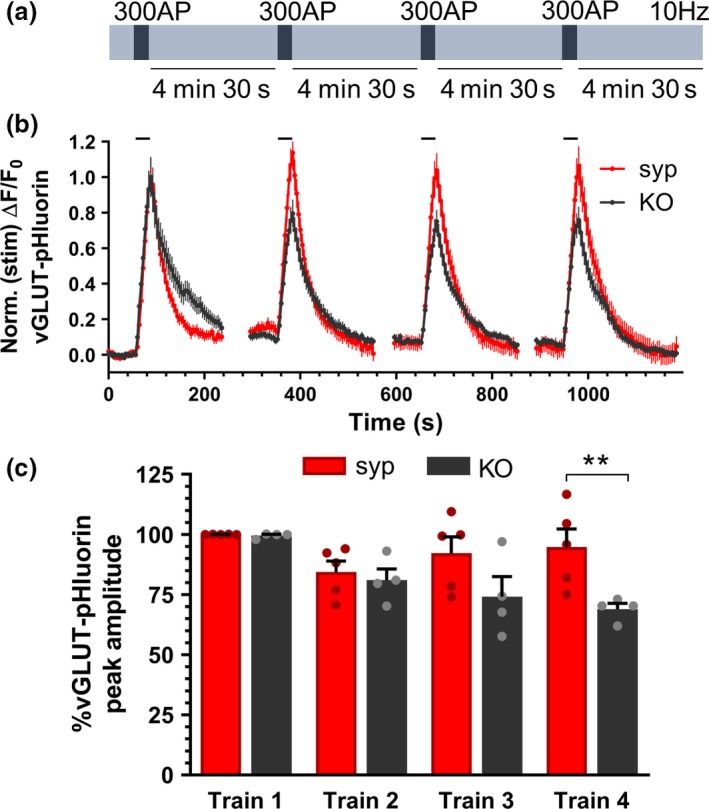
Synaptophysin is required to sustain SV fusion events during repetitive stimulation. Primary cultures of synaptophysin knockout hippocampal neurons were transfected with vGLUT1‐pHluorin and either empty mCerulean (KO) vector or synaptophysin‐mCerulean (syp). Neurons were challenged with four trains of 300 action potentials delivered at 10 Hz at 5‐min intervals as shown in (a). (b) Representative traces of the evoked vGLUT1‐pHluorin response (ΔF/F0 ± SEM) in synaptophysin knockout neurons expressing either mCer (KO, black) or syp (syp, red) are displayed. Traces are normalised to the peak amplitude response during the first‐action potential train. Bar indicates period of stimulation. (c) Bar graph displaying the mean peak amplitudes of the vGLUT1‐pHluorin peak for each of the four trains of stimuli, calculated as a proportion of the first train. Bars indicate SEM (n = 4 KO, n = 5 syp; repeated measures two‐way anova with Sidak’s multiple comparison test, **p < 0.01; train 1 p > 0.9999, train 2 p = 0.9859, train 3 p = 0.0985, train 4 p = 0.0086. n indicates a single field of view from a single coverslip; assays were repeated across two independent cultures from independent animals).
Loss of synaptophysin results in a progressive reduction of sybII from nerve terminals and SVs during repeated stimulation
We next investigated whether the inability of synaptophysin knockout neurons to sustain exocytic capacity was because of a specific depletion of sybII from both nerve terminals and SVs during repeated SV turnover. To examine this, we employed both optical and biochemical approaches. First, we used immunofluorescence to examine whether sybII was selectively depleted from nerve terminals during repetitive stimulation in synaptophysin knockout neurons. Neurons were fixed either at rest, or after being challenged with an identical repetitive stimulation paradigm as above (Fig 2a), and then immunolabelled for sybII. As a control, neurons were immunolabelled for another abundant transmembrane SV protein, synaptotagmin‐1 (Fig 2b). Regions of interest were placed over immunofluorescent puncta (corresponding to nerve terminals), and the ratio of fluorescence in synaptophysin knockout neurons relative to those expressing syp‐mCer in the same field of view was determined (Fig 2c). At rest, the intensity of synaptotagmin‐1 immunofluorescence in synaptophysin knockout neurons was approximately equal to that in neurons expressing syp‐mCer, with no significant change in this ratio observed following repeated stimulation (Fig. 2d; synaptotagmin‐1, 96.91 ± 6.84 % (at rest) and 104.78 ± 5.98 % (post‐stimulation) of syp‐mCer‐expressing neurons; p > 0.05). Therefore, the localisation of synaptotagmin‐1 at nerve terminals is not affected by either the absence of synaptophysin or the repeated trains of stimulation. In contrast, sybII immunofluorescence in synaptophysin knockout neurons at rest was only approximately 75% of that in neurons expressing syp‐mCer (Fig 2d; sybII, 75.27 ± 7.46 % (at rest) of syp‐mCer–expressing neurons). Importantly, repeated stimulation induced a significant reduction in the amount of sybII at nerve terminals in knockout neurons (Fig 2d; sybII, 44.76 ± 3.35 % (post‐stimulation)) of syp‐mCer–expressing neurons; p < 0.05 versus sybII (at rest)), which was not evident for synaptotagmin‐1. This suggests that there is a specific de‐enrichment of sybII at nerve terminals in the absence of synaptophysin, which is exacerbated by the periods of neuronal activity.
Figure 2.
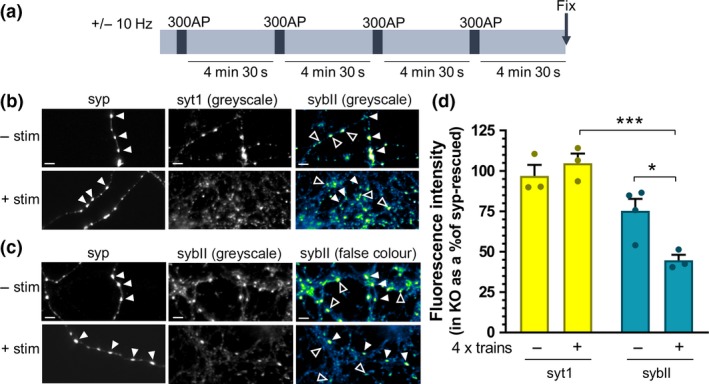
SybII is de‐enriched from synaptophysin knockout nerve terminals during repetitive stimulation. Primary cultures of synaptophysin knockout (KO) hippocampal neurons were transfected with synaptophysin‐mCer (syp). Neurons were challenged with four trains of 300 action potentials delivered at 10 Hz at 5‐min intervals as shown in (a) or left to rest for an equivalent period. At the end of the final rest period, neurons were fixed. (b and c) Representative images of synaptophysin knockout neurons, transfected with syp and immunolabelled for anti‐GFP (which also labels mCer, to label syp‐transfected cells, left, greyscale) and either synaptotagmin‐1 (syt1; b) or sybII (c) (middle, greyscale image; right, false colour, with warmer colours indicating increased fluorescence intensity). Closed arrows indicate transfected nerve terminals, open arrows indicate untransfected nerve terminals. Scale bar indicates 5 µm. (d) Bar graph displaying the mean relative fluorescent intensity of either synaptotagmin‐1 (yellow) or sybII (teal) puncta of untransfected (KO) nerve terminals as a percentage of syp‐transfected (syp‐rescued) terminals from same field of view. Bars indicate SEM. (−) indicates neurons left at rest, and (+) indicates neurons that have undergone four trains of stimuli (all n = 3 independent experiments, one‐way anova with Sidak’s multiple comparison test; ***p < 0.001, *p < 0.05. syt1 (−) vs. (+) p = 0.8952; sybII (−) vs. (+) p = 0.0302; syt1 (−) vs. sybII (−) p = 0.1448; syt1 (+) vs. sybII (+) p = 0.0006. n indicates a single field of view from a single coverslip, assays were repeated across two independent cultures from independent animals).
To confirm the activity‐dependent de‐enrichment of sybII from SVs in synaptophysin knockout neurons, we performed a biochemical fractionation study. Primary cultures of either wild‐type or synaptophysin knockout neurons were challenged with a similar stimulation protocol, the only exception being replacement of action potential trains with elevated KCl (50 mM) challenges (Fig. 3a). Following the final recovery period, neurons were fractionated using a low‐speed centrifugation step to separate plasma membrane from SVs and thus allow determination of sybII levels in both subcellular compartments (Di Paolo et al. 2002; Cousin et al. 2003). Western blotting of both fractions was performed to determine the relative amount of SV proteins present (Fig. 3b). Abundant transmembrane SV proteins, such as sybII and synaptotagmin‐1 can be either localised to SVs, or the presynaptic plasma membrane, because of the dynamic cycling of SVs depositing these proteins into the membrane, and to form a pool of readily retrievable protein for endocytosis (Sankaranarayanan and Ryan 2000; Fernandez‐Alfonso et al. 2006; Gordon et al. 2011; Zhang et al. 2015). Following repeated depolarisation, there was no significant change in the proportion of sybII or synaptotagmin‐1 in the plasma membrane and SV fractions of wild‐type neurons compared to unstimulated control neurons (WT synaptotagmin‐1 mean ± SEM: 1.439 ± 0.205 (unstimulated), 2.126 ± 0.453 (KCl), p = 0.9857; WT sybII: 4.279 ± 0.718 (unstimulated), 7.034 ± 1.384 (KCl), p = 0.5198; two‐way anova with Tukey’s multiple comparison test). However, in synaptophysin knockout neurons, repeated depolarisation induced a redistribution of sybII to the plasma membrane fraction resulting in a significantly larger proportion of sybII in the plasma membrane fraction relative to the SV sybII fraction when compared to synaptophysin knockout (KO) neurons at rest (KO sybII: 6.828 ± 1.804 (unstimulated), 12.827 ± 3.134 (KCl), p = 0.0249; two‐way anova with Tukey’s multiple comparison test). In addition, this ratio was significantly greater than that of repeatedly depolarised wild‐type neurons (KO sybII (KCl) vs. WT sybII (KCl), p = 0.0319; two‐way anova with Tukey’s multiple comparison test). This effect was specific for sybII, with no significant effect of genotype or repeated stimulation on the distribution of synaptotagmin‐1 (KO synaptotagmin‐1: 1.583 ± 0.312 (unstimulated), 1.338 ± 0.243 (KCl), p = 0.9993; two‐way anova with Tukey’s multiple comparison test). Examination of paired stimulated versus unstimulated neurons reveals a significant difference only for plasma membrane/SV fractions of sybII in synaptophysin KO neurons (Fig. 3c, p < 0.05). Taken together with the optical data presented above, this demonstrates that repeated neuronal activity results in a selective de‐enrichment of sybII from SVs in synaptophysin knockout neurons.
Figure 3.

SybII is redistributed from SVs to the plasma membrane during repetitive stimulation in synaptophysin knockout neurons. Primary hippocampal cultures from either wild‐type (WT) or synaptophysin knockout (KO) mice were challenged with four periods of depolarization with 50 mM KCl for 10 s at 5‐min intervals as shown in (a) or left to rest for an equivalent period. After the final 5‐min recovery period, neurons were hypotonically lysed and fractionated to obtain a plasma membrane (PM) or vesicular (SV) fraction. (b) Representative Western blots illustrating the partitioning of either synaptotagmin‐1 (syt1) or synaptobrevin‐II (sybII) in PM and SV fractions from unstimulated (−) and KCl‐stimulated (+) neurons. (c) Bar graph displaying the paired ratio of the relative proportions of these cargoes as PM/SV of resting neurons (−) compared to those that have undergone four trains of depolarisation with KCl (+) (N = 5 for wild‐type, N = 5 for KO, repeated measures two‐way anova with Sidak’s multiple comparison test, ***p < 0.001. All (−) vs. (+); WT syt1 p = 0.9630, WT sybII p = 0.1155; KO syt1 p = 0.9993; KO sybII p = 0.0004. N indicates an independent culture from independent animals).
Exogenous sybII fully restores presynaptic performance in synaptophysin knockout neurons
We have shown that periods of repeated activity in synaptophysin knockout neurons result in (i) a depression in exocytic capacity and (ii) a selective de‐enrichment of sybII from SVs. One simple hypothesis that can be tested is that the progressive reduction in SV fusion is because of the inefficient retrieval of sybII during multiple cycles of endocytosis. To test this hypothesis, we attempted to restore SV fusion capacity in synaptophysin knockout neurons by expressing additional sybII (Fig. 4a). Our rationale was, that by providing excess substrate for an inefficient process, sufficient sybII would be internalised to restore normal function. To achieve this, synaptophysin knockout neurons were transfected with either flag‐tagged sybII (sybII‐flag) or an empty flag vector (Figure S1A). Expression of sybII‐flag resulted in an approximately 2‐fold increase in sybII levels at nerve terminals as monitored by immunofluorescence, with vGLUT‐pHluorin used as a marker of presynaptic terminals (Figure S1B). To determine whether this additional sybII load could safeguard sustained SV fusion, we co‐transfected synaptophysin knockout neurons with vGLUT1‐pHluorin and either sybII‐flag or empty flag vector. In neurons expressing the empty flag vector, there was a progressive decrease in the maximal amplitude of evoked vGLUT1‐pHluorin fluorescence across multiple trains of activity (Fig. 4b) as observed previously (Fig. 1b). Notably, the expression of sybII‐flag in synaptophysin knockout neurons fully corrected this defect (Fig. 4b). This was confirmed when the amplitude of the peak vGLUT1‐pHluorin response was calculated across all stimuli, with a significant difference across multiple stimulus trains emerging when synaptophysin knockout neurons transfected with the flag empty vector were compared to those expressing sybII‐flag (Fig. 4c; third stimulus train: flag 68.84 ± 6.12 %, sybII‐flag 94.63 ± 5.20 %, p < 0.001; fourth stimulus train flag 67.08 ± 6.71 %, sybII‐flag 92.74 ± 5.13 %, p < 0.001). Therefore, the reduction in SV fusion events in synaptophysin knockout neurons during repetitive stimulation can be fully corrected by the titration of additional copies of sybII. This indicates that the primary presynaptic role for synaptophysin is to ensure the efficient retrieval of sybII during SV endocytosis.
Figure 4.
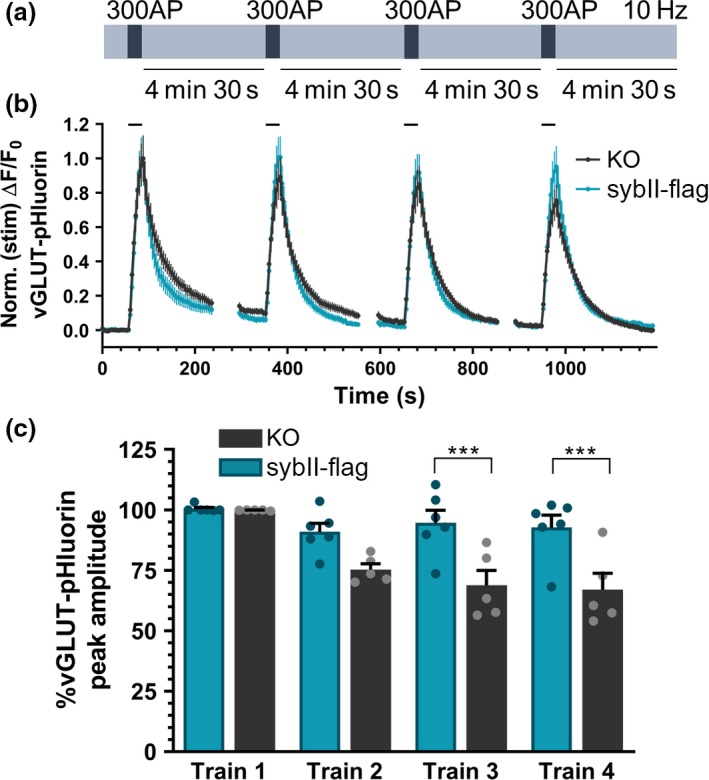
Additional copies of sybII correct depression of SV fusion events in synaptophysin knockout neurons. Primary cultures of synaptophysin knockout hippocampal neurons were transfected with vGLUT1‐pHluorin and either empty flag vector (KO) or synaptobrevin‐II‐flag (sybII‐flag). Neurons were challenged with four trains of 300 action potentials delivered at 10 Hz at 5‐min intervals as shown in (a). (b) Representative traces of the evoked vGLUT1‐pHluorin response (ΔF/F0 ± SEM) in synaptophysin knockout neurons expressing either flag (black, KO) or sybII‐flag (teal) is displayed. Traces are normalised to the peak amplitude response during the first‐action potential train. Bar indicates period of stimulation. (c) Bar graph displaying the mean peak amplitudes of the vGLUT‐pHluorin peak for each of the four trains of stimuli, calculated as a proportion of the first train. Bars indicate SEM (n = 5 KO, n = 6 sybII‐flag; repeated measures two‐way anova with Sidak’s multiple comparison test, ***p < 0.001; train 1 p > 0.9999, train 2 p = 0.0609, train 3 p = 0.0007, train 4 p = 0.0008. n indicates a single field of view from a single coverslip; assays were repeated across three independent cultures from independent animals).
Discussion
Synaptophysin is essential for the efficient retrieval of the essential soluble NSF attachment protein receptor (SNARE) sybII (Gordon et al. 2011); however, the effects of its absence on neurotransmission are relatively minor (McMahon et al. 1996; Janz et al. 1999; Kwon and Chapman 2011). We demonstrate here that synaptophysin performs a key role in sustaining SV fusion during periods of repeated neuronal activity by ensuring a sufficient complement of sybII molecules on SVs. The absence of synaptophysin resulted in a progressive reduction in SV exocytosis with a concomitant depletion of sybII from SVs. This reduction in presynaptic performance was a direct result of inefficient sybII retrieval, since titration of additional sybII fully restored function even in the absence of synaptophysin.
Our data demonstrate that repeated trains of action potential stimuli result in a depression of the evoked peak amplitude of vGLUT1‐pHluorin. Since this parameter also has a small contribution from SV retrieval occurring during stimulation, it is possible that this reduction is because of an increased endocytosis during stimulation. However, when these experiments were repeated in the presence of the V‐type ATPase inhibitor bafilomycin A1 (which arrests SV acidification and therefore allows direct monitoring of SV fusion events (Sankaranarayanan and Ryan 2001)), this depression was still apparent in synaptophysin knockout neurons expressing mCer empty vector, but not those expressing syp‐mCer (Figure S2). Therefore, the progressive reduction in vGLUT1‐pHluorin peak amplitude in synaptophysin knockout neurons was a result of reduced SV fusion. This reduction in SV fusion was also not a consequence of a slowing in SV endocytosis, since when the time constant (tau, τ) of endocytosis was calculated, there was no difference between wild‐type and synaptophysin knockout neurons under these conditions (Figure S3).
We demonstrate a significant redistribution of sybII from SVs to the plasma membrane following repetitive stimulation in synaptophysin knockout neurons. At first glance, it would appear that both sybII and synaptotagmin‐1 are enriched on the plasma membrane rather than on SVs; however, this is a consequence of sample preparation. Plasma membrane pellets were resuspended in a small volume of sample buffer, whereas the SV fraction originated from a supernatant, thus making the latter much more dilute. To control for this, samples were normalised to an internal β‐actin control in addition to an internal ratio of plasma membrane and SV fractions.
Considering that as few as 1‐3 sybII molecules are required for SV fusion (Mohrmann et al. 2010, van den Bogaart et al. 2010, Sinha et al. 2011), and there are approximately 70 copies of sybII per vesicle (Takamori et al. 2006; Wilhelm et al. 2014), it is perhaps unexpected that a moderate (50 %) reduction in vesicular sybII would impact on exocytic capacity. However, previous studies support the observation that a partial depletion of sybII from SVs has deleterious effects on neurotransmitter release. SybII+/− mice have a 40% reduction in basal neurotransmission; notably, this defect in neurotransmission is phenocopied in mice lacking the sybII endocytosis adaptor AP180, which have 40% less sybII on SVs (Koo et al. 2015). Interestingly, these neurotransmission defects are exacerbated when these mice are crossed (Koo et al. 2015). More recent experiments have shown that reducing sybII levels has a disproportionate effect on SV fusion. Titration of tetanus toxin into neurons to cleave sybII, which resulted in a 50% reduction in the level of functional sybII, decreased miniature excitatory postsynaptic current frequency to approximately 10% of controls (Bao et al. 2018). Together, these results highlight the importance of functional sybII copy number for sustaining the capacity of neurotransmitter release.
The question remains, why does a 50% reduction in sybII levels (i.e. from approximately 70 copies to 35 copies per vesicle) result in depressed exocytosis, when the fusion of a single SV can be elicited with 1‐3 copies of the v‐SNARE? The most likely scenario is that SV fusion becomes inefficient because of a dilution of sybII molecules on SVs. In this scenario, a high surface coverage of sybII allows the rapid and immediate formation of SNARE complexes, whereas a reduction in copy number would increase the variance in the time to form a fusion‐competent, stable trans‐SNARE complex. Thus, while fusion could still be supported with fewer sybII molecules, the chance of this occurring is reduced, resulting in an overall drop in the fidelity of exocytosis. This is supported by studies showing cooperativity in SNARE complex formation (Hernandez et al. 2014), and that increasing the copy number of v‐SNAREs increases the stability of trans‐SNARE complexes (Bao et al. 2018). What seems to be apparent is that although SV fusion can proceed with very few sybII molecules, the efficiency of the process scales in a non‐linear manner with vesicular sybII copy number.
Synaptophysin has been proposed to control clearance of sybII from the active zone directly after SV fusion (Rajappa et al. 2016). This potential role should be complementary to the subsequent role for synaptophysin in sybII retrieval; however, when short‐term plasticity (STP, which would be affected by inefficient sybII clearance) is monitored in synaptophysin knockout neurons, only very minor effects were observed (McMahon et al. 1996; Janz et al. 1999; Kwon and Chapman 2011). However, an effect was observed using an optical correlate of STP (Rajappa et al. 2016). In this protocol, an initial train of action potentials was used as a reference before the delivery of a second stimulus train in which the amplitude of the sybII‐pHluorin response was monitored. In these experiments, a 20% reduction in sybII‐pHluorin amplitude was observed only during high frequency stimulation (Rajappa et al. 2016). Unfortunately, this assay was not repeated with a different pHluorin reporter whose trafficking was unaffected by the absence of synaptophysin. This would have strengthened the assertion that this was an effect on STP, and not simply an increase in sybII stranding at the plasma membrane owing to the prior reference stimulus. Regardless, the key conclusion from our work is that the principal presynaptic role for synaptophysin is the efficient retrieval of sybII to SVs undergoing endocytosis, and not the clearance of sybII from the active zone. We can come to this conclusion since increased dosage of sybII was sufficient to fully restore SV fusion events in synaptophysin knockout neurons. If clearance of sybII from the active zone was the primary role for synaptophysin, this increased dosage should have resulted in an exacerbation of the depression of SV fusion events in synaptophysin knockout neurons. Moreover, enrichment of sybII in the plasma membrane in synaptophysin knockout neurons after intense stimulation does not interfere with SV endocytosis. SV endocytosis kinetics are also unaffected in synaptophysin knockout neurons expressing exogenous sybII (Figure S3). Therefore, it is the reduced retrieval of sybII to newly endocytosed vesicles that underlies the progressive decrease in exocytic efficiency, not alterations in either SV endocytosis or reduced sybII clearance from the active zone.
As stated above, a reduction in SV fusion events is not apparent in synaptophysin knockout neurons within a single‐action potential train, but is revealed during repeated stimulus trains. Therefore synaptophysin acts as a safeguard to sustain presynaptic performance during patterns of intense neuronal activity. Our results predict that any neuron or circuit that undergoes such patterns of repetitive firing would experience a reduction in neurotransmission in the event of synaptophysin dysfunction. This may be particularly relevant during early brain development, where bursts of intense synaptic activity consolidate synaptic connections (Sztainberg and Zoghbi 2016). In agreement, a series of mutations within the SYP gene have been identified in patients with intellectual disability presenting with or without epilepsy (Tarpey et al. 2009; Harper et al. 2017). Importantly, all of these mutations fail to support wild‐type levels of sybII retrieval in synaptophysin knockout neurons (Gordon and Cousin 2013; Harper et al. 2017).
In summary, we have demonstrated that the principal role of synaptophysin is to sustain presynaptic performance by maintaining a vesicular pool of sybII. This role is particularly important in neurons that experience extended periods of activity; loss of synaptophysin under these conditions results in the plasma membrane stranding of sybII and depression of neurotransmitter release.
Supporting information
Figure S1. SybII expression in synaptophysin knockout neurons.
Figure S2. Presynaptic performance defects in synaptophysin knockout neurons are due to less SV fusion events.
Figure S3. Depression in SV fusion events in synaptophysin knockout neurons is not due to defects in endocytosis.
Acknowledgments and conflict of interest disclosure
This work was supported by a grant from the Biotechnology and Biological Sciences Research Council to MAC (BB/L019329/1) and a grant and Career Development Fellowship from The National Health and Medical Research Council (NHMRC) of Australia to SLG (GNT1085483). ACK was supported by a Marie‐Curie Initial Training Network award (Project number 289581 ‐ NPlast) to MAC. JRKM and KJS were supported by a grant from the Medical Research Council (G1002117). The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. We thank members of the Gordon and Cousin labs for helpful discussions. The authors declare no conflict of interest.
All experiments were conducted in compliance with the ARRIVE guidelines.
Contributor Information
Michael A. Cousin, Email: m.cousin@ed.ac.uk.
Sarah L. Gordon, Email: sarah.gordon@florey.edu.au.
References
- Baker K., Gordon S. L., Grozeva D., et al (2015) Identification of a human synaptotagmin‐1 mutation that perturbs synaptic vesicle cycling. J. Clin. Investig. 125, 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H., Daniels R. W., MacLeod G. T., Charlton M. P., Atwood H. L. and Zhang B. (2005) AP180 maintains the distribution of synaptic and vesicle proteins in the nerve terminal and indirectly regulates the efficacy of Ca2+‐triggered exocytosis. J. Neurophysiol. 94, 1888–1903. [DOI] [PubMed] [Google Scholar]
- Bao H., Das D., Courtney N. A., et al (2018) Dynamics and number of trans‐SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G., Holt M. G., Bunt G., Riedel D., Wouters F. S. and Jahn R. (2010) One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 17, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin M. A. (2017) Integration of synaptic vesicle cargo retrieval with endocytosis at central nerve terminals. Front. Cell Neurosci. 11, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin M. A., Malladi C. S., Tan T. C., Raymond C. R., Smillie K. J. and Robinson P. J. (2003) Synapsin I‐associated phosphatidylinositol 3‐kinase mediates synaptic vesicle delivery to the readily releasable pool. J. Biol. Chem. 278, 29065–29071. [DOI] [PubMed] [Google Scholar]
- Dunning C. J., Black H. L., Andrews K. L., et al (2016) Multisite tyrosine phosphorylation of the N‐terminus of Mint1/X11alpha by Src kinase regulates the trafficking of amyloid precursor protein. J. Neurochem. 137, 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshkind L. G. and Leube R. E. (1995) Mice lacking synaptophysin reproduce and form typical synaptic vesicles. Cell Tissue Res. 282, 423–433. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Alfonso T., Kwan R. and Ryan T. A. (2006) Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron 51, 179–186. [DOI] [PubMed] [Google Scholar]
- Gordon S. L. and Cousin M. A. (2013) X‐linked intellectual disability‐associated mutations in synaptophysin disrupt synaptobrevin II retrieval. J. Neurosci. 33, 13695–13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. L., Leube R. E. and Cousin M. A. (2011) Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J. Neurosci. 31, 14032–14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. B., Mancini G. M. S., van Slegtenhorst M. and Cousin M. A. (2017) Altered synaptobrevin‐II trafficking in neurons expressing a synaptophysin mutation associated with a severe neurodevelopmental disorder. Neurobiol Dis 108, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J. M., Kreutzberger A. J., Kiessling V., Tamm L. K. and Jahn R. (2014) Variable cooperativity in SNARE‐mediated membrane fusion. Proc. Natl Acad. Sci. USA 111, 12037–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz R., Sudhof T. C., Hammer R. E., Unni V., Siegelbaum S. A. and Bolshakov V. Y. (1999) Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron 24, 687–700. [DOI] [PubMed] [Google Scholar]
- Kaempf N. and Maritzen T. (2017) Safeguards of neurotransmission: Endocytic adaptors as regulators of synaptic vesicle Composition and function. Front. Cell Neurosci. 11, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali E. T. and Jorgensen E. M. (2014) Visualizing presynaptic function. Nat. Neurosci. 17, 10–16. [DOI] [PubMed] [Google Scholar]
- Kelly B. T. and Owen D. J. (2011) Endocytic sorting of transmembrane protein cargo. Curr. Opin. Cell Biol. 23, 404–412. [DOI] [PubMed] [Google Scholar]
- Koo S. J., Markovic S., Puchkov D., et al (2011) SNARE motif‐mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl Acad. Sci. USA 108, 13540–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo S. J., Kochlamazashvili G., Rost B., et al (2015) Vesicular Synaptobrevin/VAMP2 levels guarded by AP180 control efficient neurotransmission. Neuron 88, 330–344. [DOI] [PubMed] [Google Scholar]
- Kwon S. E. and Chapman E. R. (2011) Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. T., Bolshakov V. Y., Janz R., Hammer R. E., Siegelbaum S. A. and Sudhof T. C. (1996) Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc. Natl Acad. Sci. USA 93, 4760–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. E., Sahlender D. A., Graham S. C., Honing S., Robinson M. S., Peden A. A. and Owen D. J. (2011) The molecular basis for the endocytosis of small R‐SNAREs by the clathrin adaptor CALM. Cell 147, 1118–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R., de Wit H., Verhage M., Neher E. and Sorensen J. B. (2010) Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330, 502–505. [DOI] [PubMed] [Google Scholar]
- Nonet M. L., Holgado A. M., Brewer F., et al (1999) UNC‐11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol. Biol. Cell 10, 2343–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., Sankaranarayanan S., Wenk M. R., et al (2002) Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 33, 789–804. [DOI] [PubMed] [Google Scholar]
- Rajappa R., Gauthier‐Kemper A., Boning D., Huve J. and Klingauf J. (2016) Synaptophysin 1 clears synaptobrevin 2 from the presynaptic active zone to prevent short‐term depression. Cell Rep. 14, 1369–1381. [DOI] [PubMed] [Google Scholar]
- Rao Y., Ruckert C., Saenger W. and Haucke V. (2012) The early steps of endocytosis: from cargo selection to membrane deformation. Eur. J. Cell Biol. 91, 226–233. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S. and Ryan T. A. (2000) Real‐time measurements of vesicle‐SNARE recycling in synapses of the central nervous system. Nat. Cell Biol. 2, 197–204. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S. and Ryan T. A. (2001) Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat. Neurosci. 4, 129–136. [DOI] [PubMed] [Google Scholar]
- Schoch S., Deak F., Konigstorfer A., Mozhayeva M., Sara Y., Sudhof T. C. and Kavalali E. T. (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294, 1117–1122. [DOI] [PubMed] [Google Scholar]
- Sinha R., Ahmed S., Jahn R. and Klingauf J. (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc. Natl Acad. Sci. USA 108, 14318–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y. and Zoghbi H. Y. (2016) Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 19, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Takamori S., Holt M., Stenius K., et al (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- Tarpey P. S., Smith R., Pleasance E., et al (2009) A systematic, large‐scale resequencing screen of X‐chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandingham P. A., Barmchi M. P., Royer S., Green R., Bao H., Reist N. and Zhang B. (2014) AP180 couples protein retrieval to clathrin‐mediated endocytosis of synaptic vesicles. Traffic 15, 433–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier S. M., Kam K., Yang H., Fortin D. L., Hua Z., Nicoll R. A. and Edwards R. H. (2006) Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51, 71–84. [DOI] [PubMed] [Google Scholar]
- Wilhelm B. G., Mandad S., Truckenbrodt S., et al (2014) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. [DOI] [PubMed] [Google Scholar]
- Zhang N., Gordon S. L., Fritsch M. J., et al (2015) Phosphorylation of synaptic vesicle protein 2A at Thr84 by casein kinase 1 family kinases controls the specific retrieval of synaptotagmin‐1. J. Neurosci. 35, 2492–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SybII expression in synaptophysin knockout neurons.
Figure S2. Presynaptic performance defects in synaptophysin knockout neurons are due to less SV fusion events.
Figure S3. Depression in SV fusion events in synaptophysin knockout neurons is not due to defects in endocytosis.