

Abstract
This multicenter, double‐blind, placebo‐controlled, randomized study was designed to evaluate the efficacy and safety of pseudoephedrine hydrochloride 30‐mg tablets in children aged 6 to 11 years for the temporary relief of nasal congestion due to the common cold. The primary efficacy end point was the weighted sum of the change from baseline in instantaneous nasal congestion severity score over the period from 1 to 8 hours following the first dose of study drug on day 1. Safety assessments included adverse events, sleepiness ratings, and vital signs. Pseudoephedrine was superior to placebo in reducing instantaneous nasal congestion severity in pediatric children over the first 8 hours after dosing on day 1 (least squares mean difference between treatment groups was 1.2; P = .029). Overall, secondary end points associated with nasal congestion were supportive on day 1, whereas secondary end points on day 2 were only numerically favorable. Somnolence was reported in a greater percentage of children on pseudoephedrine compared to placebo (71.9% vs 63.9%), while similar percentages of children in the same respective groups reported insomnia (34.4% and 38.9%) and nervousness (20.0% and 23.6%).Pseudoephedrine provides temporary relief of nasal congestion associated with the common cold in children 6 to <12 years of age at the current over‐the‐counter monograph dose. Multiple dosing of pseudoephedrine for up to 7 days, when given as needed for symptom relief, was generally safe in this population of children with the common cold.
Keywords: children, common cold, efficacy, nasal congestion, pediatrics, pseudoephedrine, safety
Pseudoephedrine hydrochloride is a widely used nasal decongestant in over‐the‐counter (OTC) adult and pediatric cough and cold medicines. Notably, pseudoephedrine‐containing medicines are sold behind the counter in the United States by pharmacists to reduce diversion of these products for manufacture of illegal drugs (Combat Methamphetamine Epidemic Act of 2005). Pseudoephedrine is commercially available in liquid and tablet formulations for oral administration and is indicated for use by adults and children in the temporary relief of nasal congestion due to the common cold, hay fever, or other upper respiratory allergies, and for sinus congestion and pressure. The current OTC monograph dosing regimen for children 6 to 11 years of age is 1 dose of pseudoephedrine 30 mg every 4 to 6 hours, not to exceed 120 mg (4 doses) in 24 hours. Adults and children, 12 years of age and older, may take 1 dose of pseudoephedrine 60 mg every 4 to 6 hours, not to exceed 240 mg (4 doses) in 24 hours. Complete information is available in the “Cold, Cough, Allergy, Bronchodilator, and Antiasthmatic Drug Products for Over‐the‐Counter Use” monograph, Code of Federal Regulations (CFR) Title 21, Part 341.80.
Pseudoephedrine has direct agonist activity at α‐ and β2‐adrenergic receptors. The vasoconstriction that pseudoephedrine produces is believed to be principally an α‐adrenergic receptor response.1 Stimulation of α1‐adrenergic receptors located on capacitance blood vessels of the nasal mucosa (postcapillary venules) results in vasoconstriction, decreased blood volume, and a decrease in the volume of the nasal mucosa (nasal decongestion).2 Constricted blood vessels allow less fluid to enter the nose, throat, and sinus linings, which results in decreased inflammation of nasal membranes, as well as decreased mucosal secretions.2 Thus, by constriction of blood vessels, mainly those located in the nasal passages, pseudoephedrine causes a decrease in nasal congestion.3, 4
In 2007, a citizen petition was sent to the US Food and Drug Administration (FDA) requesting that the FDA relabel cough and cold products sold OTC in the United States because they had not been shown to be safe and effective in children under 6 years of age. Although published clinical studies evaluated the safety and effectiveness of pseudoephedrine in children with nasal congestion due to the common cold, these studies are limited5, 6 and most only evaluated pseudoephedrine in combination with other active ingredients.
At a subsequently convened joint meeting of the FDA Nonprescription Drugs Advisory Committee and the Pediatric Advisory Committee,7 it was agreed that the known efficacy profile for OTC cough and cold medicines in children was generally recognized from studies in adults using a fraction of the adult dose based on body weight (Clark's Rule). Efficacy was generally recognized not just for children <6 years of age (as per the petition) but more broadly for children <18 years of age. The joint committee recommended that clinical studies in children <12 years of age be conducted to evaluate the efficacy of single‐ingredient cough and cold medicines. In response, and as part of a clinical research program under the aegis of the Consumer Health Products Association, the current study was designed to assess safety and confirm effectiveness of single‐ingredient pseudoephedrine in children 6 through 11 years of age for the temporary relief of nasal congestion due to the common cold, an indication described under CFR Title 21, Part 341.80(b)(1).
Methods
Human Subject Protection
The clinical study described herein was conducted during the winter cold seasons from November 2012 through April 2016 in accordance with Good Clinical Practices. A central institutional review board (IRB) (Schulman IRB, Cincinnati, Ohio) approved the study protocol and the informed consent and assent documents before initiation. The parents (or legal guardians) of all potential children read and signed the IRB‐approved informed consent document and each subject signed (or otherwise marked) the IRB‐approved assent form before participation in the study.
Selection of Pseudoephedrine Dose
In preparation for the efficacy and safety study, a population pharmacokinetic model was developed to characterize the pharmacokinetics of pseudoephedrine in children.8 Pooled pediatric and adult pharmacokinetic data were used for simulations of pediatric OTC doses and to confirm the OTC monograph dosing rule. Overall, pseudoephedrine plasma concentrations in children displayed a mono‐exponential decay over time in this model, with typical population pharmacokinetic parameters (given reference covariates of a 70‐kg adult and fasted state) showing an oral clearance (CL/F) of 34.9 (95% confidence interval [CI], 33.7‐36.2) L/h, a volume of distribution (V/F) of 260 (95%CI, 252‐269) L, and an absorption rate constant of 1.67 (95%CI, 1.48‐1.90) h−1. The median half‐life estimate was 4.7 hours, with a range of 2.5 to 8.2 hours.
Variability in pseudoephedrine CL/F and V/F values was primarily explained in the pharmacokinetic model by allometrically scaled weight and was not explained by age. This suggests that the pharmacokinetic parameters of pseudoephedrine are similar in the pediatric population compared with adults when appropriately scaled by weight to the 0.75th power as a measure of body size; there were no developmental differences in CL/F. This finding is consistent with pseudoephedrine being mainly excreted unchanged in the urine in children and adults, and renal function being fully developed by 2 years of age.9, 10, 11 While the 30‐mg dose and 4‐ to 6‐hour dosing intervals of pseudoephedrine selected for evaluation in this clinical study were based on the OTC monograph, the preparatory modeling and simulations of pediatric pseudoephedrine doses also supported the selection.
Study Design and Children
This was a multicenter, double‐blind, placebo‐controlled, randomized study in male and female children aged 6 through 11 years, who presented at the investigational site with common cold symptoms and were able to swallow oral tablets without chewing. Eligible children must have been experiencing symptoms that started within the 2.5 days before screening and must have had a self‐reported nasal congestion severity score equating to stuffy or very stuffy (score = 3 or 4 on a 5‐point categorical scale: 0 [not stuffy at all]; 1 [a tiny bit stuffy]; 2 [a little stuffy]; 3 [stuffy]; 4 [very stuffy]). Parents must also have reported that the children had at least 2 of the following cold symptoms: runny nose, sneezing, sore throat, headache, body achiness, and/or cough. The children were otherwise healthy, as determined by study personnel based on a review of the children medical histories, vital sign measurements, and focused physical examination (ie, eyes, ears, nose, throat, neck, heart, lungs) results on day 1. In addition, children were excluded from the study if, at the time of enrollment, they were experiencing symptoms of seasonal or perennial allergic rhinitis, had symptoms or diagnoses of sinusitis, pneumonia, strep throat, acute otitis media, or influenza, or were from homes where there was smoking. The use of prescription or nonprescription drugs and dietary supplements, including herbal supplements, was prohibited during the study, with the exception of the continued use of daily vitamins or multivitamins/multiminerals and the use of medication for hyperexcitability disorder if the regimen had been stable for at least 3 months. This limitation in the use of concomitant medications was to ensure children were not administering therapies that could have interfered with the activity of pseudoephedrine or otherwise confounded the efficacy and safety assessments in the study.
Children who met the entry criteria were enrolled and stratified by age (younger children, 6‐8 years; older children, 9‐11 years) and self‐reported baseline nasal congestion severity score (3 [stuffy]; 4 [very stuffy]). The minimum enrollment for each age cohort (6‐7 years, 8‐9 years, and 10‐11 years) was to have been no less than 20% of the total study enrollment.
Study Drug
Within each stratification group, children were randomized to 1 of 2 treatments: pseudoephedrine 30‐mg tablet or matching placebo tablet in labeled blister cards (Bilcare Global Clinical Supplies, Americas [now Sharp Clinical Services, Inc]). Children received daily doses of the assigned study drug orally for up to 7 days: 1 tablet 3 times daily for 2 days (4 hours apart on day 1 and 6 hours apart on day 2); and then 1 tablet as needed on days 3 through 7 (every 4‐6 hours up to a maximum of 4 doses in each 24‐hour period). All children were instructed to swallow the study drug with water.
Study Assessments
Following randomization and the first use of study drug, children remained at the study center for at least 4 hours to provide hourly subjective ratings of instantaneous nasal congestion severity and related nasal functions (ie, nasal breathing and nasal clearing), as well as hourly measurements of heart rate and blood pressure.
Children were released from the study center after the second dose of study drug at hour 4 and then, using a diary, continued to assess their instantaneous nasal congestion severity and related nasal functions at hours 6, 7, and 8. Separately, children provided a reflective nasal congestion relief score just before the second dose of study drug on day 1 (ie, a 4‐hour nasal congestion relief score) and just prior to the last dose of study drug on day 1 (ie, an 8‐hour nasal congestion relief score).
On day 2, the children, with assistance from the parent when needed for reading and comprehension, reported their instantaneous nasal congestion severity score and assessed related nasal functions within 1 hour of awakening, just before the first dose of study drug at hour 0. The children subsequently scored their reflective nasal congestion severity relief just before the second dose of study drug at hour 6 and again just before the third dose of study drug at hour 12 (if awake, or just before bedtime if it was earlier than hour 12). Note that the third dose of study drug on day 2 was administered only if the subject was awake.
On days 3 through 7, the children, with assistance from the parent when needed for reading and comprehension, reported their instantaneous nasal congestion severity score and assessed related nasal functions within 1 hour of awakening, just before the first dose of study drug at hour 0. On these days, children received the study drug as needed, every 4 to 6 hours, up to a maximum of 4 doses in 24 hours. The parent determined the duration of dosing based on the subject's need for continued symptom relief. The instantaneous nasal congestion severity scores and related nasal function assessments were reported at hour 0 each day through day 7, regardless of whether the child received additional doses of study drug or was no longer symptomatic.
Each of the efficacy assessments (instantaneous nasal congestion severity, nasal congestion relief, nasal congestion severity relief, instantaneous nasal breathing, and instantaneous nasal clearing) was scored using a 5‐point scale. Descriptors were assigned to the numerical values for analysis purposes, as shown in Table 1, which also lists the specific question posed to children for each assessment. The scales were presented to the children with both written descriptors and graphic representations to aid in their understanding. These self‐report questions and response scales were previously developed and validated using standard concept elicitation and cognitive debriefing techniques in 2 qualitative studies of children experiencing nasal symptoms associated with a cold (Study 1, data on file; Study 2).12 The questions and response options used wording appropriate for the cognitive abilities of children 6 to 11 years of age. Parents were provided standard instructions for administering the assessment questions and were given detailed explanations for response options associated with the degree of nasal congestion severity.
Table 1.
Efficacy Assessment Variables and Related Scales
| Assessment | Definition of Assessment and Specific Question Posed to Children |
|---|---|
| NCSi | Nasal congestion severity (instantaneous)
|
| NCSr | Nasal congestion severity (reflective)
|
| NCR | Nasal congestion relief (reflective)
|
| NSF | Nasal symptom and function composite score: includes the NCSi, nasal breathing (instantaneous), and nasal clearing (instantaneous) scores
|
| Value | NCSi and NCSr | NCR | Nasal Breathing | Nasal Clearing |
|---|---|---|---|---|
| 0 | Not stuffy at all | Not any better | Not hard | Clear |
| 1 | A tiny bit stuffy | A tiny bit better | A tiny bit hard | Partly clear |
| 2 | A little stuffy | A little better | A little hard | A little clear |
| 3 | Stuffy | Better | Hard | A tiny bit clear |
| 4 | Very stuffy | A lot better | Very hard | Not clear at all |
NCR, nasal congestion relief; NCSi, nasal congestion severity (instantaneous); NCSr, nasal congestion severity relief (reflective); NSF, nasal symptom and functioning composite score.
All scales included graphical representations (such as increasingly large circles or increasingly filled boxes) along with the descriptors to help children better assess the severity of their symptoms and differentiate the scores.
To evaluate safety, study personnel monitored for adverse events (AEs) and measured blood pressure and heart rate hourly after the first dose of study drug at the study center on day 1. Additionally, the children scored their level of sleepiness using faces on the validated Maldonado scale at baseline and again at approximately hours 2 and 6 after receiving the first 2 doses of the study drug on day 1.13
Each day during the study, regardless of whether or not the subject received a dose of study drug, the children parents used an AE Assessment Tool to grade daytime drowsiness/sleepiness, dizziness, and nervousness/agitation, as well as difficulty sleeping during the previous night. Parents received training from qualified study staff on how to complete the AE Assessment Tool in which the grades were reported using a scale ranging from 0 (complete absence of condition) to 3 (severe, condition is present all day, often impacts daily activities). Parents could also document any additional AEs not otherwise listed in the AE Assessment Tool.
A final study visit occurred within 48 hours of the subject completing the treatment period on day 7. During this visit, blood pressure, oral temperature, heart rate, and respiratory rate were measured, and the children were discharged from the study.
Statistical Methodology
All efficacy and safety parameters were evaluated, respectively, using the efficacy and safety analysis sets. Both analysis sets were limited to include only children who received at least 1 dose of the study drug. The efficacy analysis set was further limited to include only children who completed both the baseline assessment and at least 1 postbaseline assessment for the primary efficacy variable (instantaneous nasal congestion severity score).
The primary efficacy end point was the weighted sum of the change from baseline (baseline minus postbaseline) in the instantaneous nasal congestion severity score over the first 8 hours of treatment on day 1. The weights used were equal to the elapsed time in hours since the previous assessment time point. In addition to descriptive statistics, the study drug groups were compared using an analysis of variance with baseline congestion severity, age category (6‐8 and 9‐11 years), and treatment as factors (with no interaction terms). The difference between study drug groups was presented using least squares means (LSM), along with standard errors, 95%CIs, and P values; statistical significance was declared if the 2‐sided P value for the treatment group difference was ≤.05.
The secondary efficacy end points included the weighted sum of the changes from baseline in nasal congestion severity scores from 1 to 4 hours and from 6 to 8 hours on day 1, the instantaneous nasal congestion severity score at each time point from 1 to 8 hours on day 1, the sum of the nasal congestion relief scores at 4 and 8 hours on day 1, the nasal congestion severity relief score at 6 and 12 hours on day 2, and the sum of the nasal congestion severity relief scores at 6 and 12 hours on day 2. Another efficacy end point included the weighted sum of the changes from baseline in the nasal symptom and functioning composite score (ie, sum of the instantaneous nasal congestion severity score and instantaneous scores for nasal breathing and nasal clearing) from 1 to 8 hours, 1 to 4 hours, and 6 to 8 hours after the first study drug dose on day 1. Descriptive statistics were presented for the results of each of the secondary and other efficacy end points. Additionally, for each end point, the study drug groups were compared using an analysis of variance with baseline congestion severity (instantaneous nasal congestion severity and nasal congestion relief only), age category, and treatment as factors; the differences between study drug groups were presented using LSM, standard errors, 95%CIs, and P values.
For the analysis of safety, the number and percentage of children experiencing an AE or serious AE was presented by system organ class and preferred term for each study drug group. Mean daily ratings on the AE Assessment Tool were presented by study drug and day; the study drugs were compared using an analysis of variance at each time point with age category and treatment as factors. Descriptive statistics were used to summarize results of blood pressure (systolic and diastolic) and radial pulse measurements on day 1 (before dosing; 1, 2, 3, and 4 hours after the first dose; and at study completion), as well as body temperature and respiratory rate (before dosing and at study completion). Faces on the Maldonado scale were converted to the Thurston scale, with scores of 0, 0.94, 2.08, 2.83, and 3.60 representing images with increasing sleepiness.13 At each time point (baseline, hour 2 and hour 6 on day 1), the differences between study drug groups were compared using an analysis of variance with age category and treatment as factors.
Results
Subject Demographics and Disposition
In this study, 568 children were randomized to study drug, including 286 in the pseudoephedrine group and 282 in the placebo group. Overall, 556 of the 568 randomized children (97.9%) completed the study, including 282 (98.6%) in the pseudoephedrine group and 274 (97.2%) in the placebo group. Of the 568 randomized children, 563 (99.1%) were included in the efficacy analysis set and 565 (99.5%) were included in the safety analysis set (Figure 1).
Figure 1.
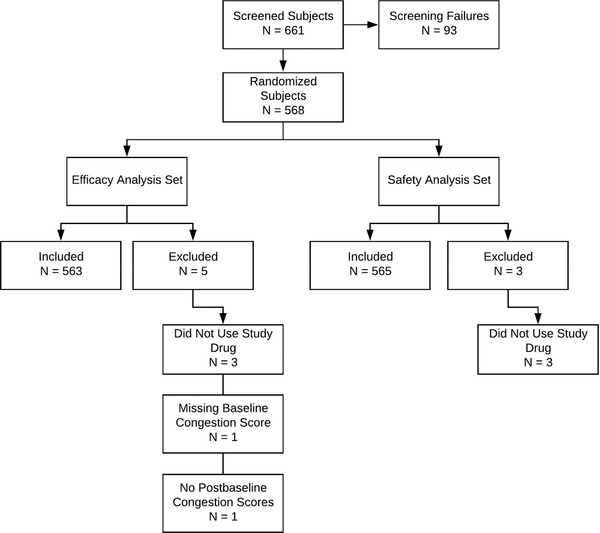
Analysis set composition.
The 568 randomized children had a mean age of 8.2 years (range, 5‐11 years), with 40.3%, 31.7%, and 27.8% of the children being 6 to 7, 8 to 9, and 10 to 11 years of age, respectively (Table 2). Children were evenly divided by sex (50.2% male and 49.8% female), and were primarily white (47.5%) or black (44.0%). Approximately two‐thirds of the children (67.4%) entered the study with a nasal congestion score of 3 (stuffy), while the remaining one‐third of the children (32.4%) entered the study with a nasal congestion score of 4 (very stuffy). The children had a mean height, weight, and body mass index of 134.7 cm, 34.9 kg, and 18.8 kg/m2, respectively. No important differences were observed between study drug groups in regard to any demographic parameter or baseline characteristic (Table 2).
Table 2.
Summary of Demographic and Baseline Characteristics by Treatment Group (All Randomized Children)
| Pseudoephedrine (N = 286) | Placebo (N = 282) | Total (N = 568) | |
|---|---|---|---|
| Age, y | |||
| Mean (SD) | 8.2 (1.70) | 8.2 (1.69) | 8.2 (1.70) |
| Min, max | 5, 11 | 6, 11 | 5, 11 |
| Age cohorts at enrollment (y), n (%) | |||
| <6 | 1 (0.3) | 0 | 1 (0.2) |
| 6‐7 | 118 (41.3) | 111 (39.4) | 229 (40.3) |
| 8‐9 | 83 (29.0) | 97 (34.4) | 180 (31.7) |
| 10‐11 | 84 (29.4) | 74 (26.2) | 158 (27.8) |
| Sex, n (%) | |||
| Male | 149 (52.1) | 136 (48.2) | 285 (50.2) |
| Female | 137 (47.9) | 146 (51.8) | 283 (49.8) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 44 (15.4) | 41 (14.5) | 85 (15.0) |
| Not Hispanic or Latino | 242 (84.6) | 241 (85.5) | 483 (85.0) |
| Race, n (%) | |||
| White | 133 (46.5) | 137 (48.6) | 270 (47.5) |
| Black | 126 (44.1) | 124 (44.0) | 250 (44.0) |
| Asian | 13 (4.5) | 11 (3.9) | 24 (4.2) |
| American Indian or Alaska Native | 1 (0.3) | 0 | 1 (0.2) |
| Native Hawaiian or other Pacific Islander | 0 | 1 (0.4) | 1 (0.2) |
| Other | 13 (4.5) | 9 (3.2) | 22 (3.9) |
| Nasal congestion severity, n (%) | |||
| 3 = stuffy | 191 (66.8) | 192 (68.1) | 383 (67.4) |
| 4 = very stuffy | 94 (32.9) | 90 (31.9) | 184 (32.4) |
| Missing | 1 (0.3) | 0 | 1 (0.2) |
| Height (cm), n | 284 | 280 | 564 |
| Mean (SD) | 135.4 (14.0) | 134.0 (12.3) | 134.7 (13.2) |
| Min, max | 101.1, 175.3 | 104.1, 172.7 | 101.1, 175.3 |
| Weight (kg), n | 284 | 281 | 565 |
| Mean (SD) | 35.4 (13.5) | 34.3 (12.7) | 34.9 (13.1) |
| Min, max | 17.9, 85.9 | 15.9, 88.9 | 15.9, 88.9 |
| Body mass index (kg/m2), n | 284 | 280 | 564 |
| Mean (SD) | 18.8 (4.4) | 18.7 (4.7) | 18.8 (4.6) |
| Min, max | 10.0, 35.1 | 10.2, 43.9 | 10.0, 43.9 |
max, maximum; min, minimum; SD, standard deviation.
Efficacy
On day 1, pseudoephedrine was superior to placebo in reducing the severity of nasal congestion over 8 hours following the first 2 doses at hours 0 and 4. Specifically, the LSM difference between study drug groups for the weighted sum of the change from baseline in instantaneous nasal congestion severity score from hours 1 to 8 on day 1 (ie, the primary efficacy end point) was 1.2 (95%CI, 0.12‐2.24); this difference was statistically significant (P = .029) (Table 3).
Table 3.
Weighted Sum of the Change from Baseline in NCSi Scores from Hours 1 to 8 on Day 1 (Primary End Point, Efficacy Analysis Set, N = 563)
| Pseudoephedrine (N = 284) | Placebo (N = 279) | |||
|---|---|---|---|---|
| Day 1 | Actual Result | Weighted Sum of Change From Baseline | Actual Result | Weighted Sum of Change From Baseline |
| Baseline | ||||
| Mean (SD) | 3.3 (0.47) | 3.3 (0.47) | ||
| Median | 3.0 | 3.0 | ||
| Min, max | 3, 4 | 3, 4 | ||
| Sum of hours 1‐8 | ||||
| Mean (SD) | 11.9 (6.41) | 10.7 (6.98) | ||
| Median | 12.0 | 11.0 | ||
| Min, max | –4, 30 | –7, 30 | ||
| LS mean (SE) | 12.6 (0.40) | 11.4 (0.40) | ||
| Study drug difference | ||||
| LS mean difference (SE) | 1.2 (0.54) | |||
| 95%CI | (0.12‐2.24) | |||
| P value | .029 | |||
CI, confidence interval; LS, least squares; max, maximum; min, minimum; NCSi, Nasal Congestion Severity (instantaneous); SD, standard deviation; SE, standard error.
The NCSi score was based on a scale ranging from 0 (not stuffy at all) to 4 (very stuffy). The weights used were equal to the elapsed time in hours since the previous time point. Baseline was defined as the last available assessment prior to the first dose of study drug. The difference between treatment groups was compared with an analysis of variance for each time interval with baseline congestion severity, age category, and treatment as factors.
Regarding the secondary efficacy end points that included the weighted sum of the changes from baseline in instantaneous nasal congestion severity scores over the periods of 1 to 4 hours and, separately, 6 to 8 hours after the first dose of study drug on day 1, the LSM differences between study drug groups were 0.6 (95% CI, 0.08‐1.21; P = .026) and 0.5 (95%CI, −0.09 to 1.16; P = .091), respectively (Table 4). The difference was statistically significant in the first period from 1 to 4 hours; the differences between treatment groups numerically favored pseudoephedrine over placebo in the second period from 6 to 8 hours.
Table 4.
Weighted Sum of the Change From Baseline in NCSi Scores From Hours 1 to 4 and Hours 6 to 8 on Day 1 (Secondary End Points, Efficacy Analysis Set, N = 563)
| Pseudoephedrine (N = 284) | Placebo (N = 279) | |||
|---|---|---|---|---|
| Day 1 | Actual Result | Weighted Sum of Change From Baseline | Actual Result | Weighted Sum of Change From Baseline |
| Baseline | ||||
| Mean (SD) | 3.3 (0.47) | 3.3 (0.47) | ||
| Median | 3.0 | 3.0 | ||
| Min, max | 3, 4 | 3, 4 | ||
| Sum of hours 1‐4 | ||||
| Mean (SD) | 5.5 (3.48) | 4.9 (3.60) | ||
| Median | 6.0 | 5.0 | ||
| Min, max | −2, 16 | −3, 16 | ||
| LS mean (SE) | 5.9 (0.21) | 5.3 (0.21) | ||
| Study drug difference | ||||
| LS mean difference (SE) | 0.6 (0.29) | |||
| 95%CI | (0.08‐1.21) | |||
| P value | .026 | |||
| Sum of hours 6‐8 | ||||
| Mean (SD) | 6.4 (3.77) | 5.8 (4.02) | ||
| Median | 6.0 | 6.0 | ||
| Min, max | –2, 16 | –4, 16 | ||
| LS mean (SE) | 6.7 (0.23) | 6.2 (0.23) | ||
| Study drug difference | ||||
| LS mean difference (SE) | 0.5 (0.32) | |||
| 95%CI | (–0.09 to 1.16) | |||
| P value | .091 | |||
CI, confidence interval; LS, least squares; max, maximum; min, minimum; NCSi, Nasal Congestion Severity (instantaneous); SD, standard deviation; SE, standard error.
The NCSi score was based on a scale ranging from 0 (not stuffy at all) to 4 (very stuffy). The weights used were equal to the elapsed time in hours since the previous time point.
Baseline was defined as the last available assessment before the first dose of study drug. The change from baseline was calculated as baseline minus postbaseline. Positive differences between study drug groups indicated a greater effect for pseudoephedrine relative to placebo.
P values were based on an analysis of variance for each time interval with baseline congestion severity, age category, and treatment as factors.
At every postbaseline time point on day 1 (ie, at hours 1, 2, 3, 4, 6, 7, and 8), the mean instantaneous nasal congestion severity score was lower (indicating a better result) and the change from baseline was higher (indicating greater improvement) in the pseudoephedrine group compared with the placebo group (data not shown in tables). The LSM differences between study drug groups ranged from 0.1 to 0.2 throughout the day. The differences favoring pseudoephedrine over placebo at hours 7 and 8 demonstrated significant improvements in nasal congestion severity (at both time points, the LSM difference between study drug groups was 0.2; 95%CI, 0.0‐0.4; at hour 7, P = .028; and at hour 8, P = .016).
Based on the sum of the nasal congestion relief scores at hours 4 and 8 on day 1, greater relief from nasal congestion (reflective) was observed in the pseudoephedrine group when compared with the placebo group. The LSM values for the sum of the scores in the pseudoephedrine and placebo groups were 4.7 and 4.3, respectively. The LSM difference between study drug groups (0.4; 95%CI, 0.1‐0.8) was statistically significant (P = 0.013) (Table 5).
Table 5.
Sum of the NCR Scores at Hours 4 and 8 on Day 1 (Secondary End Points, Efficacy Analysis Set, N = 563)
| Day 1 | Pseudoephedrine (N = 284) | Placebo (N = 279) |
|---|---|---|
| Hour 4, n | 284 | 278 |
| Mean (SD) | 2.5 (1.14) | 2.3 (1.28) |
| Median | 2.0 | 2.0 |
| Min, max | 0, 4 | 0, 4 |
| LS mean (SE) | 2.4 (0.07) | 2.2 (0.08) |
| Study drug difference | ||
| LS mean difference (SE) | 0.2 (0.10) | |
| 95%CI | (0.0‐0.4) | |
| P value | .029 | |
| Hour 8, n | 282 | 274 |
| Mean (SD) | 2.3 (1.10) | 2.1 (1.16) |
| Median | 2.0 | 2.0 |
| Min, max | 0, 4 | 0, 4 |
| LS mean (SE) | 2.3 (0.07) | 2.1 (0.07) |
| Study drug difference | ||
| LS mean difference (SE) | 0.2 (0.10) | |
| 95%CI | (0.0‐0.4) | |
| P value | .034 | |
| Sum of hours 4 and 8, n | 282 | 274 |
| Mean (SD) | 4.8 (1.88) | 4.4 (2.11) |
| Median | 5.0 | 4.0 |
| Min, max | 0, 8 | 0, 8 |
| LS mean (SE) | 4.7 (0.12) | 4.3 (0.12) |
| Study drug difference | ||
| LS mean difference (SE) | 0.4 (0.17) | |
| 95%CI | (0.1‐0.8) | |
| P value | .013 |
CI, confidence interval; LS, least squares; max, maximum; min, minimum; NCR, Nasal Congestion Relief; SD, standard deviation; SE, standard error.
The NCR score was based on a scale ranging from 0 (not any better) to 4 (a lot better). Positive differences between study drug groups indicated a greater effect for pseudoephedrine relative to placebo.
P values were based on an analysis of variance at each time point with baseline congestion severity, age category, and treatment as factors.
On day 2 of the study, individual nasal congestion severity relief scores at hours 6 and 12, along with the sum of those scores, were analyzed. At hour 6, the LSM nasal congestion severity relief score was 1.8 in both the pseudoephedrine and placebo groups (Table 6). At hour 12, the LSM nasal congestion severity relief score was 1.6 in the pseudoephedrine group and 1.7 in the placebo group. The LSM values for the sums of the nasal congestion severity relief scores at hours 6 and 12 in the pseudoephedrine and placebo groups were 3.4 and 3.5, respectively. The LSM difference between study drug groups, which favored pseudoephedrine over placebo, was −0.1 (95%CI, −0.5 to 0.2); the difference, however, was not statistically significant (P = .376).
Table 6.
NCSr Scores at Hours 6 and 12 on Day 2 and the Sum of the NCSr Scores at Hours 6 and 12 on Day 2 (Secondary End Points, Efficacy Analysis Set, N = 563)
| Day 2 | Pseudoephedrine (N = 284) | Placebo (N = 279) |
|---|---|---|
| NCSr: hour 6, n | 279 | 274 |
| Mean (SD) | 1.7 (1.04) | 1.8 (1.11) |
| Median | 2.0 | 2.0 |
| Min, max | 0, 4 | 0, 4 |
| LS mean (SE) | 1.8 (0.06) | 1.8 (0.07) |
| Study drug difference | ||
| LS mean difference (SE) | –0.0 (0.09) | |
| 95%CI | (–0.2 to 0.1) | |
| P value | .661 | |
| NCSr: hour 12, n | 276 | 273 |
| Mean (SD) | 1.6 (1.04) | 1.7 (1.09) |
| Median | 2.0 | 2.0 |
| Min, max | 0, 4 | 0, 4 |
| LS mean (SE) | 1.6 (0.06) | 1.7 (0.06) |
| Study drug difference | ||
| LS mean difference (SE) | –0.1 (0.09) | |
| 95%CI | (–0.3 to 0.1) | |
| P value | .293 | |
| NCR: sum of hours 6 and 12, n | 273 | 271 |
| Mean (SD) | 3.3 (1.88) | 3.5 (2.01) |
| Median | 3.0 | 4.0 |
| Min, max | 0, 8 | 0, 8 |
| LS mean (SE) | 3.4 (0.12) | 3.5 (0.12) |
| Study drug difference | ||
| LS mean difference (SE) | –0.1 (0.17) | |
| 95%CI | (–0.5 to 0.2) | |
| P value | .376 |
CI, confidence interval; LS, least squares; max, maximum; min, minimum; NCR, Nasal Congestion Relief; NCSr, Nasal Congestion Severity Relief (reflective); SD, standard deviation; SE, standard error.
The NCSr score was based on a scale ranging from 0 (not stuffy at all) to 4 (very stuffy). Negative differences between study drug groups indicated a greater effect for pseudoephedrine relative to placebo.
P values were based on an analysis of variance at each time point with baseline congestion severity, age category, and treatment as factors.
For the weighted sum of the changes from baseline in the nasal symptom and functioning composite score (comprising the instantaneous nasal congestion severity, nasal breathing, and nasal clearing scores) over the periods of 1 to 8 hours, 1 to 4 hours, and 6 to 8 hours after the first dose of study drug on day 1, the LSM differences between study drug groups were 0.9 (95%CI, −2.3 to 4.0; P = .577), 0.8 (95%CI, −0.8 to 2.3; P = .317), and 0.1 (95%CI, −1.7 to 2.0; P = .910), respectively. Although not statistically significant, the differences between treatment groups numerically favored pseudoephedrine over placebo for all 3 periods.
Safety
In this study, 429 of the 565 children in the safety analysis set (75.9% overall, 77.2% of children in the pseudoephedrine group and 74.6% of children in the placebo group) experienced at least 1 AE (Table 7). There were no serious AEs reported in this study. Three children had their study drug withdrawn due to AEs; only 1 of those 3 children discontinued the study. The 3 children who had their study drug withdrawn included the following: 1 subject in the pseudoephedrine group who completed the study and experienced an event of generalized rash, which was considered possibly study drug related; 1 subject in the placebo group who completed the study and experienced events of headache, otitis media, and pyrexia, none of which were considered study drug related; and 1 additional subject in the placebo group who discontinued the study and experienced events of peripheral edema, peripheral swelling, pruritus, and urticaria, all of which were considered study drug related.
Table 7.
Adverse Events Occurring in ≥1% of all Children by System Organ Class and Preferred Term (Safety Analysis Set, N = 565)
| System Organ Class Preferred Term | Pseudoephedrine (N = 285), n (%) | Placebo (N = 280), n (%) |
|---|---|---|
| Number of children with at least 1 AE | 220 (77.2) | 209 (74.6) |
| Gastrointestinal disorders | 9 (3.2) | 16 (5.7) |
| Abdominal pain, upper | 5 (1.8) | 4 (1.4) |
| Diarrhea | 0 | 8 (2.9) |
| Vomiting | 2 (0.7) | 5 (1.8) |
| General disorders and administration site conditions | 19 (6.7) | 23 (8.2) |
| Fatigue | 14 (4.9) | 11 (3.9) |
| Pyrexia | 3 (1.1) | 8 (2.9) |
| Nervous system disorders | 206 (72.3) | 180 (64.3) |
| Somnolence | 205 (71.9) | 179 (63.9) |
| Dizziness | 36 (12.6) | 35 (12.5) |
| Headache | 12 (4.2) | 9 (3.2) |
| Psychiatric disorders | 121 (42.5) | 134 (47.9) |
| Insomnia | 98 (34.4) | 109 (38.9) |
| Nervousness | 57 (20.0) | 66 (23.6) |
| Agitation | 3 (1.1) | 3 (1.1) |
| Respiratory, thoracic, and mediastinal disorders | 10 (3.5) | 12 (4.3) |
| Epistaxis | 3 (1.1) | 4 (1.4) |
AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities.
MedDRA coding dictionary version 18.1.
If a subject experienced >1 AE, the subject was counted only once in a category. If a subject experienced >1 AE in a system organ class, the subject was counted only once in that system organ class.
Numbers and percentages of events by system organ class include all reported events within the classification, not just those individual events that occurred in ≥1% of all children.
While somnolence was reported in a greater percentage of children in the pseudoephedrine group than in the placebo group (71.9% vs 63.9%, respectively), similar percentages of children in the same respective groups reported insomnia (34.4% and 38.9%) and nervousness (20.0% and 23.6%) (Table 7).
Study drug–related events that were reported by 1% or more of all children included somnolence, insomnia, nervousness, dizziness, and fatigue (data not shown in tables). Of these events, a greater percentage of children in the pseudoephedrine group compared with the placebo group experienced study drug–related somnolence (57.5% vs 50.0%, respectively), while the other study drug–related events occurred at generally similar frequencies in the pseudoephedrine and placebo groups, respectively: insomnia (29.1% vs 31.4%), nervousness (16.5% vs 20.0%), dizziness (9.5% vs 8.9%, respectively), and fatigue (4.9% vs 3.6%).
Regarding the parent‐completed AE Assessment Tool, across all assessments and study days, the LSM scores in the pseudoephedrine group ranged from 0.0 to 1.0, while the LSM scores in the placebo group ranged from 0.0 to 0.8 (data not shown in tables). Significantly higher drowsiness/sleepiness was observed in the pseudoephedrine group relative to the placebo group only on day 1 (LSM difference, 0.2; P = .006), while significantly lower nervousness/agitation was observed in the pseudoephedrine group relative to the placebo group on days 2 (LSM difference, −0.1; P = .039), 4 (LSM difference, −0.0; P = .024), 5 (LSM difference, −0.0; P = .028), and 7 (LSM difference, −0.0; P = .046). No other statistically significant differences were noted between study drug groups for any assessment on any study day.
The child‐assessed sleepiness evaluation was based on the Maldonado scale following conversion to a numeric score for analysis. At baseline on day 1, the mean scores in both study drug groups were similar (∼1.4), falling roughly between the second and third faces (data not shown in tables). At both hours 2 and 6 on day 1, increases in sleepiness were observed in the pseudoephedrine group, while slight decreases in sleepiness were observed in the placebo group. The LSM difference between study drug groups was significant at both time points (difference at hour 2, 0.382; P < .001]; difference at hour 6, 0.247; P = .032).
Based on a review of vital sign measurements, no cardiovascular safety signals or trends associated with pseudoephedrine use were reported; there were no AEs of tachycardia in either study drug group. During the study, mean systolic blood pressures ranged from 105.9 to 107.4 mm Hg in the pseudoephedrine group and from 105.2 to 107.5 mm Hg in the placebo group. Mean diastolic blood pressures ranged from 67.5 to 68.6 mm Hg in the pseudoephedrine group and from 66.5 to 67.7 mm Hg in the placebo group. Finally, mean heart rate measurements ranged from 83.1 to 86.5 beats/min in the pseudoephedrine group and from 83.4 to 86.5 beats/min in the placebo group.
Discussion
Published studies of cough and cold medicines marketed for pediatric use under the OTC monograph have not conclusively demonstrated efficacy in children. This is mainly due to the small number of available studies and certain methodological limitations of those studies, which include inadequate sample sizes, limited statistical power, and the use of inappropriate and unvalidated end points. This study aimed to address these limitations and focused on a single‐ingredient product with clinical end points that are pathophysiologically related to what the drug is expected to do (ie, end points relevant to the specific cold symptoms that the product is intended to alleviate). Before initiating the study, the population pharmacokinetics of pseudoephedrine in children was characterized, and model simulations with pooled adult data confirmed that the OTC monograph dose was adequate to evaluate.8
This study was designed to assess the efficacy and safety of pseudoephedrine 30‐mg tablets in children aged 6 to 11 years for the temporary relief of nasal congestion due to the common cold. It employed relevant inclusion and exclusion criteria for subject selection, ultimately randomizing 568 children to double‐blind, placebo‐controlled treatment. Efficacy was evaluated for clinically meaningful aspects of nasal symptoms (particularly congestion severity and relief) that were assessed frequently at several time points over the first day when cold symptoms are generally most severe. Additional assessments were scheduled on day 2. These assessments were evaluated using robust statistical methods, including a sample size sufficient to achieve 90% power at a 5% significance level. Standard assessments of safety commonly employed in clinical research were performed throughout the study period.
The primary efficacy end point was purposely constructed to assess efficacy over the first 2 doses, given 4 hours apart, on day 1. This duration was selected for 3 reasons: (1) to be consistent with pseudoephedrine's acute indication in providing temporary symptom relief with dosing as needed; (2) to adequately demonstrate the efficacy of pseudoephedrine in a manner analogous to demonstrating efficacy in other single‐dose clinical models of transient acute conditions (eg, headache pain); and (3) to minimize potential challenges for parents to comply with study drug administration, such as the need to administer the study drug during the school day.
In addition to efficacy, this study also assessed the safety of multiple doses of pseudoephedrine when administered for up to 7 days. The duration of dosing was based on the maximum duration of dosing permitted by the OTC monograph for pseudoephedrine and depended on the need for continued symptom relief as determined by the parent.
Importantly, the assessment of pseudoephedrine efficacy was based on patient‐reported outcomes rather than assessments by caregivers or health care professionals. The primary and secondary end points were derived from symptom‐focused questions and response scales that were developed for children aged 6 to 11 years, using qualitative techniques outlined in the FDA guidance for patient‐reported outcome development and validation and modified for children.14, 15 Question development was informed by literature review, concept elicitation interviews with children and parents, concept selection and modification (ie, various recall periods and time anchors) and content evaluation (Study 1, data on file; Study 2).12 Although the severity of nasal congestion had been scored previously using 4‐ to 8‐point verbal response scales in published allergy and common cold studies, descriptors for the 5‐point response scales in this study were based on the validated severity scale for the assessment of common cold symptoms in adults: none, very mild, mild, moderate, and severe.16 Parallel responses were modified using age‐related vocabulary (Table 1), and tested for comprehension and severity rank order through a card‐sorting exercise. The final versions of end point questions and responses were found to have face and content validity and were understandable, although parental assistance in reporting was needed for some younger children (6‐8 years).
The current study met its primary efficacy end point and demonstrated that pseudoephedrine is superior to placebo in reducing the severity of nasal congestion over 8 hours following the first 2 doses of study drug on day 1. Regarding the secondary efficacy end points that included the weighted sum of the change from baseline in instantaneous nasal congestion severity score from 1 to 4 hours and, separately, from 6 to 8 hours on day 1, the differences between study groups favored pseudoephedrine over placebo in both periods and were significant from 1 to 4 hours. At all time points on day 1, the analyses suggested greater reductions from baseline in the instantaneous severity of nasal congestion for pseudoephedrine when compared with placebo. The sum of the nasal congestion relief scores at hours 4 and 8 on day 1 showed statistically significant differences in favor of pseudoephedrine relative to placebo. Overall, results of the secondary efficacy end point analyses associated with nasal congestion on day 1 supported the primary efficacy end point analysis, further indicating greater effectiveness of pseudoephedrine when compared with placebo in reducing the severity of nasal congestion.
No safety signals or trends were observed in this study that would adversely alter the known safety profile of pseudoephedrine when used in children for the temporary relief of nasal congestion. The frequencies and types of AEs reported by children in the pseudoephedrine and placebo groups were generally similar. No important differences were observed between study groups for any of the most commonly occurring study drug related AEs, except for somnolence, which occurred more frequently in the pseudoephedrine group. One subject in the pseudoephedrine group experienced generalized rash, which led to study drug withdrawal. This AE was considered possibly related and resolved without having to discontinue the subject from the study. No unexpected AEs were reported. These results provide evidence of the efficacy and safety data of pseudoephedrine in an otherwise healthy pediatric population experiencing nasal congestion from the common cold.
Study Limitations
This pediatric study was designed to evaluate the efficacy and safety of a fixed dose of 30‐mg pseudoephedrine, which was shown to provide adequate exposure for children aged 6 to 11 years. However, a possible limitation to the fixed dose is that weight‐based differences in pseudoephedrine effectiveness may exist within and between the age groups. Another limitation of this study is that efficacy on day 2 was not demonstrated, although trends favoring pseudoephedrine over placebo were observed. Given that the common cold is a generally mild condition that is transient and self‐limiting and that nasal congestion appears to follow diurnal variation, being worse in the morning and better in the early evening, distinguishing between a high placebo response and drug‐induced changes in symptom severity and relief is challenging. Similarly, efficacy of pseudoephedrine compared with placebo in adult studies of the common cold has been demonstrated over the initial dose or doses on the first study day, but inconsistently demonstrated efficacy on subsequent days.17, 18, 19, 20
Conclusions
Pseudoephedrine provided temporary relief of nasal congestion associated with the common cold in children aged 6 to 11 years, at the current OTC monograph dose. In particular, the study met its primary efficacy end point and demonstrated that pseudoephedrine is superior to placebo in reducing the instantaneous nasal congestion severity score over 8 hours following the first 2 doses of study drug on day 1. Overall, the secondary end points were supportive on day 1. The safety results suggest that multiple dosing of pseudoephedrine for up to 7 days, when given at the OTC monograph dose on an as‐needed basis for symptom relief, is generally safe. Somnolence occurred more frequently in the pseudoephedrine group than in the placebo group. No safety signals indicative of increased risk associated with the administration of pseudoephedrine were observed in otherwise healthy children 6 to 11 years of age who presented with the common cold.
Conflicts of Interest
Drs. Gelotte and Albrecht were paid consultants of Consumer Healthcare Products Association (CHPA). Ms. Hynson is an employee of Concentrics Research, which received consulting fees from Perrigo Company paid on behalf of CHPA. Ms. Gallagher is an employee of Perrigo Company.
The study described herein was registered on ClinicalTrials.gov as NCT01744106. Schulman IRB, a central institutional review board, reviewed and approved the protocol and associated informed consent/assent forms. Concentrics Research was the contract research organization that planned and conducted the study.
Funding
This study was sponsored and supported by Perrigo Company in collaboration with the Consumer Healthcare Products Association (CHPA) Pediatric Cough Cold Pseudoephedrine Task Group, which provided all research funding. Medical writing support was provided by Illuminated Research, LLC.
Data Accessibility
The data presented herein were obtained through a clinical study and the raw data are not publicly available.
References
- 1. Drew CD, Knight GT, Hughes DT, Bush M. Comparison of the effects of D‐(−)‐ephedrine and L‐(+)‐pseudoephedrine on the cardiovascular and respiratory systems in man. Br J Clin Pharmacol. 1978;6(3):221‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson DA, Hricik JG. The pharmacology of alpha‐adrenergic decongestants. Pharmacotherapy. 1993;13(6 Pt 2):110S‐115S; discussion 143S‐146S. [PubMed] [Google Scholar]
- 3. Empey DW, Medder KT. Nasal decongestants. Drugs. 1981;21(6):438‐443 [DOI] [PubMed] [Google Scholar]
- 4. Hoffman BB. Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists In: Hardman JG, Limbird LE, eds. Goodman & Gilman's The Pharmacologic Basis of Therapeutics. 10th ed New York: McGraw‐Hill, Medical Publishing Division; 2001:215‐268. [Google Scholar]
- 5. Martinez Gallardo F, Lopez Fiesco A, Zamora G. Symptomatic treatment of common cold in children with a new combination of naproxen sodium plus pseudoephedrine hydrochloride: a comparative trial against pseudoephedrine syrup. Proc West Pharmacol Soc. 1994;37:157‐158. [PubMed] [Google Scholar]
- 6. Weippl G. Therapeutic approaches to the common cold in children. Clin Ther. 1984;6(4):475‐482. [PubMed] [Google Scholar]
- 7. FDA Advisory Meeting . Joint Meeting of the Nonprescription Drugs Advisory Committee and the Pediatric Advisory Committee 2007. https://wayback.archive-it.org/7993/20170404050526/ https://www.fda.gov/ohrms/dockets/ac/07/minutes/2007-4323m1-Final.pdf. Accessed November 4, 2018.
- 8. Gastonguay MR, Knebel W, Gelotte CK, Kramer T. Evaluation of the performance of pediatric OTC monograph dosing guidance for pseudoephedrine via population pharmacokinetic modeling and simulation. Clin Pharmacol Therap. 2011;89(suppl. 1):Abstract. [Google Scholar]
- 9. Hayton WL. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS PharmSci. 2000;2(1):E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nieder M, Jaeger H. Sensitive quantification of pseudoephedrine in human plasma and urine by high‐performance liquid chromatography. J Chromatogr. 1988;424(1):73‐82. [DOI] [PubMed] [Google Scholar]
- 11. Simons FE, Gu X, Watson WT, Simons KJ. Pharmacokinetics of the orally administered decongestants pseudoephedrine and phenylpropanolamine in children. J Pediatr. 1996;129(5):729‐734. [DOI] [PubMed] [Google Scholar]
- 12. Halstead, P , Arbuckly, R , Marshall, C , et al. Development and content validity testing of patient‐reported outcome items for children to self‐assess symptoms of the common cold. In press. [DOI] [PMC free article] [PubMed]
- 13. Maldonado CC, Bentley AJ, Mitchell D. A pictorial sleepiness scale based on cartoon faces. Sleep. 2004;27(3):541‐548. [DOI] [PubMed] [Google Scholar]
- 14. Arbuckle R, Abetz‐Webb L. “Not just little adults”: qualitative methods to support the development of pediatric patient‐reported outcomes. Patient. 2013;6(3):143‐159. [DOI] [PubMed] [Google Scholar]
- 15. FDA guidance for industry: patient‐reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes. 2006;4:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barrett B, Locken K, Maberry R, et al. The Wisconsin Upper Respiratory Symptom Survey (WURSS): a new research instrument for assessing the common cold. J Fam Pract. 2002;51(3):265. [PubMed] [Google Scholar]
- 17. Taverner D, Danz C, Economos D. The effects of oral pseudoephedrine on nasal patency in the common cold: a double‐blind single‐dose placebo‐controlled trial. Clin Otolaryngol. 1999;24:47‐51. [DOI] [PubMed] [Google Scholar]
- 18. Latte J, Taverner D, Slobodian P, Shakib S. A randomized, double‐blind, placebo‐controlled trial of pseudoephedrine in coryza. Clin Exper Pharmacol Physiol. 2004;31:429‐432. [DOI] [PubMed] [Google Scholar]
- 19. Latte J, Traverner D. Clinical trial of 3 days of treatment with oral pseudoephedrine for the common cold in the southern hemisphere. Am J Rhinol. 2007;21:452‐455. [DOI] [PubMed] [Google Scholar]
- 20. Eccles R, Jawad MS, Jawad SS, et al. Efficacy and safety of single and multiple doses of pseudoephedrine in the treatment of nasal congestion associated with common cold. Am J Rhinol. 2005;19:25‐31. [PubMed] [Google Scholar]