

Abstract
Key points
Neuropathic pain spreads spatially beyond the injured sites, and the mechanism underlying the spread has been attributed to inflammation occurring in the spinal cord.
However, the spatial spread of spinal/cortical potentiation induced by conduction block of the peripheral nerves can be observed prior to inflammation.
In the present study, we found that spreading potentiation and hypersensitivity acutely induced by unilateral hindpaw ischaemia are nitric oxide (NO)‐dependent and that NO is produced by ischaemia and quickly diffuses within the spinal cord.
We also found that NO production induced by ischaemia is not observed in the presence of an antagonist for group II metabotropic glutamate receptors (mGluRs) and that neuronal NO synthase‐positive dorsal horn neurons express group II mGluRs.
These results suggest strongly that NO‐mediated spreading potentiation in the spinal cord is one of the trigger mechanisms for neuropathic pain.
Abstract
Cortical/spinal responses to hindpaw stimulation are bilaterally potentiated by unilateral hindpaw ischaemia in mice. We tested the hypothesis that hindpaw ischaemia produces nitric oxide (NO), which diffuses in the spinal cord to induce spatially spreading potentiation. Using flavoprotein fluorescence imaging, we confirmed that the spreading potentiation in hindpaw responses was induced during ischaemia in the non‐stimulated hindpaw. This spreading potentiation was blocked by spinal application of l‐NAME, an inhibitor of NO synthase (NOS). Furthermore, no spreading potentiation was observed in neural NOS (nNOS) knockout mice. Spinal application of an NO donor was enough to induce cortical potentiation and mechanical hypersensitivity. The spatial distribution of NO during unilateral hindpaw ischaemia was visualized using 4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein (DAF‐FM). An increase in fluorescence derived from the complex of DAF‐FM with NO was observed on the ischaemic side of the spinal cord. A similar but smaller increase was also observed on the contralateral side. Somatosensory potentiation after hindpaw ischaemia is known to be inhibited by spinal application of LY354740, an agonist of group II metabotropic glutamate receptors (mGluRs). We confirmed that the spinal DAF‐FM fluorescence increases during hindpaw ischaemia were not observed in the presence of LY354740. We also confirmed that approximately half of the nNOS‐positive neurons in the superficial laminae of the dorsal horn expressed mGluR2 mRNA. These results suggest that disinhibition of mGluR2 produces NO which in turn induces a spreading potentiation in a wide area of the spinal cord. Such spreading, along with the consequent non‐specific potentiation in the spinal cord, may trigger neuropathic pain.
Keywords: Neuropathic pain, Nitric oxide, Spinal cord
Key points
Neuropathic pain spreads spatially beyond the injured sites, and the mechanism underlying the spread has been attributed to inflammation occurring in the spinal cord.
However, the spatial spread of spinal/cortical potentiation induced by conduction block of the peripheral nerves can be observed prior to inflammation.
In the present study, we found that spreading potentiation and hypersensitivity acutely induced by unilateral hindpaw ischaemia are nitric oxide (NO)‐dependent and that NO is produced by ischaemia and quickly diffuses within the spinal cord.
We also found that NO production induced by ischaemia is not observed in the presence of an antagonist for group II metabotropic glutamate receptors (mGluRs) and that neuronal NO synthase‐positive dorsal horn neurons express group II mGluRs.
These results suggest strongly that NO‐mediated spreading potentiation in the spinal cord is one of the trigger mechanisms for neuropathic pain.
Introduction
Peripheral nerve injuries produce neuropathic pain in animal models (Bennett & Xie, 1988; Kim & Chung, 1992; Decosterd & Woolf, 2000). Neuropathic pain develops chronically over 2–3 weeks after the peripheral nerve injury, and is modified by inflammatory responses occurring in the spinal cord and by the resulting epigenetic regulation (Campbell & Meyer, 2006; Penas & Navarro, 2018). At the same time, somatosensory cortical plasticity is acutely induced after nerve injuries (Calford & Tweedale, 1988) and nerve conduction block (Björkman et al. 2009). Acute cortical potentiation occurs within several hours after partial nerve cutting and before the inflammatory responses occur; therefore, it may be the initial pathological step for the development of neuropathic pain (Komagata et al. 2011). An abnormal sensation of irritation frequently occurs after the ischaemic conduction block of peripheral nerves (Mogyoros et al. 2000), and hindpaw ischaemia and the resulting conduction block of peripheral nerves induce cortical/spinal potentiation within 30 min (Watanabe et al. 2015). Therefore, it is natural to assume that similar neural mechanisms are responsible for both types of acute potentiation after partial nerve cutting and ischaemic conduction block. Because hindpaw ischaemia for 3 h is enough to induce neuropathic pain lasting for 4 weeks (Coderre et al. 2004), the neural mechanisms that trigger neuropathic pain might be understood by investigating the acute neural changes that occur during hindpaw ischaemia.
Cortical potentiation after partial nerve cutting is observed in uninjured nerves (Komagata et al. 2011). Similarly, spatial spreading is observed in the potentiation that follows hindpaw ischaemia: potentiation is induced on the contralateral side of ischaemic conduction block, as well as on the ipsilateral side (Watanabe et al. 2015). Spatial spreading is also a characteristic feature of neuropathic pain: it is observed in sites distant from the site of nerve injury (Milligan et al. 2003; Racz et al. 2008; Shenker et al. 2008; Carlton et al. 2009). Spatial invasion of the inflammatory responses within the spinal cord may be responsible for the spread of information between a particular spinal neuron that fails to receive afferents and the surrounding spinal neurons that receive normal afferents. It is also possible that the spreading information may be distributed via neural networks (Fitzgerald, 1982; Koltzenburg et al. 1999) or diffusion of chemical mediators. The mouse may be an ideal experimental animal model for testing the third possibility, because the mouse spinal cord is sufficiently small that influences of unilaterally produced diffusible mediators can be detected on the other side of the spinal cord (Watanabe et al. 2015).
Nitric oxide (NO) is a diffusible mediator involved in synaptic plasticity (Garthwaite & Boulton, 1995; Garthwaite, 2016) and neuropathic pain (Schmidtko et al. 2009). Neuronal NO synthase (nNOS) is distributed in the superficial laminae of the spinal dorsal horn (Zhang et al. 1993; Reuss & Reuss, 2001). Therefore, we tested the hypothesis that the spreading potentiation induced by unilateral hindpaw ischaemia is mediated by NO. It is also known that group II metabotropic glutamate receptors (mGluRs) are involved in the potentiation after partial nerve cutting (Komagata et al. 2011) and ischaemic conduction block (Watanabe et al. 2015), as in neuropathic pain (Simmons et al. 2002; Jones et al. 2005; Osikowicz et al. 2013). A simple explanation for the relationship between NO signalling and group II mGluRs is that the role of group II mGluRs on cortical/spinal potentiation is mediated by NO production. Therefore, we tested the effects of an agonist of group II mGluRs on spinal NO production. We also investigated whether group II mGluRs and nNOS are co‐localized in the same spinal neurons.
Methods
Ethical approval
The experiments in the present study were approved by the ethics committee for animal experiments in Niigata University (approved number: 233‐4 and 372‐7) and were carried out in accordance with the approved guidelines and the policy and regulations on animal experimentation of The Journal of Physiology.
Animals
Male C57BL/6 mice of 7–10 weeks of age, purchased from Charles River Japan (Yokohama, Japan), were used in the present study. Male and age‐matched mice with a targeted disruption of the nNOS gene (Huang et al. 1993) obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in our laboratory were also used for the experiments.
Surgery
Mice were operated on as described previously (Watanabe et al. 2015). They were anaesthetized with urethane (1.65 g/kg, i.p.), and a tracheotomy was performed to facilitate respiration. Body temperature was monitored using a rectal probe and maintained at 38°C using a silicon rubber heater. These surgical operations were usually completed within 60 min. Recordings were started 30 min after the surgical operations. Additional doses of urethane (0.1–0.2 g/kg, s.c.) were administered when necessary. To investigate the cortical responses to hindpaw stimulation, the disinfected head skin of the mice was incised, and the skull over the right somatosensory cortex was exposed. The surface of the skull was cleaned with sterile saline, and a small piece of metal was attached to the skull with a dental acrylic resin (Super Bond; Sun Medical, Shiga, Japan) to fix the head under a binocular epifluorescence microscope (M165 FC; Leica Microsystems, Wetzlar, Germany). The surface of the skull was covered with a mixture of petroleum jelly and liquid paraffin to keep the skull transparent. When spinal responses to hindpaw stimulation were investigated, the vertebral arch was removed at the T13 and L1 level, and the dorsal surface of the spinal cord with the intact dura mater was exposed. The surface was cleaned with saline, and covered with 2% agarose to prevent spinal movement. The surface of the agarose gel was covered with a mixture of petroleum jelly and liquid paraffin to prevent drying. The spinal cord was fixed under the binocular epifluorescence microscope using a clamp (STS‐A; Narishige, Tokyo, Japan). Spontaneous respiration was maintained during the imaging experiment, because movement of the spinal cord caused by respiration is typically minimal.
Flavoprotein fluorescence imaging
Flavoprotein fluorescence imaging was performed to investigate the cortical and spinal responses to hindpaw stimulation, as previously described (Watanabe et al. 2015). Brush vibration with an amplitude of 0.2 mm at 50 Hz was applied for 600 ms to the sole of the hindpaw using a solenoid mechanical stimulator (DPS‐290; Dia Medical, Tokyo, Japan). Endogenous green fluorescence (wavelength: 500–550 nm) was recorded in blue light (wavelength: 450–490 nm). Images (128 × 168 pixels) of the primary somatosensory cortex or the spinal cord were recorded at 9 frames/s using a cooled charge coupled device camera (ORCA‐R2; Hamamatsu Photonics, Hamamatsu, Japan). The camera was attached to the binocular epifluorescence microscope with a 75‐W xenon light source and a 1× objective lens. Serial images were taken in recording sessions repeated at 50‐s intervals. Fluorescence changes elicited by the stimulation were averaged over 24 trials. Because approximately 20 min was needed to obtain data from the 24 trials, the recording time of the averaged data was defined as the middle point of the recording period. Spatial averaging in 5 × 5 pixels and temporal averaging in three consecutive frames were used to smoothen and improve the image quality. The images were normalized, pixel by pixel, with respect to a reference image (F 0), which was obtained by averaging five images taken immediately before stimulation. In the figures, the selected parts of the normalized images are shown in a pseudocolour scale representing the fractional fluorescence changes (ΔF/F 0). The response amplitude of the cortical responses was evaluated with respect to ΔF/F 0 in a square window of 60 × 60 pixels or 1.55 × 1.55 mm at 0.6–1.0 s after the onset of the stimulus. The response amplitude in spinal responses was evaluated with respect to ΔF/F 0 in a square window of 100 × 25 pixels or 3.84 × 0.96 mm at 0.6‐1.0 s after the stimulus onset. The location of the window was adjusted to maximize the response amplitude with respect to ΔF/F 0. After the recordings were completed, the mice were killed with an overdose of pentobarbital (300 mg/kg, i.p.).
Hindpaw ischaemia
A small rubber cuff was set around the right thigh and covered with a hard‐plastic tube, as previously described (Watanabe et al. 2015). Air pressure at 250 mmHg was applied for 2 h to the tubing connected to the cuff using a mercury manometer. The pressure was directed to the thigh, because inflation of the rubber cuff was limited by the hard‐plastic tube. When the effects of a transient ischaemia were tested, the rubber cuff was set around the left thigh, and an air pressure of 250 mmHg was applied for 30 min. We confirmed that this pressure was sufficient to eliminate cortical responses induced by hindpaw stimulation during the ischaemia.
Visualization of NO production in the spinal cord during hindpaw ischaemia
To visualize NO production in the spinal cord, 4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein (DAF‐FM; Goryo Chemical, Sapporo, Japan) was used (Kojima et al. 1999). DAF‐FM is converted to a fluorescent substance when combined with NO. The surface of the spinal cord was exposed at the T13–L1 level, and fixed under the same binocular epifluorescence microscope used for flavoprotein fluorescence imaging. To avoid the influence of the surgical operation, the surface of the spinal cord was coated with a 2% agarose gel containing 20 μM DAF‐FM 30 min after the operation. An image (256 × 336 pixels, exposure time: 500 ms) of the spinal cord was recorded in green fluorescence (wavelength: 500–550 nm) excited with blue light (wavelength: 450–490 nm) before the ischaemic treatment. Then, the ischaemic treatment was applied to the right hindpaw, and images of the spinal cord were acquired at 30‐min intervals during the hindpaw ischaemia. The obtained data were quantified by selecting a region of interest (ROI) of 100 × 20 pixels or 1.92 × 0.38 mm on the right ischaemic side and the left non‐ischaemic side, respectively. The position of the right ROI was determined so that the amount of increase in fluorescence intensity at 120 min after the onset of ischaemia was maximized in the ROI, and the left ROI on the non‐ischaemic side was positioned symmetrically with the right ROI. For comparison of the data between the different mice, the increase in fluorescence intensity at 120 min after the start of ischaemia on the right ischaemic side was taken as 100%, and the data at the other time points and the data on the non‐ischaemic side were expressed relative to this value. In the control group of mice, a rubber cuff was set around the right thigh, but no pressure was applied.
Intrathecal application of drugs
For flavoprotein fluorescence imaging, the spinal cord with the intact dura mater was exposed at the L5–L6 level, and 5 μl of 100 mM l‐NAME (FUJIFILM Wako, Osaka, Japan), 2 mM NOR3 ((±)‐(E)‐4‐ethyl‐2‐[(E)‐hydroxyimino]‐5‐nitro‐3‐hexanamide, FUJIFILM Wako) or saline was intrathecally injected using a hypodermic needle of 30 gauge under direct visual control. When the effects of group II mGluRs on ischaemia‐induced NO production were examined, 5 μl of 10 nM LY354740 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), an agonist of group II mGluRs, was intrathecally injected 30 min before visualization of NO production. For behavioural experiments, 5 μl of 2 mM NOR3 or saline was blindly injected into the intrathecal space at the L5–L6 level of the non‐anesthetized mice. In the latter case, the position of the injection was verified by tail flick responses elicited by insertion of the needle (Hylden & Wilcox, 1980). In preliminary behavioural experiments, 5.0 μl of 1 mM NOR3 showed no significant effects.
Estimation of mechanical hypersensitivity
The mechanical thresholds for hindpaw‐withdrawal reflex were measured using the von Frey filaments (Amaya et al. 2009). The forces produced by the von Frey filaments were between 0.04 and 2 g. Mice were placed separately in a transparent plastic box with a mesh floor, and accustomed to the state for 30 min. The thresholds were determined based on the minimal force that induced a hindpaw‐withdrawal reflex at least twice in 10 trials. The thresholds were measured before and 30–180 min after intrathecal application of 5 μl of 2 mM NOR3 or saline.
Immunohistochemistry
Mice were deeply anaesthetized with isoflurane, and perfused with a buffered 10% formaldehyde solution (Mildform 10N; FUJIFILM Wako). The spinal cords obtained from the lumbar enlargements were collected as soon as possible. The tissues were immersed in the same fixative at 4°C for 4 h or overnight, and equilibrated in 20% sucrose overnight at 4°C before cryoprotection. Sections with a thickness of 10 μm were prepared using a cryostat microtome (CM1520, Leica Microsystems), mounted on APS‐coated slide glasses (S8441; Matsunami, Osaka, Japan), and stored at –70°C until use. Sections were washed three times with 0.1 M PBS (pH 7.6) for 10 min, then treated with 2% goat serum for 60 min. The sections were primarily incubated with a goat anti‐nNOS antibody (1:100, ab1376; Abcam, Cambridge, UK) and a rabbit anti‐mGluR2 and anti‐mGluR3 antibody (1:100, ab6438; Abcam) for 2 days at 4°C. After washing, sections were secondarily incubated with a donkey anti‐goat IgG H&L conjugated with Alexa Fluor488 (to visualize nNOS; 1:1000, ab150129; Abcam), and a donkey anti‐rabbit IgG conjugated with rhodamine (to visualize mGluR2 and mGluR3; 1:2000 AP182R; EMD Millipore, Burtlington, MA, USA) overnight at 4°C. After washing, the sections were mounted using a medium containing DAPI (H‐1200; Vector Laboratories, Burlingame, CA, USA).
In situ hybridization
Double staining with in situ hybridization and immunohistochemistry was performed on frozen sections, as previously described (Toda et al. 2018). To generate the probes for in situ hybridization, rat mGluR2 and mGluR3 plasmids were obtained from RIKEN BRC (Tsukuba, Japan). Digoxigenin‐labelled probes were generated using the appropriate polymerases (T7 or T3 polymerases). Sense probes were used as negative controls. Sections were washed twice with 0.01 M PBS for 5 min, treated with 1 μg/ml proteinase K in 50 mM Tris‐HCl at pH 7.6 and 5 mM EDTA for 60 min, and then rinsed in 0.01 M PBS. After fixation in 4% paraformaldehyde in 0.01 M PBS, and acetylation in 0.1 M triethanolamine at pH 8.0 containing 0.25% acetic anhydride for 10 min, the sections were prehybridized at 65°C for 3 h with a hybridization solution that contained 50% formamide, 0.75 M NaCl, 0.075 M sodium citrate in diethylpyrocarbonate‐treated water, 0.2 mg/ml yeast tRNA, 0.1 mg/ml heparin, 1× Denhardt's solution, 0.2% Tween 20 and 5 mM EDTA. Then, the sections were incubated with a hybridization solution containing a diluted digoxigenin‐labelled RNA probe at 65°C overnight. Hybridized sections were washed twice with 50% formamide, 0.15 M NaCl, 0.015 M sodium citrate in diethylpyrocarbonate‐treated water at 65°C for 15 min (first time) and 30 min (second time), and finally with 0.15 M NaCl, 0.015 M sodium citrate in diethylpyrocarbonate‐treated water at 65°C for 30 min. The sections were washed twice in maleic acid buffer (0.1 M maleic acid at pH 7.5, 0.15 M NaCl and 0.1% Tween 20) for 30 min at room temperature and incubated with alkaline phosphatase‐conjugated sheep anti‐digoxigenin antibody diluted with 0.5% skimmed milk (1: 2000; Roche Diagnostics, Manheim, Germany) overnight at 4°C. Then, they were washed three times with PBS for 30 min each, and incubated with the colour development solution [50 μg/ml 4‐nitro blue tetrazolium chloride (NBT) and 175 μg/ml 5‐bromo‐4‐chloro‐3‐indolyl‐phosphate (BCIP); Roche Diagnostics] in alkaline phosphatase buffer (0.1 M Tris‐HCl at pH 9.5, 0.05 M MgCl2, 0.1 M NaCl and 0.1% Tween 20) for 3–10 h in the dark. After in situ hybridization, immunohistochemistry was performed using a goat anti‐nNOS antibody (1:100, ab1376; Abcam) and HRP‐conjugated anti‐goat antibody as a secondary antibody (1: 200; MBL, Nagoya, Japan).
Statistical analysis
Unpaired data obtained from experimental mice administered with l‐NAME or NOR3 and control mice administered with saline vehicle were evaluated using a two‐way ANOVA in Easy R, a free software program for statistical analysis (Kanda, 2013). The data obtained from nNOS knockout mice were compared with those obtained from wild‐type mice of the same age and sex. In post hoc analyses, unpaired data obtained in different groups of mice were evaluated using the t test with the Bonferroni correction for multiple comparisons. Paired data obtained on the ischaemic side and the non‐ischaemic side of the spinal cord of the same mice were evaluated using a paired t test. Unpaired data obtained from different mice were evaluated using a t test. Values presented in the figures represent the mean and SEM in the groups of mice, unless otherwise specified. The number of mice in each group was adjusted to be between 5 and 10 to obtain significant results within this range. P values less than 0.05 are usually not shown. The difference in the frequency of mGluR2 or mGluR3 expression in nNOS‐positive neurons was evaluated by using the χ2‐test in Excel.
Results
NO‐mediated cortical spreading potentiation induced by hindpaw ischaemia
Cortical responses to hindpaw stimulation are bilaterally potentiated by hemilateral hindpaw ischaemia (Watanabe et al. 2015). To test the hypothesis that this cortical spreading potentiation is mediated by endogenous NO, we administered 5 μl of 100 mM l‐NAME, an inhibitor of NOS, to the spinal cord (Fig. 1 A). When saline was administered to the spinal cord as a control, cortical spreading potentiation appeared at 30 min after the onset of hindpaw ischaemia, and the potentiation persisted for 120 min during the ischaemia (Fig. 1 B–D). The amplitude of cortical potentiation induced by 120 min of ischaemia was 254 ± 27% (n = 8) when saline was administered, and 134 ± 6% (n = 8) or 114 ± 10% (n = 8) when 50 or 100 mM l‐NAME was administered, respectively. Therefore, we used 100 mM l‐NAME in the present study. A two‐way ANOVA showed significant effects of 100 mM l‐NAME (P = 1.9 × 10−8) and time after the onset of ischaemia (P = 0.0099), but not in the interaction. In the post hoc analysis, significant differences in the potentiation were observed between the two groups at 30, 60 and 120 min after the onset of ischaemia (Fig. 1 D).
Figure 1. Cortical spreading potentiation during right hindpaw ischaemia with or without the spinal application of l‐NAME.
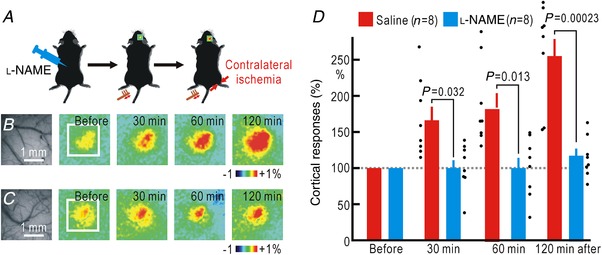
A, experimental method. B, cortical responses in the right somatosensory cortex elicited by brush vibration applied to the sole of the left hindpaw. In these mice, 5 μl saline was intrathecally applied to the spinal cord before the recordings. Original and pseudocolour images in ΔF/F 0 of the right somatosensory cortices are shown. The time after the onset of ischaemia in the right hindpaw is shown in each pseudocolour image. The white square in the left‐most pseudocolour image represents the ROI for the measurement of the response amplitude in ΔF/F 0. The pseudocolour scale shows the percentages of ΔF/F 0. C, cortical somatosensory responses elicited by hindpaw stimulation before and after the onset of ischaemia in the right hindpaw. In these mice, l‐NAME (5 μl, 100 mM) was applied to the spinal cord. Original and pseudocolour images in ΔF/F 0 are shown. D, relative amplitudes of the cortical responses normalized by those recorded before the onset of ischaemia. Mean and SEM are shown. Individual data are also shown by dots. The scale in the bar graphs represents the relative amplitude of the responses as a percentage. [Color figure can be viewed at http://wileyonlinelibrary.com]
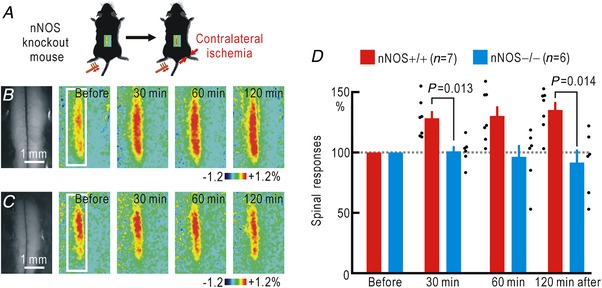
There are three types of NOS (Bredt & Snyder, 1992). Because cortical potentiation was induced within 30 min of hindpaw ischaemia, the involvement of inducible NOS (iNOS) is unlikely. Because nNOS is present in the dorsal horn of the spinal cord (Zhang et al. 1993; Reuss & Reuss, 2001), nNOS is likely to be responsible for inducing the cortical potentiation. To confirm this hypothesis, experiments were carried out in mice lacking nNOS (Fig. 2 A). In wild‐type mice, hindpaw ischaemia in the non‐stimulated side induced cortical potentiation (Fig. 2 B, D). However, almost no potentiation was observed following ischaemia in the nNOS knockout mice (Fig. 2 C, D). A two‐way ANOVA showed significant effects of nNOS knockout (P = 1.4 × 10−7), but not of the time after the onset of ischaemia and their interaction. In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30, 60 and 120 min after the onset of ischaemia (Fig. 2 D).
Figure 2. Cortical potentiation during hindpaw ischaemia contralateral to the stimulated hindpaw in wild‐type and nNOS knockout mice.
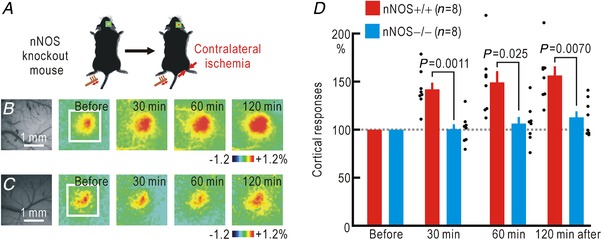
A, experimental method. B, cortical responses elicited by hindpaw stimulation in wild‐type (nNOS+/+) mice before and after the onset of hindpaw ischaemia. Original and pseudocolour images in ΔF/F 0 of the right somatosensory cortices are shown. C, cortical somatosensory responses elicited by hindpaw stimulation in knockout (nNOS−/−) mice before and after the onset of ischaemia in the right hindpaw. Original and pseudocolour images in ΔF/F 0 are shown. D, relative amplitudes of the cortical responses normalized by those recorded before the onset of hindpaw ischaemia. [Color figure can be viewed at http://wileyonlinelibrary.com]
Somatosensory responses to stimuli applied to an ischaemic hindpaw cannot be elicited because of ischaemic conduction block of the peripheral nerves. However, comparison of the somatosensory responses before ischaemia and after recovery from ischaemia reveals that cortical potentiation also occurs on the ischaemic side (Watanabe et al. 2015). It is very likely that the two types of potentiation in both sides share common mechanisms. To confirm this hypothesis, we used nNOS knockout mice and compared the somatosensory responses before ischaemia and after recovery from ischaemia to test whether cortical potentiation in the responses to stimuli applied to the hindpaw was induced by ischaemia (Fig. 3 A). In wild‐type mice, potentiation was observed 30 min after hindpaw ischaemia was terminated, and this potentiation persisted for 120 min (Fig. 3 B, D). However, no such potentiation was recorded in the nNOS knockout mice (Fig. 3 C, D). A two‐way ANOVA showed significant effects of nNOS knockout (P = 1.9 × 10−5), but not of the time after the onset of ischaemia or their interaction. In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30 and 120 min after the onset of ischaemia (Fig. 3 D). These results suggest strongly that NO induces cortical spreading potentiation on both sides, possibly due to diffusion of NO within the spinal cord.
Figure 3. Cortical potentiation after ischaemia in the stimulated hindpaw in wild‐type and nNOS knockout mice.
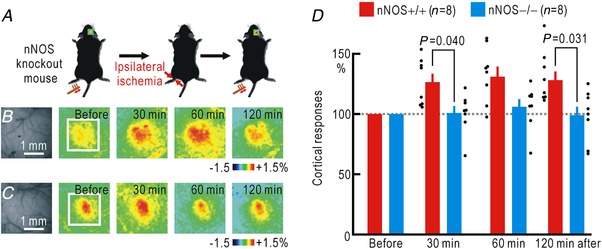
A, experimental method. B, right cortical responses elicited by left hindpaw stimulation in wild‐type (nNOS+/+) mice before and after ischaemia in the left hindpaw for 30 min. Original and pseudocolour images in ΔF/F 0 of the right somatosensory cortices are shown. The time after termination of hindpaw ischaemia is shown in each pseudocolour image. C, right cortical responses elicited by left hindpaw stimulation in knockout (nNOS−/−) mice before and after ischaemia in the left hindpaw for 30 min. D, relative amplitudes of the cortical responses normalized by those recorded before hindpaw ischaemia. [Color figure can be viewed at http://wileyonlinelibrary.com]
NO‐mediated spinal spreading potentiation induced by hindpaw ischaemia
Spatial spread of the somatosensory potentiation induced by hindpaw ischaemia is observed not only at the cortical but also at the spinal level (Watanabe et al. 2015). Therefore, it is very likely that the spinal spreading potentiation is also mediated by NO. To confirm this hypothesis, we investigated the spinal responses using flavoprotein fluorescence imaging. After spinal application of saline or l‐NAME into the intrathecal space, the surface of the spinal cord was uncovered, and the spinal cord responses to left hindpaw stimulation were recorded before and during ischaemia of the opposite right hindpaw (Fig. 4 A). When saline was administered, spinal potentiation appeared 30 min after the onset of the hindpaw ischaemia, and the potentiation persisted for at least 2 h (Fig. 4 B, D). However, no such potentiation was observed in the mice treated with l‐NAME (Fig. 4 C, D). A two‐way ANOVA showed significant effects of l‐NAME (P = 1.7 × 10−5), but not of the time after the onset of ischaemia or their interaction. In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30, 60 and 120 min after the onset of ischaemia (Fig. 4 D).
Figure 4. Spinal spreading potentiation during right hindpaw ischaemia with or without spinal application of l‐NAME.
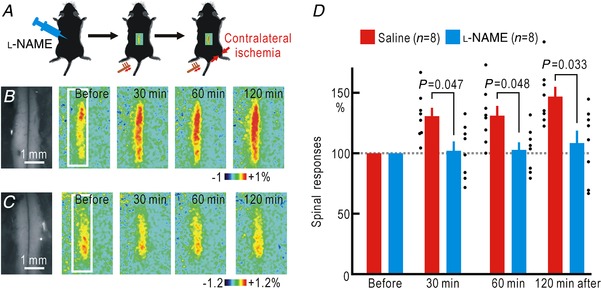
A, experimental method. B, spinal responses in the left spinal cord elicited by brush vibration applied to the sole of the left hindpaw. In these mice, 5 μl saline was applied to the spinal cord before the recordings. Original and pseudocolour images in ΔF/F 0 of the spinal cord are shown. The time after the onset of ischaemia in the right hindpaw is shown in each pseudocolour image. The white rectangle in the left‐most pseudocolour image represents the ROI for the measurement of the response amplitude in ΔF/F 0. C, spinal responses elicited by left hindpaw stimulation before and after the onset of ischaemia in the right hindpaw. In these mice, l‐NAME (5 μl, 100 mM) was applied to the spinal cord. Original and pseudocolour images in ΔF/F 0 are shown. D, relative amplitudes of the spinal responses normalized by those recorded before the onset of hindpaw ischaemia. [Color figure can be viewed at http://wileyonlinelibrary.com]
Cortical spreading potentiation during hindpaw ischaemia was not observed in nNOS knockout mice. Therefore, we tested whether spinal spreading potentiation was blocked in nNOS knockout mice (Fig. 5 A). The amplitudes (ΔF/F 0) of the spinal responses to hindpaw stimulation were 0.56 ± 0.4% (n = 8) in wild‐type mice and 0.64 ± 0.5% (n = 7) in nNOS knockout mice. The difference was not statistically significant (P = 0.13). Even though spinal spreading potentiation was observed in control wild‐type mice (Fig. 5 B, D), no such potentiation was observed in nNOS knockout mice (Fig. 5 B, D). A two‐way ANOVA showed significant effects of nNOS knockout (P = 4.8 × 10−6), but not of the time after the onset of ischaemia or their interaction. In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30 and 120 min after the onset of ischaemia (Fig. 5 D). The differences between both groups of mice were significant between 30 min and 2 h after the onset of ischaemia (Fig. 5 D). Taken together, these results suggest strongly that NO‐mediated cortical spreading potentiation induced by hindpaw ischaemia is a result of NO‐mediated spreading potentiation at the spinal cord level.
Figure 5. Spinal spreading potentiation during right hindpaw ischaemia in wild‐type and nNOS knockout mice.
A, experimental method. B, spinal responses elicited by left hindpaw stimulation in wild‐type (nNOS+/+) mice before and after the onset of ischaemia in the right hindpaw. Original and pseudocolour images in ΔF/F 0 of the spinal cord are shown. C, spinal responses elicited by left hindpaw stimulation in knockout (nNOS−/−) mice before and after the onset of ischaemia in the right hindpaw. Original and pseudocolour images in ΔF/F 0 are shown. D, relative amplitudes of the spinal responses normalized by those recorded before the onset of ischaemia. [Color figure can be viewed at http://wileyonlinelibrary.com]
Cortical potentiation and mechanical hypersensitivity induced by exogenous NO
The left and right sides of the spinal cord are connected via neural circuits (Fitzgerald, 1982; Koltzenburg et al. 1999). If the spatial spread of cortical/spinal potentiation is attributed to neural activities mediated via such neural circuits, the presence of NO within the spinal cord may not be enough to induce somatosensory potentiation. To test this possibility, we investigated whether cortical responses are potentiated after application of NOR3, an NO donor, into the spinal intrathecal space (Fig. 6 A). As a result, cortical potentiation was observed between 30 min and 2 h after application of NOR3 (Fig. 6 B). Such changes were not observed after intrathecal administration of saline. A two‐way ANOVA showed significant effects of NOR3 (P = 5.6 × 10−8), but not of the time after application of NOR3 or their interaction. In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30, 60 and 120 min after application of NOR3 (Fig. 6 B).
Figure 6. Cortical potentiation and mechanical hypersensitivity induced by spinal application of NOR3.
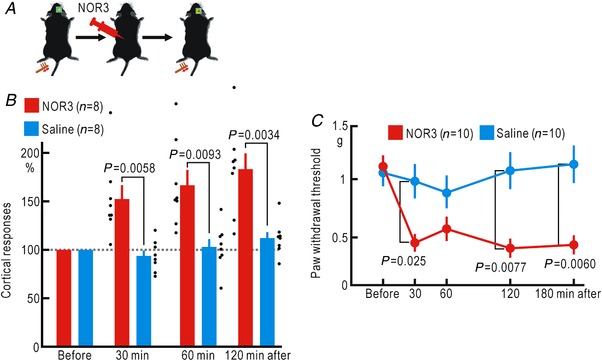
A, experimental method. B, relative amplitudes of the cortical responses normalized by those recorded before spinal application of 5 μl saline or 2 mM NOR3. The abscissa shows the time after the spinal application of saline or NOR3. C, mechanical threshold of the hindpaw withdrawal reflex measured by the von Frey test before spinal application of 5 μl saline or 2 mM NOR3. [Color figure can be viewed at http://wileyonlinelibrary.com]
Cortical potentiation after hindpaw ischaemia is accompanied by mechanical hypersensitivity (Watanabe et al. 2015). Therefore, mechanical hypersensitivity may also occur after spinal application of NOR3. When we investigated the hindpaw withdrawal reflex with the von Frey test, the mechanical threshold decreased 30 min after the spinal application of NOR3 and remained low until 3 h after the application (Fig. 6 C). On the other hand, the mechanical threshold was unchanged when saline was administered to the spinal cord. A two‐way ANOVA showed significant effects of NOR3 (P = 1.9 × 10−7), the time after application of NOR3 (P = 0.015), and their interaction (P = 0.015). In the post hoc analysis, significant differences in potentiation were observed between the two groups at 30, 120 and 180 min after application of NOR3 (Fig. 6 C). These results indicate that the presence of NO is enough to induce somatosensory spreading potentiation and mechanical hypersensitivity.
Visualization of spinal NO distribution during unilateral hindpaw ischaemia
Spinal application of l‐NAME prevented cortical potentiation and spinal potentiation during hindpaw ischaemia. These results strongly suggest that NO is produced in the spinal cord during hindpaw ischaemia possibly in the ischaemic side, and diffuses to the non‐ischaemic side. To confirm this hypothesis, we visualized the endogenous NO produced in the mouse spinal cord. We used DAF‐FM, which binds to NO and is converted to a fluorescent form (Kojima et al. 1999; Mabuchi et al. 2003). DAF‐FM was mixed into an agarose gel covering the spinal cord surface, and the changes in fluorescence intensity during hindpaw ischaemia were recorded (Fig. 7 A). In mice with hindpaw ischaemia, gradual increases in fluorescence intensity during hindpaw ischaemia were observed on the ischaemic side (Fig. 7 B). The fluorescence intensity increased almost linearly with time (Fig. 7 D). Increases in fluorescence intensity were observed not only on the ischaemic side but also on the non‐ischaemic side. However, when compared at 2 h after the onset of ischaemia, the fluorescence increase on the ischaemic side was significantly stronger than that on the non‐ischaemic side (Fig. 7 D). In sham operated mice, only small increases in fluorescence intensity were observed in the spinal cord. These results suggest strongly that NO is continuously produced in the spinal cord on the ischaemic side during hindpaw ischaemia, and a considerable amount of the NO produced rapidly diffuses to the non‐ischaemic side.
Figure 7. Visualized spinal NO production during right hindpaw ischaemia using DAF‐FM.
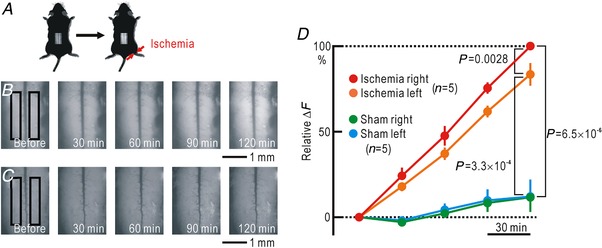
A, experimental method. B, images of the spinal cord covered with 2% agarose gel containing 20 μM DAF‐FM. The time after the onset of ischaemia in the right hindpaw is shown in each image. The intensity of the fluorescence is shown on an arbitrary grey scale. The black rectangles represent ROIs for measurement of fluorescence intensity. C, images of the spinal cord covered with 2% agarose gel containing DAF‐FM. In this experiment, sham treatment without pressure was applied to the right hindpaw. D, relative amplitudes of the increases in fluorescence normalized by those recorded 2 h after the onset of right hindpaw ischaemia in the right side of the spinal cord. [Color figure can be viewed at http://wileyonlinelibrary.com]
Interaction between mGluR2 and nNOS in spinal neurons
Activation of group II mGluRs, mGluR2 and mGluR3 (Tanabe et al. 1992), is known to alleviate neuropathic pain (Simmons et al. 2002; Jones et al. 2005; Osikowicz et al. 2013). In our previous studies, spinal application of LY354740 (10 nM, 5 μl), an agonist of group II mGluRs, blocked cortical potentiation induced by partial cutting of the peripheral nerves (Komagata et al. 2011) and cortical/spinal potentiation induced by hindpaw ischaemia (Watanabe et al. 2015). Therefore, we tested the effect of LY354740 (10 nM, 5 μl) on the changes in spinal DAF‐FM fluorescence induced by hindpaw ischaemia. DAF‐FM fluorescence was increased by 53 ± 7% (n = 5) during a 120 min period of ischaemia, while it was slightly decreased by 15 ± 5% (n = 6) in the presence of LY354740. As these values were significantly different (P = 5.0 × 10−5), these findings suggest strongly that the role of group II mGluRs on cortical/spinal potentiation is mediated by NO production.
A simple explanation of the interaction between mGluR2 and NO production is that group II mGluRs and nNOS co‐localize in spinal neurons that receive glutamatergic inputs from peripheral nerves, so that ischaemic conduction block of basal firing in Aβ afferents (Komagata et al. 2011) produces disinhibition via group II mGluRs, resulting in an elevation of nNOS activity (Fig. 8 A). Although other possible interactions between group II mGluRs and nNOS cannot be excluded, the distribution of group II mGluRs and nNOS in the spinal cord is an important clue for understanding this relationship. We performed immunostaining of group II mGluRs and nNOS (Fig. 8 B, C). Group II mGluRs were enriched in the superficial laminae of the dorsal horn (Fig. 8 B). Cellular bodies with immunoreactivity to nNOS were also distributed in the superficial laminae of the dorsal horn (Fig. 8 B, C). Unlike nNOS, group II mGluRs are distributed mainly outside of the cell bodies (Fig. 8 C), possibly in the dendritic postsynaptic sites or presynaptic nerve terminals.
Figure 8. Colocalization of mGluR2 and nNOS in spinal neurons.
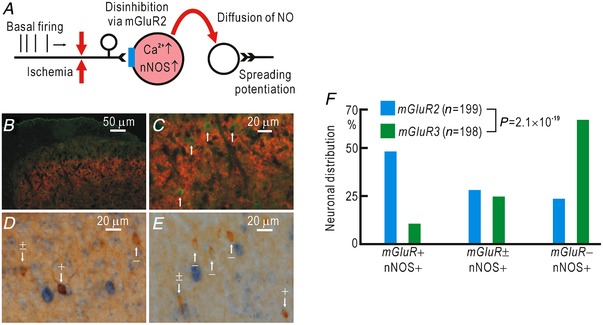
A, hypothetical relationship between signalling pathways mediated via mGluR2 and NO. B, immunohistochemical staining of group II mGluRs (mGluR2 and mGluR3, red) and nNOS (green). Colocalization of group II mGluRs and nNOS was observed in the superficial laminae of the dorsal horn. C, the same image shown in B at higher magnification. The cell bodies with nNOS (white arrows) were not stained with antibodies for group II mGluRs. D, immunostaining for nNOS (brown) and in situ hybridization of mRNA for mGluR2 (blue). Some cell bodies with nNOS were clearly (+), faintly (±) or not stained (–) with mRNA for mGluR2. White arrows show examples. E, immunostaining for nNOS (brown) and in situ hybridization of mRNA for mGluR3 (blue). F, relative distribution of nNOS‐positive spinal neurons that were clearly (+), faintly (±) or not stained (–) with mRNA for mGluR2 or mGluR3. [Color figure can be viewed at http://wileyonlinelibrary.com]
Our immunostaining experiments did not reveal whether group II mGluRs co‐localize with nNOS in the same spinal neurons or not. Furthermore, the antibody used in our experiments binds to both mGluR2 and mGluR3 in group II mGluRs, and it is not clear which of them coexists in nNOS‐positive spinal neurons. Therefore, we separately visualized the mRNAs of mGluR2 and mGluR3 by in situ hybridization (Fig. 8 D, E). Approximately half of the nNOS‐positive neurons were clearly stained with the mGluR2 mRNA while only approximately 10% of nNOS‐positive neurons were clearly stained with the mGluR3 mRNA (Fig. 8 F). These results are compatible with the possibility shown in Fig. 8 A and suggest that spinal dorsal horn neurons expressing both mGluR2 and nNOS have a crucial role in the cortical/spinal spreading potentiation induced by hindpaw ischaemia.
Discussion
Technical merits of the present mouse model
In the present study, we used a mouse model to test the hypothesis that the spatial spread of cortical/spinal potentiation induced by nerve conduction block is attributed to NO, a diffusible messenger (Garthwaite & Boulton, 1995; Garthwaite, 2016). One of the merits of our mouse model is that transcranial imaging of cortical responses using flavoprotein fluorescence is possible. Activity‐dependent fluorescence changes derived from mitochondrial flavoproteins are sufficiently rapid to capture the dynamic neural activity in vivo (Shibuki et al. 2003; Reinert et al. 2004). We measured the amplitudes of the flavoprotein fluorescence responses at 0.6–1.0 s after the stimulus onset. The haemodynamic responses are induced after this time window (Kitaura et al. 2007). A close relationship between the neuronal responses and fluorescence imaging has been demonstrated in previous studies (Sibuki et al 2003; Reinert et al. 2004; Tohmi et al. 2006). Various types of cortical plasticity including somatosensory potentiation induced by hindpaw ischaemia have been demonstrated using this method (Takahashi et al. 2006; Tohmi et al. 2006; Komagata et al. 2011; Yoshitake et al. 2013; Watanabe et al. 2015). We also recorded the potentiation of spinal responses during hindpaw ischaemia, as previously reported (Watanabe et al. 2015).
We succeeded in visualizing endogenous production of NO in the spinal cord in vivo using DAF‐FM, which emits green fluorescence when combined with NO in spinal slices (Mabuchi et al. 2003; Xu et al. 2006). We were also able to record the signals derived from DAF‐FM in the spinal cord of anaesthetized mice for the first time. The linear increase in the intensity of the fluorescence derived from DAF‐FM indicated that NO was constantly produced during hindpaw ischaemia. A fluorescence increase was also recorded on the opposite side approximately 1 mm from the ischaemic side. The intensity on the non‐ischaemic side was significantly weaker than that on the ischaemic side, suggesting that NO is produced on the ischaemic side and quickly diffuses into the non‐ischaemic side. Diffusion of NO has also been observed in spinal slices: when a spinal slice loaded with DAF‐FM is stimulated with NMDA, fluorescence increases derived from NO are similarly observed in both superficial and deep laminae, while only superficial laminae are enriched with NOS (Xu et al. 2006).
Involvement of NO in somatosensory potentiation
NO is a diffusible mediator that is characterized by a small molecular weight and no electrical charge. In addition to its vasodilatory effect, NO has important roles in the induction of various types of synaptic plasticity in the CNS (Garthwaite & Boulton, 1995; Garthwaite, 2016). It has also been shown that NO is involved in the pathogenesis of neuropathic pain at the spinal cord level (Wu et al. 2001; Schmidtko et al. 2009; Tanabe et al. 2009). NO promotes the generation of inflammatory mediators (Salvemini et al. 1993) and is known to induce neuropathic pain via various messengers (Schmidtko et al. 2009). Several studies have shown that NO produced by nNOS is important for neuropathic pain (Cope et al. 2010; Roh et al. 2011). On the other hand, other studies have reported that iNOS expression is important for neuropathic pain (De Alba et al. 2006; Quan et al. 2013). In the present study, spinal application of NOR3 produced cortical potentiation and mechanical hypersensitivity, thus indicating that the presence of spinal NO is enough to produce cortical potentiation and mechanical hypersensitivity, regardless of the source of NO. As we focused on the acute changes occurring within 30 min after the onset of ischaemia, the importance of nNOS was clearly demonstrated. However, our results do not exclude the possibility that the expression of iNOS, which is increased during inflammation in the chronic phase of neuropathic pain, plays an important role in the pathogenesis of neuropathic pain. Peripheral nerve injuries result in neuropathic pain and neuroplasticity not only on the side of the injury but also on the contralateral side in the chronic phase (Milligan et al. 2003). This expansion in the pain areas is attributable to invasion of the inflammatory process and the diffusion of inflammatory substances (Milligan et al. 2003; Racz et al. 2008; Carlton et al. 2009). The present results raise the possibility that NO‐mediated potentiation may contribute to the spread of neuropathic pain beyond the injured sites in the chronic phase of neuropathic pain, as well as in the acute phase. Furthermore, the spread, and consequently the non‐specific NO‐mediated spinal potentiation induced by ischaemia may modify heat or cold pain sensitivity, as it induces mechanical hypersensitivity.
Mechanisms underlying NO‐mediated spreading potentiation
When peripheral nerves lose excitability by injuries or ischaemia, spontaneous firing of the peripheral nerve fails to reach spinal neurons (Komagata et al. 2011; Watanabe et al. 2015). If NO is produced in the spinal neurons that fail to receive spontaneous inputs from peripheral nerves, it can diffuse to the surrounding neurons to produce potentiation in a non‐specific manner. Such a mechanism may explain how various spinal sites exhibit potentiation after conduction block of peripheral nerves. Somatosensory potentiation is induced within 2–3 h after partial nerve cutting (Komagata et al. 2011) and within 30 min after recovery from hindpaw ischaemia (Watanabe et al. 2015). Hindpaw ischaemia may produce a conduction block in more nerve fibres than partial nerve cutting, so that NO is produced in more spinal neurons to induce a rapid potentiation. In the present study, we observed that both nNOS and group II mGluR are present in the superficial laminae of the dorsal horn. Furthermore, the mRNA of mGluR2 was present in approximately half of the nNOS‐positive cell bodies of spinal neurons in this area. Activation of group II mGluR inhibits neurotransmitter release in the presynaptic terminals (Goudet et al. 2009) and hyperpolarizes the postsynaptic neurons by opening potassium channels (Irie et al. 2006; Lee & Sherman, 2010), thus suppressing Ca2+ influx (Koga et al. 2010). Therefore, disinhibition of group II mGluR effects by conduction block of peripheral nerves can lead to a sustained increase in intracellular Ca2+ levels, which maintains nNOS activity at a plateau level. It has also been reported that a rise in intracellular Ca2+ levels of spinal lamina I neurons is enough to induce synaptic long‐term potentiation in the absence of any presynaptic stimulation (Naka et al. 2013).
The gate control theory assumes that nociceptive spinothalamic tract neurons receive innocuous afferent inputs from various sources, and the responses to innocuous afferents are normally inhibited via interneurons in the superficial laminae of the dorsal horn (Melzack & Wall, 1965; Daniele & MacDermott, 2009). However, nociceptive spinothalamic tract neurons show increased responsiveness or potentiation to innocuous mechanical stimuli in animals with injured nerves (Paleček et al. 1992). These changes after nerve injury are explained by a reduced Cl– gradient in these neurons (Sivilotti & Woolf, 1994; Moore et al. 2002; Coull et al. 2005; Price et al. 2009). The decrease in the Cl– gradient is attributable to the downregulation of the neuron‐specific KCl cotransporter (KCC2) in nociceptive spinothalamic tract neurons (Coull et al. 2005). KCC2 activity is known to be suppressed by NO (Yassin et al. 2014). Thus, spinal neurons that fail to receive spontaneous signals from peripheral nerves can be connected to nociceptive spinothalamic tract neurons that exhibit potentiation to innocuous afferents via diffusion of NO. Even though this explanation does not exclude other possibilities for the mechanisms underlying the initial development of neuropathic pain, it is highly likely that NO plays an essential role in neuropathic pain, as reported in previous studies (Wu et al. 2001; Schmidtko et al. 2009; Tanabe et al. 2009).
Roles of the spinal cord in neuropathic pain
In the present study, spreading potentiation was observed at the cortical level as well as in the spinal cord. In subjects with neuropathic pain, profound neural plasticity has been reported in the somatosensory cortex (Flor et al. 1995; Kim et al. 2016), as well as in higher areas such as the anterior cingulate cortex (Xu et al. 2008; Zhao et al. 2018) and medial prefrontal cortex (Metz et al. 2009; Kelly et al. 2016; Sang et al. 2018). The influence of nerve injuries on the spinal cord occurs initially in the CNS. NO generation in the spinal cord and the accompanying spreading potentiation are the earliest events after peripheral nerves are injured, and may be potentially maintained even in the chronic phase via expression of iNOS (De Alba et al. 2006; Quan et al. 2013). Therefore, it is natural to assume that the changes in the spinal cord will sequentially spread to other parts of the brain via ascending pathways in the pathogenesis of neuropathic pain. Therefore, NO‐mediated spreading potentiation in the spinal cord is an important target for the treatment of neuropathic pain, because it is the initial event and occurs at the entrance of afferents to higher brain areas involved in neuropathic pain.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Conception and/or design of the study – T.O., K.S. Acquisition of the data – T.O., M.S., M.H. Analysis or interpretation of the data – T.O., T.W., M.S., Y.K., M.H., H.T., R.H., T.K., H.T., H.B., K.S. Writing of the manuscript – T.O., T.K., K.S. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Grant‐in‐Aid for Scientific Research (no. 22115011, no.16H01892 to K.S., and no. 17H06692 to T.O.).
Acknowledgements
We thank S. Maruyama, A. Matsushima, M. Isogai and N. Yoshioka for technical assistance.
Biography
Takeshi Onishi is an anaesthesiologist who studied as a PhD student at the Brain Research Institute, Niigata University. Together with Katsuei Shibuki, he investigated the role of nitric oxide in cortico‐spinal plasticity induced by hindpaw ischaemia. Currently, he is working as a specially appointed assistant professor in the Department of Anesthesiology, Niigata University. He is interested in chronic pain from viewpoints of basic neuroscience and clinical medicine.

Edited by: Janet Taylor & Weifang Rong
REFERENCES
- Amaya F, Samad TA, Barrett L, Broom DC & Woolf CJ (2009). Periganglionic inflammation elicits a distally radiating pain hypersensitivity by promoting COX‐2 induction in the dorsal root ganglion. Pain 142, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ & Xie YK (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. [DOI] [PubMed] [Google Scholar]
- Björkman A, Weibul A, Rosén B, Svensson J & Lundborg G (2009). Rapid cortical reorganisation and improved sensitivity of the hand following cutaneous anaesthesia of the forearm. Eur J Neurosci 29, 837–844. [DOI] [PubMed] [Google Scholar]
- Bredt DS & Snyder SH (1992). Nitric oxide, a novel neuronal messenger. Neuron 8, 3–11. [DOI] [PubMed] [Google Scholar]
- Calford MB & Tweedale R (1988). Immediate and chronic changes in responses of somatosensory cortex in adult flying‐fox after digit amputation. Nature 332, 446–448. [DOI] [PubMed] [Google Scholar]
- Campbell JN & Meyer RA (2006). Mechanisms of neuropathic pain. Neuron 52, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD & Hulsebosch CE (2009). Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 147, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Xanthos DN, Francis L & Bennett GJ (2004). Chronic post‐ischemia pain (CPIP): a novel animal model of complex regional pain syndrome‐type I (CRPS‐I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain 112, 94–105. [DOI] [PubMed] [Google Scholar]
- Cope JL, Chung E, Ohgami Y & Quock RM (2010). Antagonism of the antinociceptive effect of nitrous oxide by inhibition of enzyme activity or expression of neuronal nitric oxide synthase in the mouse brain and spinal cord. Eur J Pharmacol 626, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW & De Koninck Y (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. [DOI] [PubMed] [Google Scholar]
- Daniele CA & MacDermott AB (2009). Low‐threshold primary afferent drive onto GABAergic interneurons in the superficial dorsal horn of the mouse. J Neurosci 29, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Alba J, Clayton NM, Collins SD, Colthup P, Chessell I & Knowles RG (2006). GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain 120, 170–181. [DOI] [PubMed] [Google Scholar]
- Decosterd I & Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M (1982). The contralateral input to the dorsal horn of the spinal cord in the decerebrate spinal rat. Brain Res 236, 275–287. [DOI] [PubMed] [Google Scholar]
- Flor H, Elbert T, Knecht S, Wienbruch C, Pantev C, Birbaumer N, Larbig W & Taub E (1995). Phantom‐limb pain as a perceptual correlate of cortical reorganization following arm amputation. Nature 375, 482–484. [DOI] [PubMed] [Google Scholar]
- Garthwaite J (2016). From synaptically localized to volume transmission by nitric oxide. J Physiol (Lond) 594, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J & Boulton CL (1995). Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57, 683–706 [DOI] [PubMed] [Google Scholar]
- Goudet C, Magnaghi V, Landry M, Nagy F, Gereau RW & Pin JP (2009). Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev 60, 43–56. [DOI] [PubMed] [Google Scholar]
- Huang PL, Dawson TM, Bredt DS, Snyder SH & Fishman MC (1993). Targeted disruption of the neuronal nitric oxide synthase gene. Cell 75, 1273–1286. [DOI] [PubMed] [Google Scholar]
- Hylden JL & Wilcox GL (1980). Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67, 313–316. [DOI] [PubMed] [Google Scholar]
- Irie T, Fukui I & Ohmori H (2006). Activation of GIRK channels by muscarinic receptors and group II metabotropic glutamate receptors suppresses Golgi cell activity in the cochlear nucleus of mice. J Neurophysiol 96, 2633–2644. [DOI] [PubMed] [Google Scholar]
- Jones CK, Eberle EL, Peters SC, Monn JA & Shannon HE (2005). Analgesic effects of the selective group II (mGlu2/3) metabotropic glutamate receptor agonists LY379268 and LY389795 in persistent and inflammatory pain models after acute and repeated dosing. Neuropharmacology 49 Suppl 1, 206–218. [DOI] [PubMed] [Google Scholar]
- Kanda Y (2013). Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant 48, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Huang M, Meltzer H & Martina M (2016). Reduced glutamatergic currents and dendritic branching of layer 5 pyramidal cells contribute to medial prefrontal cortex deactivation in a rat model of neuropathic pain. Front Cell Neurosci 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH & Chung JM (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363. [DOI] [PubMed] [Google Scholar]
- Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, Inada H, Roh SE, Kim SJ, Lee G, Bae H, Moorhouse AJ, Mikoshiba K, Fukazawa Y, Koizumi S & Nabekura J (2016). Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest 126, 1983–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura H, Uozumi N, Tohmi M, Yamazaki M, Sakimura K, Kudoh M, Shimizu T & Shibuki K (2007). Roles of nitric oxide as a vasodilator in neurovascular coupling of mouse somatosensory cortex. Neurosci Res 59, 160–171. [DOI] [PubMed] [Google Scholar]
- Koga K, Iwahori Y, Ozaki S & Ohta H (2010). Regulation of spontaneous Ca2+ spikes by metabotropic glutamate receptors in primary cultures of rat cortical neurons. J Neurosci Res 88, 2252–2262. [DOI] [PubMed] [Google Scholar]
- Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y & Nagano T (1999). Fluorescent indicators for imaging nitric oxide production. Angew Chem Int Ed Engl 38, 3209–3212. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Wall PD & McMahon SB (1999). Does the right side know what the left is doing? Trends Neurosci 22, 122–127. [DOI] [PubMed] [Google Scholar]
- Komagata S, Chen S, Suzuki A, Yamashita H, Hishida R, Maeda T, Shibata M & Shibuki K (2011). Initial phase of neuropathic pain within a few hours after nerve injury in mice. J Neurosci 31, 4896–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC & Sherman SM (2010). Topography and physiology of ascending streams in the auditory tectothalamic pathway. Proc Natl Acad Sci U S A 107, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T, Matsumura S, Okuda‐Ashitaka E, Kitano T, Kojima H, Nagano T, Minami T & Ito S (2003). Attenuation of neuropathic pain by the nociceptin/orphanin FQ antagonist JTC‐801 is mediated by inhibition of nitric oxide production. Eur J Neurosci 17, 1384–1392. [DOI] [PubMed] [Google Scholar]
- Melzack R & Wall PD (1965). Pain mechanisms: a new theory. Science 150, 971–979. [DOI] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV & Martina M (2009). Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S A 106, 2423–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier SF & Watkins LR (2003). Spinal glia and proinflammatory cytokines mediate mirror‐image neuropathic pain in rats. J Neurosci 23, 1026–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogyoros I, Bostock H & Burke D (2000). Mechanisms of paresthesias arising from healthy axons. Muscle Nerve 23, 310–320. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H & Woolf CJ (2002). Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22, 6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka A, Gruber‐Schoffnegger D & Sandkühler J (2013). Non‐Hebbian plasticity at C‐fiber synapses in rat spinal cord lamina I neurons. Pain 154, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osikowicz M, Mika J & Przewlocka B (2013). The glutamatergic system as a target for neuropathic pain relief. Exp Physiol 98, 372–384. [DOI] [PubMed] [Google Scholar]
- Paleček J, Dougherty PM, Kim SH, Palecková V, Lekan H, Chung JM, Carlton SM & Willis WD (1992). Responses of spinothalamic tract neurons to mechanical and thermal stimuli in an experimental model of peripheral neuropathy in primates. J Neurophysiol 68, 1951–1966. [DOI] [PubMed] [Google Scholar]
- Penas C & Navarro X (2018). Epigenetic modifications associated to neuroinflammation and neuropathic pain after neural trauma. Front Cell Neurosci 12, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Cervero F, Gold MS, Hammond DL & Prescott SA (2009). Chloride regulation in the pain pathway. Brain Res Rev 60, 149–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan HH, Kang KS, Sohn YK & Li M (2013). Tempol reduces injury area in rat model of spinal cord contusion injury through suppression of iNOS and COX‐2 expression. Neurol Sci 34, 1621–1628. [DOI] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, Pintado B, Gutierrez‐Adan A, Sanguino E, Manzanares J, Zimmer A & Maldonado R (2008). Crucial role of CB2 cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci 28, 12125–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert KC, Dunbar RL, Gao W, Chen G & Ebner TJ (2004). Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol 92, 199–211. [DOI] [PubMed] [Google Scholar]
- Reuss MH & Reuss S (2001). Nitric oxide synthase neurons in the rodent spinal cord: distribution, relation to Substance P fibers, and effects of dorsal rhizotomy. J Chem Neuroanat 21, 181–196. [DOI] [PubMed] [Google Scholar]
- Roh DH, Choi SR, Yoon SY, Kang SY, Moon JY, Kwon SG, Han HJ, Beitz AJ & Lee JH (2011). Spinal neuronal NOS activation mediates sigma‐1 receptor‐induced mechanical and thermal hypersensitivity in mice: involvement of PKC‐dependent GluN1 phosphorylation. Br J Pharmacol 163, 1707–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG & Needleman P (1993). Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A 90, 7240–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang K, Bao C, Xin Y, Hu S, Gao X, Wang Y, Bodner M, Zhou YD & Dong XW (2018). Plastic change of prefrontal cortex mediates anxiety‐like behaviors associated with chronic pain in neuropathic rats. Mol Pain 14, 1744806918783931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Tegeder I & Geisslinger G (2009). No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci 32, 339–346. [DOI] [PubMed] [Google Scholar]
- Shenker NG, Haigh RC, Mapp PI, Harris N & Blake DR (2008). Contralateral hyperalgesia and allodynia following intradermal capsaicin injection in man. Rheumatology (Oxford) 47, 1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, Watanabe M, Watanabe S, Kouuchi T & Tanaka R (2003). Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol (Lond) 549, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivilotti L & Woolf CJ (1994). The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch‐evoked allodynia in the spinal cord. J Neurophysiol 72, 169–179. [DOI] [PubMed] [Google Scholar]
- Simmons RM, Webster AA, Kalra AB & Iyengar S (2002). Group II mGluR receptor agonists are effective in persistent and neuropathic pain models in rats. Pharmacol Biochem Behav 73, 419–427. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Hishida R, Kubota Y, Kudoh M, Takahashi S & Shibuki K (2006). Transcranial fluorescence imaging of auditory cortical plasticity regulated by acoustic environments in mice. Eur J Neurosci 23, 1365–1376. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Masu M, Ishii T, Shigemoto R & Nakanishi S (1992). A family of metabotropic glutamate receptors. Neuron 8, 169–79. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Nagatani Y, Saitoh K, Takasu K & Ono H (2009). Pharmacological assessments of nitric oxide synthase isoforms and downstream diversity of NO signaling in the maintenance of thermal and mechanical hypersensitivity after peripheral nerve injury in mice. Neuropharmacology 56, 702–708. [DOI] [PubMed] [Google Scholar]
- Toda H, Kawasaki K, Sato S, Horie M, Nakahara K, Bepari AK, Sawahata H, Suzuki T, Okado H, Takebayashi H & Hasegawa I (2018). Locally induced neuronal synchrony precisely propagates to specific cortical areas without rhythm distortion. Sci Rep 8, 7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohmi M, Kitaura H, Komagata S, Kudoh M & Shibuki K (2006). Enduring critical period plasticity visualized by transcranial flavoprotein imaging in mouse primary visual cortex. J Neurosci 26, 11775–11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Sasaki M, Komagata S, Tsukano H, Hishida R, Kohno T, Baba H & Shibuki K (2015). Spinal mechanisms underlying potentiation of hindpaw responses observed after transient hindpaw ischemia in mice. Sci Rep 5, 11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Fang L, Lin Q & Willis WD (2001). Nitric oxide synthase in spinal cord central sensitization following intradermal injection of capsaicin. Pain 94, 47–58. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW & Zhuo M (2008). Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci 28, 7445–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Matsumura S, Mabuchi T, Takagi K, Abe T & Ito S (2006). In situ measurement of neuronal nitric oxide synthase activity in the spinal cord by NADPH‐diaphorase histochemistry. J Neurosci Methods 150, 174–184. [DOI] [PubMed] [Google Scholar]
- Yassin L, Radtke‐Schuller S, Asraf H, Grothe B, Hershfinkel M, Forsythe ID & Kopp‐Scheinpflug C (2014). Nitric oxide signaling modulates synaptic inhibition in the superior paraolivary nucleus (SPN) via cGMP‐dependent suppression of KCC2. Front Neural Circuits 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake K, Tsukano H, Tohmi M, Komagata S, Hishida R, Yagi T & Shibuki K (2013). Visual map shifts based on whisker‐guided cues in the young mouse visual cortex. Cell Rep 5, 1365–1374. [DOI] [PubMed] [Google Scholar]
- Zhang X, Verge V, Wiesenfeld‐Hallin Z, Ju G, Bredt D, Synder SH & Hökfelt T (1993). Nitric oxide synthase‐like immunoreactivity in lumbar dorsal root ganglia and spinal cord of rat and monkey and effect of peripheral axotomy. J Comp Neurol 335, 563–575. [DOI] [PubMed] [Google Scholar]
- Zhao R, Zhou H, Huang L, Xie Z, Wang J, Gan WB & Yang G (2018). Neuropathic pain causes pyramidal neuronal hyperactivity in the anterior cingulate cortex. Front Cell Neurosci 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]