

Summary
The black yeast Aureobasidium pullulans is a textbook example of a generalistic and ubiquitous fungus thriving in a wide variety of environments. To investigate whether A. pullulans is a true generalist, or alternatively, whether part of its versatility can be attributed to intraspecific specialization masked by cryptic diversification undetectable by traditional phylogenetic analyses, we sequenced and analysed the genomes of 50 strains of A. pullulans from different habitats and geographic locations. No population structure was observed in the sequenced strains. Decay of linkage disequilibrium over shorter physical distances (<100 bp) than in many sexually reproducing fungi indicates a high level of recombination in the species. A homothallic mating locus was found in all of the sequenced genomes. Aureobasidium pullulans appears to have a homogeneous population genetics structure, which is best explained by good dispersal and high levels of recombination. This means that A. pullulans is a true generalist that can inhabit different habitats without substantial specialization to any of these habitats at the genomic level. Furthermore, in the future, the high level of A. pullulans recombination can be exploited for the identification of genomic loci that are involved in the many biotechnologically useful traits of this black yeast.
Introduction
The black yeast Aureobasidium pullulans (Ascomycota, Dothideomycetes, Dothideales) is best known for its considerable biotechnological potential (Chi et al., 2009; Prasongsuk et al., 2018). It is used for the production of pullulan, a neutral polysaccharide that is composed of maltotriose units, and that has numerous applications in medicine, pharmacy and the food industry (Shabtai and Mukmenev, 1995; Leathers, 2003; Cheng et al., 2011). One A. pullulans strain is used to produce the antimycotic aureobasidin A (Takesako et al., 1993). Other bioproducts of A. pullulans that have been proposed, although not yet fully exploited, range from antibacterial compounds and liamocins (Price et al., 2013), to a wide range of extracellular enzymes that A. pullulans produces (Chi et al., 2009; Molnarova et al., 2014). Furthermore, A. pullulans is a commercially available biocontrol species that is effective against both bacterial and fungal plant pathogens (Johnson and Temple, 2013; Spadaro and Droby, 2016).
Although the ecology of A. pullulans is not as extensively studied as its biotechnological potential, it is at least as versatile. A. pullulans is found on (and in) plants (Andrews et al., 2002; Grube et al., 2011), in coastal hypersaline waters (Gunde‐Cimerman et al., 2000; Oren and Gunde‐Cimerman, 2012), in glacial ice (Zalar et al., 2008; Branda et al., 2010), and in various indoor habitats (Kaarakainen et al., 2009; Samson et al., 2010; Zalar et al., 2011). Aureobasidium pullulans often occurs under stress conditions that are generally considered to be limiting to microbial growth, such as in refrigerated, frozen, salt‐preserved and dried foods (Pitt and Hocking, 1999; Nisiotou et al., 2010), in aviation fuel tanks (Rauch et al., 2006), and on synthetic polymer surfaces (Cappitelli and Sorlini, 2008). The ability of A. pullulans to survive in such unconventional habitats and to outcompete other species has been linked to its great adaptability to novel environments, to its polyextremotolerance – the ability to tolerate a plethora of different stresses (Gostinčar et al., 2011) – and to its nutritional versatility, which is associated with its wide enzymatic profile (Di Francesco et al., 2017). Its production of antimicrobial compounds and siderophores, and its biofilm formation and other factors are also likely to contribute to its adaptability (Takesako et al., 1993; Wang et al., 2009; Klein and Kupper, 2018).
Many of these habitats of A. pullulans share common challenges, which can result in common adaptations, such as accumulation of intracellular glycerol, which serves as a compatible solute under hypersaline conditions, and a cryoprotectant under freezing conditions. Melanization, biofilm formation and oxidative stress responses are also believed to be useful in a number of stress scenarios. As a consequence, there is substantial overlap in microbial diversity between these habitats (Butinar et al., 2007; Gostinčar et al., 2010; 2011). Other parameters, however, differ considerably between habitats, such as UV exposure and temperatures between Mediterranean coastal hypersaline ponds and subglacial Arctic ice. While A. pullulans is a species with exceptional phenotypic plasticity (Slepecky and Starmer, 2009), the question remains whether a single genomic configuration can allow for the flexibility required to adapt to conditions on plant surfaces, inside an Arctic glacier, in concentrated sea water, in house dust, and in other habitats, some of which are very particular.
This question can be at least partially answered through investigations into the population genomics of A. pullulans. Despite the increasing accessibility of such approaches, the number of fungal population genomics studies in the literature outside of the medical field remains relatively modest. Also, the term ‘generalism’ in fungi is often used in the scope of host selection of pathogenic fungi (e.g. in both plants and animals). Habitat generalism has been less‐well studied, especially from the population genetics, or genomics, perspective.
Population genomics analyses on different fungi have often reached different conclusions, although many have described previously undiscovered diversification within different species. An analysis of Candida krusei/Pichia kudriavzevii, for example, segregated clinical and environmental isolates into separate clades (Douglass et al., 2018). Saccharomyces cerevisiae has also shown separate clustering of wild and domesticated strains with several domestication events, despite the single origin of the species in East Asia (Duan et al., 2018; Peter et al., 2018). Two non‐recombining African lineages have been described for Cryptococcus neoformans, one of which has accumulated adaptations presumably (coincidentally) predisposing it for human virulence (Desjardins et al., 2017). Hemileia vastatrix is the cause of coffee leaf rust, and it was initially believed to be a genetically unstructured and cosmopolitan species, but was later revealed to be a complex of cryptic species with marked host tropism (Silva et al., 2018). Population structuring (with no significant geographic structure) has been reported for the pathogenic Pneumocystis species (Cissé et al., 2018). The same was noted for the yeast Metschnikowia reukaufii, where the lineages detected were reported to be metabolically distinct (Dhami et al., 2018). Population genomics of an arbuscular mycorrhizal fungus Rhizophagus irregularis recognized four main genetic groups within the species and also showed that this divergence is not linked to geographic origin, which indicated that the genotypes were dispersed at an intercontinental scale (Savary et al., 2018).
In two of the earliest population genomics studies, the mushroom Suillus brevipes (Branco et al., 2015) and the model mould Neurospora crassa (Ellison et al., 2011) were shown to contain cryptic populations that had not been recognized prior to genome sequencing. As noted in these studies, unrecognized cryptic diversity can mask host–symbiont specificity and change the inference of evolutionary processes. It is not difficult to imagine how the same reasoning might apply to habitat specificity. Some fungal species can inhabit a wide variety of fundamentally different habitats. Their polyextremotolerance and adaptability combine into a generalistic fungal phenotype, which confers upon them the ability to rapidly adapt to novel habitats, such as those created by human activities (Gostinčar et al. 2010; 2011; 2012; 2015). The question is whether these apparently generalistic species are actually collages of cryptic specialists that have blurred into seemingly uniform entities through our inability to recognize their cryptic diversity. Our previous studies on A. pullulans contained some indications of such a scenario (Zalar et al., 2008; Gostinčar et al., 2014).
Biological classification of Aureobasidium spp. is not trivial. Their large phenotypic plasticity prevents the use of many morphological markers. Furthermore, sexual recombination in A. pullulans has not been observed to date. When molecular markers and the phylogenetic species concept were used, three groups of A. pullulans strains were identified as distinct from the core A. pullulans (Zalar et al., 2008). These were first described as varieties (Zalar et al., 2008), and later, after sequencing and comparison of the genomes of one strain per variety, they were classified as separate species (Gostinčar et al., 2014). While the numbers of habitats remained large in the case of A. pullulans, Aureobasidium melanogenum showed a (non‐exclusive) preference for aquatic environments, and Aureobasidium subglaciale appeared to be limited to glacial habitats (Zalar et al., 2008; Gostinčar et al., 2014). Only one strain of Aureobasidium namibiae has been described so far, which indicates its much narrower distribution.
Would the ubiquitous and generalistic species A. pullulans further disintegrate into intraspecific phylogenetic lineages adapted to different types of environmental conditions if sufficiently high resolution was used in investigations of its population(s)? Since the use of conventional phylogenetic markers has not provided such high resolution, we sequenced the whole genomes of 50 A. pullulans isolates from various habitats and geographical locations (Table 1). Using population genomic tools, we tested the hypothesis that the great ecological versatility of A. pullulans is a consequence of cryptic diversification and specialization; this hypothesis is rejected.
Table 1.
Strains sequenced in this study.
| Culture collection strain number | Present study number | Isolation habitat | Sampling site location |
|---|---|---|---|
| EXF‐674 | 1 | Indoors: air conditioner grate for entering air | Slovenia: Ljubljana |
| EXF‐676 | 2 | Indoors: air conditioner grate for entering air | Slovenia: Ljubljana |
| EXF‐1645 | 3 | Glacial: glacial ice | Norway: Ny‐Ålesund |
| EXF‐1668 | 4 | Glacial: glacial ice from sea water | Norway: Ny‐Ålesund |
| EXF‐2618 | 5 | Plant: grape surface | Slovenia: Ljubljana |
| EXF‐3358 | 6 | Other: sea water | Croatia: Mljet |
| CBS 584.75a (EXF‐3374) | 7 | Plant: grape surface | France: Beaujeu |
| CBS 146.30 (EXF‐3380) | 8 | Plant: oak slime flux | Germany: Ohlsdorf, Hamburg |
| CBS 109810 (EXF‐3403) | 9 | Indoors: fourth block wall surface, radioactivity 2.0×104 Bq/m2s | Ukraine: Chernobyl |
| EXF‐3519 | 10 | Plant: oak leaf surface | Slovenia: Ljubljana |
| EXF‐3645 | 11 | Glacial: glacial ice at the edge of glacier | Norway: Ny‐Ålesund |
| EXF‐3670 | 12 | Glacial: glacial ice at the edge of glacier | Norway: Ny‐Ålesund |
| EXF‐3750 | 13 | Glacial: glacial ice at the edge of glacier | Norway: Ny‐Ålesund |
| EXF‐3780 | 14 | Hypersaline: microbial mat, bottom of the sea water evaporation pond | Puerto Rico: Candelaria |
| EXF‐3844 | 15 | Plant: dried olives | Slovenia |
| EXF‐3863 | 16 | Hypersaline: salpans crystalization pond water | Slovenia: Sečovlje |
| EXF‐3984 | 17 | Glacial: glacial ice with sediment | Norway: Ny‐Ålesund |
| EXF‐4010 | 18 | Glacial: glacial ice with sediment | Norway: Ny‐Ålesund |
| EXF‐4256 | 19 | Glacial: glacial ice | Norway: Ny‐Ålesund |
| EXF‐5628 | 20 | Indoors: rubber seal | Slovenia: Blejska Dobrava |
| EXF‐6176b | 21 | Glacial: glacial ice | Argentina: San Carlos de Bariloche |
| EXF‐6267 | 22 | Hypersaline: salpan evaporating sea water | Slovenia: Sečovlje |
| EXF‐6298 | 23 | Indoors: washing powder tray | Slovenia: Postojna |
| EXF‐6514 | 24 | Plant: peach bone | Slovenia |
| EXF‐6519 | 25 | Other: felt on the bottom side of a metal roof tile | Slovenia: Mengeš |
| EXF‐6604 | 26 | Plant: roots of Juncus trifidus | Poland: Babia Góra massif |
| EXF‐8126 | 27 | Indoors: metal surface, basement of pumpkin seed oil pressing facility | Slovenia: Gibina |
| EXF‐8127 | 28 | Indoors: surface of metal bucket used for carrying water | Slovenia: Cuber |
| EXF‐8128 | 29 | Plant: maple leaf surface | Slovenia: Ljubljana |
| EXF‐8828 (CRUB 1715) | 30 | Glacial: glacial meltwater | Argentina: San Carlos de Bariloche |
| EXF‐8841 (CRUB 1819) | 31 | Plant: Nothofagus pumilio leaf surface | Argentina: San Carlos de Bariloche |
| EXF‐9398b | 32 | Plant: black olives fermentation | Greece: Attica |
| EXF‐9399 | 33 | Plant: grape surface | Greece: Attica |
| EXF‐9635 | 34 | Glacial: glacial ice | Italy: Calderone glacier |
| EXF‐9785 | 35 | Indoors: Interior of water supply connector | Slovenia: Kapla |
| EXF‐10080 | 36 | Indoors: kitchen sink drain | Slovenia: Ljubljana |
| EXF‐10081 | 37 | Indoors: kitchen sink drain | Slovenia: Ljubljana |
| EXF‐10085 | 38 | Indoors: kitchen cutting board surface | Slovenia: Planina pri Sevnici |
| EXF‐150c | 39 | Hypersaline: salpan evaporating sea water | Slovenia: Seča |
| EXF‐10507 | 40 | Other: marble block surface | Italy: Messina |
| EXF‐10606b | 41 | Other: creosote treated railway ties surface | Denmark |
| EXF‐10629 | 42 | Other: car petrol reservoir inlet inner surface | Slovenia: Jezero |
| EXF‐10632 | 43 | Other: car diesel reservoir inlet inner surface | Slovenia: Jezero |
| EXF‐10659 | 44 | Indoors: indoor air sample | Slovenia: Celje |
| EXF‐10751 | 45 | Other: cloud sample | France |
| EXF‐10796 | 46 | Plant: persimmon surface | Slovenia |
| EXF‐11013 | 47 | Plant: commercial biocontrol strain | N.D. |
| EXF‐11014 | 48 | Plant: commercial biocontrol strain | N.D. |
| EXF‐11318 | 49 | Plant: apple surface | Slovenia: Horjul |
| EXF‐11319 | 50 | Plant: apple surface | Slovenia: Horjul |
| EXF‐11323 | 51 | Plant: sweet chestnut leaf surface | Slovenia: Horjul |
| EXF‐11825 | 52 | Indoors: kitchen freezer rubber seal | Slovenia: Bistrica ob Sotli |
| EXF‐11900 | 53 | Indoors: kitchen refrigerator rubber seal | Croatia: Malinska, Krk |
| EXF‐11991 | 54 | Indoors: kitchen refrigerator condensation water outlet | Slovenia: Zagorje ob Savi |
Neotype strain.
Aureobasidium sp., shown to be distinct from A. pullulans in the genome analysis.
Reference genomic strain, included as control.
Results
To investigate the intraspecific phylogeny and population structure of the generalistic and ubiquitous yeast A. pullulans, the genomes of 54 A. pullulans‐like strains identified by internal transcribed spacer‐based phylogenetic analysis were sequenced. The selection of strains included the reference genome strain EXF‐150 (as control). The strains were selected to represent isolates from various habitats, with an emphasis on plant surfaces (17 strains), glacial ice (11), hypersaline waters (4) and various indoor habitats (15). They were also sampled from different geographical locations, although with a bias towards isolates obtained in Slovenia (32 isolates).
The statistics of the genome sequencing, assembly and annotation were largely similar between the strains (Table 2 and Supporting Information Table S1). The average coverage of sequenced genomes [excluding non‐A. pullulans genomes 21, 32, 41 (see below) and 39 (reference genome)] was 73× (±23.73× SD). A few possible aneuploidies were detected, for strains 8, 10, 33 and 35 (Supporting Information Fig. S1). On average, the genomes were assembled into 1629 contigs (±1523 SD), whereby the best assembly contained only 137 contigs. The average genome size was very similar between the strains, at 28.04 Mbp (±1.03 Mbp SD). The most different genome from the other genomes was genome 21 (which was later identified as being distinct from A. pullulans), with average mapping depth to the reference genome at only 9× and 36.5 Mbp assembly size. This was identified as one of the least complete assemblies according to the content of Benchmarking Universal Single‐Copy Orthologues (BUSCOs), with only genomes 8 and 50 being less complete, although it also contained 7.60% duplicated BUSCOs, while other genomes contained a maximum of 0.70%. The completeness of the other annotated genomes was high (96.28% ±2.37% SD). The numbers of predicted gene models and other statistics were comparable to the reference genome (Table 2 and Supporting Information Table S1).
Table 2.
Statistics for the sequenced A. pullulans genomes.
| Statistica | Minimumb | Meanb | Maximumb | Standard deviationb |
|---|---|---|---|---|
| Coverage | 37 | 73 | 148 | 23.73 |
| Genome assembly size (Mb) | 23.79 | 28.04 | 30.69 | 1.03 |
| Number of contigs | 137 | 1629 | 7437 | 1522.84 |
| Contig N50 | 4553 | 74165 | 547498 | 88815 |
| GC content (%) | 50.41 | 50.65 | 51.25 | 0.14 |
| Coding sequence total length (Mb) | 12.01 | 15.40 | 16.19 | 0.66 |
| Coding sequence total length (%genome) | 49.06 | 54.91 | 56.55 | 1.28 |
| Gene models (n) | 9527 | 10646 | 11081 | 238 |
| Gene average length (bp) | 1363 | 1564 | 1621 | 50 |
| Exons per gene (average) | 2.27 | 2.55 | 2.70 | 0.09 |
| Intron average length (bp) | 69.00 | 77.44 | 88.00 | 5.17 |
| Complete BUSCOs (%) | 84.80 | 96.28 | 98.20 | 2.37 |
| Complete and single‐copy BUSCOs (%) | 84.80 | 95.93 | 97.90 | 2.32 |
| Complete and duplicated BUSCOs (%) | 0.00 | 0.36 | 0.70 | 0.21 |
| Fragmented BUSCOs (%) | 0.70 | 2.52 | 10.30 | 1.63 |
| Missing BUSCOs (%) | 0.60 | 1.20 | 5.50 | 0.84 |
| SNP density (%) | 1.29 | 1.73 | 3.37 | 0.26 |
Complete data for each genome is available in the Supporting Information Table S1.
Calculated from 50 A. pullulans genomes, not including genomes 21, 32, 41 (too divergent) and 39 (reference genome).
BUSCOs, Benchmarking Universal Single‐Copy Orthologues.
The number of core genes shared between all 50 genomes and the reference A. pullulans genome was 3637 (identified by all three of the used clustering algorithms: bidirectional best hit, COGtriangle and OrthoMCL). Additional 2711 genes were present in 48–50 genomes (the so‐called ‘soft core’ genome). Neither the core nor the soft core or cloud genomes (categories defined by Contreras‐Moreira and Vinuesa (2013)) were found to be significantly enriched in any GO‐Slim Biological Process terms, with the exception of the enrichment in unclassified proteins in the cloud genome. The same was true for the group of 149 genes duplicated in at least one of the 50 genomes. Only if the Fisher's exact test was calculated with no correction, the core genome was enriched for cellular amino acid biosynthetic process; the cloud genome contained fewer genes involved in protein ubiquitination, small molecule metabolic process, formation of translation initiation ternary complex, translational termination and elongation and cellular response to stress; finally, the group of duplicated genes was enriched for biological regulation, filamentous growth, dicarboxylic acid transport and inorganic anion transmembrane transport.
Phylogenetic analyses based on both the predicted genes and single nucleotide polymorphism (SNP) data (Fig. 1) failed to detect the so‐called ‘strong phylogenetic signal’ for clonality (Tibayrenc and Ayala, 2012). The phylogenies were close to the extreme (star‐like) multifurcating tree. The phylogenies also suggested that strains 21, 32 and 41 were very divergent from the rest of A. pullulans, and as such, were probably misidentified. These were removed from the subsequent analyses, which thus included the reference genomic strain A. pullulans EXF‐150, and 50 new A. pullulans genome sequences.
Figure 1.
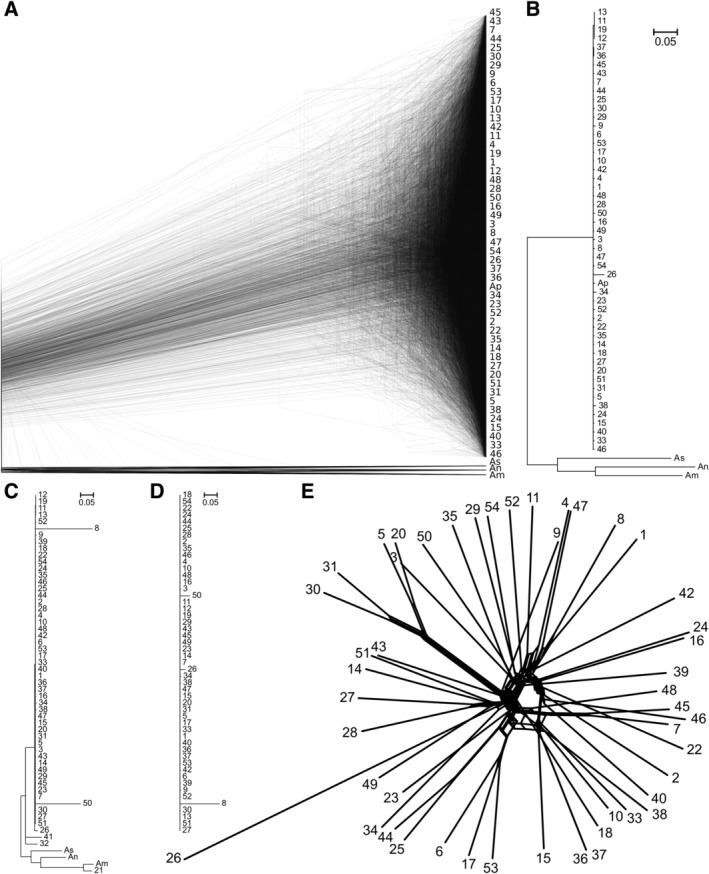
Phylogeny of Aureobasidium pullulans strains. A. Overlay of 1506 core gene trees estimated by PhyML 3.1 using the Hasegawa‐Kishino‐Yano 85 nucleotide substitution model and estimating the alpha parameter of the gamma distribution of the substitution rate categories and the proportion of invariable sites. B. Majority rule consensus tree of 1506 core gene trees described above. C. Majority rule consensus tree of 169 Benchmarking Universal Single‐Copy Orthologues with A. melanogenum (Am), A. subglaciale (As) and A. namibiae (An) orthologues used as an outgroup. D. The same without the outgroup and based on 204 gene trees, with all trees estimated as described above. E. Phylogenetic network reconstructed with the Neighbor‐Net algorithm based on the dissimilarity distance matrix calculated from the SNP data.
Compared to the reference A. pullulans genome, the other 50 sequenced genomes contained on average 1.73% (±0.26% SD) SNPs, and 0.14% (±0.03% SD) of the genome was covered by insertions/deletions. The most divergent was genome 26, with 3.37% SNPs. Strains 11, 12, 13 and 19 (which were all sampled from a geographically very limited area on Svalbard) were nearly identical to each other, and in the subsequent population analyses, of these four strains, only strain 11 was kept in the dataset, to remove any artefacts that might have arisen from the use of clones.
The software structure failed to identify any population structure based on the SNP data of the sequenced genomes, also after excluding the divergent genomes (21, 32, 41) and the clones (12, 13, 19), and even when the habitats of the strains or the sampling locations were used as prior information about the population structure (data not shown). Similar observations were made with principal component analysis of the SNP data. The first two axes (PC1, PC2) explained only 7.96% and 6.92% of the variation. There was no clustering linked to either the habitat of the 50 sequenced strains or to their sampling locations (Fig. 2).
Figure 2.
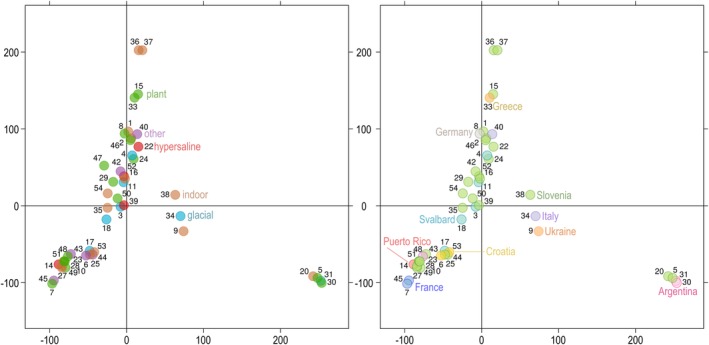
Clustering of the Aureobasidium pullulans genomes. Principal component analysis of SNP data estimated by comparing these sequenced A. pullulans genomes to the reference genome. The genomes are represented by circles, the colour of which corresponds to the habitat (left) or sampling location (right) of the sequenced strains. The first two axes explain 7.96% (horizontal) and 6.92% (vertical) of variation.
The lack of concordance between gene phylogenies and the lack of population structure might be explained by recombination between A. pullulans strains. Therefore, linkage disequilibrium (LD) was investigated in a data set of all biallelic SNP loci and a data set of biallelic SNP loci with at least 25% of each of the two alleles. Both of these data sets produced comparable results, and therefore only the analysis of the second data set is shown (Fig. 3). If the genomes are recombining, the linkage between two loci is expected to decrease as a function of the distance between the two loci in the genome (i.e. the association between the loci should approach randomness with increasing distance). In these A. pullulans genomes, the LD decay, the average physical distance over which the normalised coefficient of LD (D′) or the squared correlation coefficient (r 2) fell to half of their maximum value, was small: 93–112 bp based on D′, and 84–99 bp based on r 2 when averaging the D′ or r 2 in three nucleotide windows. The distance was even shorter if no averaging was used: 28–53 bp in case of r 2 (data not shown).
Figure 3.
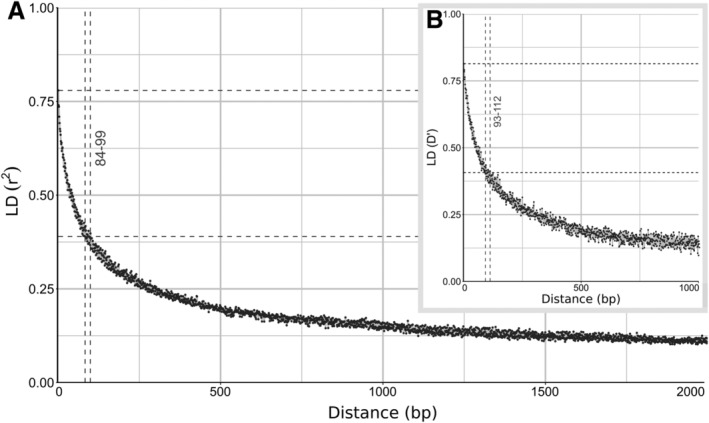
LD decay in A. pullulans estimated on all biallelic loci which were present in 25%–75% of the sequenced genomes. LD measures were averaged in three nucleotide windows. A. Squared correlation coefficient (r 2) between pairs of SNP loci plotted against the physical distance of the loci in the genome. Horizontal lines mark the maximum observed value and half of the maximum observed value. Vertical lines mark the interval of the physical distance in which the maximum value is halved. B. Same as above, with normalized coefficient of LD (D′) instead of r 2.
A putative mating locus was identified in all of these 50 sequenced A. pullulans strains (Fig. 4). It had a similar structure in all of the genomes: genes MAT1‐1 and MAT1‐2 were flanked by a gene that encodes DNA lyase on one side and a gene that encodes a pleckstrin‐homology‐domain protein on the other side. Other genes in the near vicinity included an anaphase‐promoting complex subunit, a subunit of a cytochrome oxidase (VIa), an amino‐acid permease, and three orphan genes with no matches in the GenBank database of fungal proteins.
Figure 4.
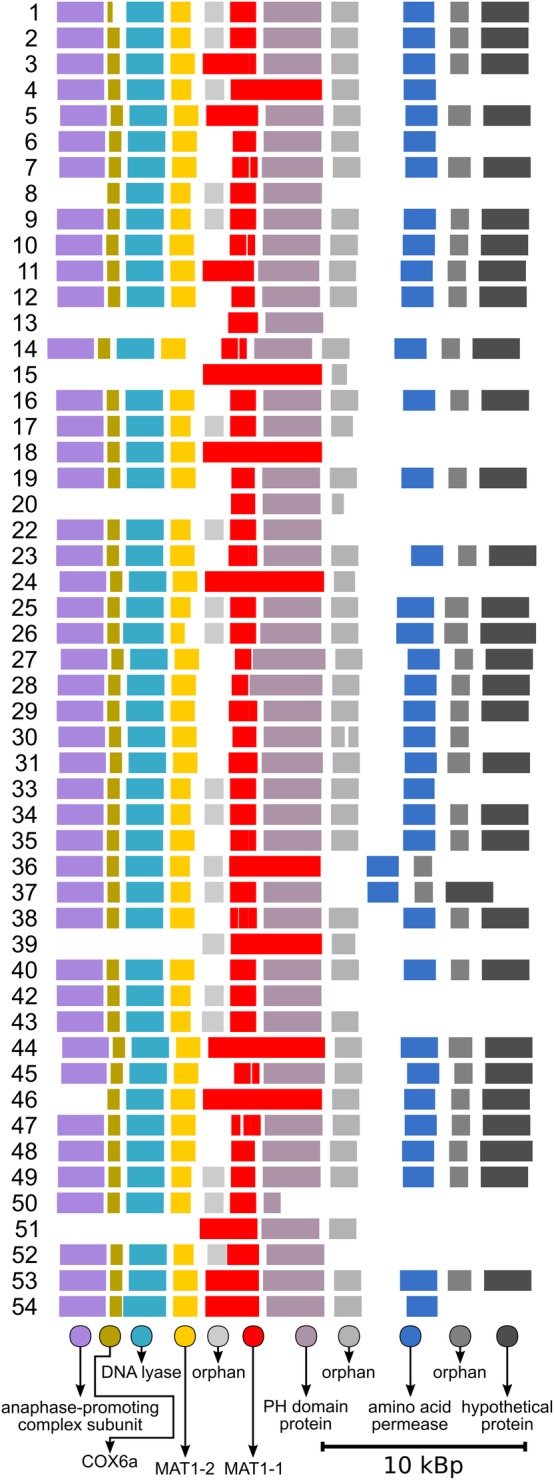
Putative mating loci in different strains of Aureobasidium pullulans. MAT1‐1: mating type‐1; MAT1‐2: mating type‐2. Strain numbers are on the left. The genes with no matches in the fungal subset of the GenBank database are indicated as ‘orphans’, and proteins with several matches in other species (but with an unknown function) are indicated as ‘hypothetical proteins’.
A preliminary search for some of the biotechnologically interesting genes of A. pullulans yielded mixed results. The putative pullulan synthase was found in all of these strains. On the other hand, an adenylation domain typical of non‐ribosomal siderophore synthetases was only found in strains 2, 3, 4, 5, 9, 11, 12, 13, 14, 18, 19, 20, 30, 31, 34, 35, 46 and 50. All genes encoding the main alkali metal cation transporters found in the reference A. pullulans genome (Gostinčar et al., 2014) were also found in all of the here sequenced genomes in at least one copy per genome each and located in the same phylogenetic lineages as their homologues from the reference genome (Supporting Information Table S2). The strains differing the most in the number of gene copies were strains 8 and 50, but most of these differences could be ascribed to the errors in annotation upon manual inspection – possibly due to a relatively poor genome assembly of these strains. The gene with the highest variation in copy number between the genomes was Ena.
Discussion
The generalistic black yeast A. pullulans can be found in many fundamentally different habitats around the world. This distribution was previously suggested to reflect a true habitat generalism that was provided by the extreme adaptability, polyextremotolerance and nutritional versatility of A. pullulans strains (Gostinčar et al., 2010; 2011; 2014). However, the mechanism behind such a wide distribution might also be cryptic diversification and specialization of unrecognized phylogenetic lineages to individual (or groups of similar) habitats. After the first population genomics studies of fungi, it became clear that many species harboured cryptic populations (Ellison et al., 2011; Branco et al., 2015; Dhami et al., 2018; Silva et al., 2018), and if these remain unidentified, they can mask the adaptation of individual populations for specific hosts, metabolic processes, types of stress, or other factors.
To investigate the population structure of A. pullulans and its potential intraspecific cryptic specialization, we sequenced the genomes of 54 A. pullulans‐like strains, which produced 50 new A. pullulans genome sequences. These 50 genomes of A. pullulans sequenced here showed little variability in terms of genome size and predicted gene content (Table 2 and Supporting Information Table S1). Only four strains contained greater differences compared to the reference genome, which indicated possible aneuploidies (Supporting Information Fig. S1). The sequenced genomes were smaller than most of the 43 representative Dothideomycetes genomes that have been deposited at GenBank to date (average genome size: 36.75 Mbp), with only 6 of these 43 with a smaller genome than the average for A. pullulans (28.04 Mbp).
The species A. pullulans previously included isolates that were later classified into the new species A. melanogenum, A. subglaciale and A. namibiae. Despite this narrower definition of A. pullulans, this species still harbours substantial diversity. The density of SNPs when compared between these new genomes and the reference genome was 1.73%. The SNP density in S. cerevisiae is 0.55% in wild strains and 0.41% in domesticated strains (Peter et al., 2018), in Neurospora. crassa this is 0.41% (Pomraning et al., 2011) and in Candida glabrata, 0.47%–0.66% (Carreté et al., 2018). The number of core genes was modest (3637) – the core genome of S. cerevisiae contains almost 5000 genes, and that estimate is based on over 1000 genomes (Peter et al., 2018). The soft core genome of A. pullulans (i.e. genes present in at least 95% strains, possibly overestimating the core genome, but also alleviating the problem of genes missing due to errors in assembly or annotation) was substantially larger (6348 genes), but still represented <55% of the reference genome, while the core genome in S. cerevisiae constitutes >75% of the reference genome (Peter et al., 2018). However, despite such large diversity observed within A. pullulans, the current delimitation of this species does not appear to be too wide, because as discussed below, there is evidence for frequent recombination between A. pullulans strains and no evidence for any structuring of the global A. pullulans population.
Genomic data can provide a wealth of information. Indeed, even for some species previously thought to have homogeneous population genetics structure, detailed analyses have uncovered cryptic populations with differences in their animal or plant virulence (Desjardins et al., 2017; Douglass et al., 2018; Silva et al., 2018), metabolism (Dhami et al., 2018), domestication patterns (Duan et al., 2018; Peter et al., 2018) and adaptation to differences in climate (Ellison et al., 2011; Branco et al., 2015; 2017). This does not appear to be the case for A. pullulans. The strains sampled here across different habitats for several continents failed to form recognizable phylogenetic lineages in the phylogenetic analysis (Fig. 1) and contained no population structure that was detectable by the STRUCTURE software or that was reflected in the principal component analysis of the SNP data (Fig. 2).
The lack of concordance between A. pullulans gene phylogenies and the lack of population structure can be explained as a consequence of recombination between the worldwide‐distributed strains of A. pullulans. This scenario is further supported by the LD decay over a very short distance that was observed for the genomes analysed here (Fig. 3). LD decay is most often measured as the distance over which LD falls to half of its maximum value (Taylor et al., 2015). In the presence of recombination, the LD between pairs of loci is a function of the distance between the two loci on the same DNA molecule, as the linkage between two loci is broken by recombination, which is more likely to occur between loci that are further apart. In the genomes of A. pullulans analysed here, the observed LD decay distance based on r 2 was between 84 bp and 99 bp when averaged in three‐nucleotide windows and even shorter (28–53 bp) when no averaging was used, which is shorter than reported for S. cerevisiae, at 500 bp (Peter et al., 2018), for N. crassa, at 780 bp (Ellison et al., 2011), and for lineages of C. neoformans var. grubii, at 5000–7500 bp (Desjardins et al., 2017). The shortest LD half‐decay distance reported by Nieuwenhuis and James (2016) was 150 for Coccidioides posadasii. Half of the species analysed in that study (Schizophyllum commune, Saccharomyces paradoxus, Neurospora crassa, Heterobasidion annosum, Lachancea kluyveri, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Batrachochytrium dendrobatidis and Candida albicans) had LD decay distances within the range of 100–1000 bp and the highly clonal species such as B. dendrobatidis and C. albicans reaching 126 420 bp and 286 740 bp respectively. This places the LD decay rate A. pullulans well within the range of sexually reproducing species.
Discosphaerina fagi was proposed to be the teleomorph of A. pullulans, although this has never been investigated in depth (de Hoog et al., 2001; Zalar et al., 2008). In the literature, A. pullulans is often regarded as an asexual species. If true, asexuality could easily be explained as a means to avoid the recombination load, a reduction in fitness due to breakage of locally adapted genotypes with recombination (Otto, 2009). In A. pullulans, which inhabits such a large number of different environments, it would be easy to imagine that local adaptation and accompanying avoidance of recombination would play an important role in improving the fitness of the species. However, this does not appear to be the case. The data obtained in the present study are the strongest indicator of recombination within A. pullulans to date, although the nature of the process remains unknown (i.e. sexual or asexual). Such population evidence for recombination in a seemingly clonal fungus has been reported before for other species. Coccidioides immitis was reportedly the first morphologically asexual fungus for which such evidence was found and was most parsimoniously explained by sexual reproduction (Burt et al., 1996; Taylor et al., 2015). Other early such examples included Aspergillus flavus (Geiser et al., 1998), Cenococcum geophilum (LoBuglio and Taylor, 2002) and Fusarium oxysporum (Koenig et al., 1997). Similar observations continued with the introduction of the population genomics, which was, for example, used to show that C. glabrata, a presumed asexual species, can recombine (Carreté et al., 2018).
However, although the perception of many fungi as predominantly or strictly clonal has been changed through population genetics and genomics studies (Taylor et al., 2015), this is not necessarily true for all species. The extremely halotolerant Hortaea werneckii, which is another black yeast from extreme environments, was described to form unusually stable diploid strains through hybridization between relatively heterozygous ancestors, although apart from this, H. werneckii appears to be limited to clonal reproduction (Gostinčar et al., 2018). Indeed, despite the apparent lack of recombination between strains of H. werneckii and the strong evidence for recombination between strains of A. pullulans, the inferred mating locus architecture of these two species is very similar: mating genes 1‐1 and 1‐2, flanked by genes that encode a pleckstrin homology domain protein and a transmembrane transporter on the one side, and an anaphase‐promoting complex subunit and a DNA lyase on the other. While the presence of a mating locus in the genome does not necessarily mean that the species can undergo sexual reproduction (Taylor et al., 2015), the reason for the different reproduction strategies of A. pullulans and H. werneckii in the presence of a similar mating locus remains unknown.
The high level of recombination observed in A. pullulans has implications beyond simply understanding its biology. It could be seen as undesirable for the use of the species in biocontrol, where the strains used for this purpose are applied in the fields or on harvested fruit, where they come in contact with wild strains and possibly recombine with them, leading to offspring with unforeseeable phenotypes (Moore, 2014). On the other hand, genome‐wide association studies have made possible the identification of loci that mediate phenotypic variation of biotechnologically important traits based on variations among different strains. However, the power of this method is correlated with the level of recombination in a population (Plissonneau et al., 2017). The high recombination of A. pullulans therefore makes it a good candidate for such studies. Furthermore, if better molecular genetic tools can be developed for A. pullulans and crossing the strains becomes a possibility in a laboratory setting, quantitative trait locus mapping can be added to the collection of tools that can be used for exploitation of this biotechnologically important yeast.
One of the most interesting traits of A. pullulans is its halotolerance. In any organism survival of high extracellular concentrations of inorganic salts requires the adjustment of intracellular osmotic pressure to avoid plasmolysis, as well as careful maintenance of physiological concentrations of alkali metal cations, such as keeping a high and stable intracellular K+ content and the elimination of toxic Na+ (Ariño et al., 2010; Plemenitaš et al., 2016). The reference genome of A. pullulans encodes for a spectrum of different types of alkali metal cation transporters (Gostinčar et al., 2014). This was confirmed here (Supporting Information Table S2) and it was furthermore discovered that the numbers of gene copies was very consistent, with one exception: the P‐type ATPase Na+ exporter Ena was present as a single‐copy gene in nine genomes (including in the reference A. pullulans EXF‐150) but was duplicated in the other 42 genomes, the extra copy representing an old phylogenetic Ena lineage also found in other Aureobasidium spp. and several other fungi (Gostinčar et al., 2014). The potentially smaller role of Ena transporters compared to the Nha Na+(K+)/H+ antiporters in hypersaline conditions has been speculated on before (Lenassi et al., 2013). The consistent presence of different types of K+ transporters is also of note. Apart from the high‐affinity channels for K+ uptake Trk (three gene copies per genome), two alternative uptake systems were found in A. pullulans (Gostinčar et al., 2014): Hak H+/K+ symporters (one copy) and Acu P‐type ATPases (two copies); while membrane depolarization activated K+ channels Tok homologues (two copies per genome) are responsible for potassium efflux. With the exception of two (poorly assembled) genomes, the copy number of all these genes was identical in the here sequenced A. pullulans strains. Considering the otherwise large amount of variability in A. pullulans, this conservation may be an indication that the functions of different types of K+ transporters are not as redundant as is generally thought (Ariño et al., 2010; Martinez et al., 2011; Ramos et al., 2011) and that maintaining all of them, in some cases even in more than one copy, benefits the cells in as yet unknown ways.
Finally, a preliminary search for some of the genes involved in the biotechnologically useful traits of A. pullulans showed that at least some traits vary considerably between the strains. While pullulan synthase was found in all of these strains, the adenylation domain of siderophore synthetases was present in only 18 of these strains. This shows that in the future these sequenced genomes can be used as a resource for informed selection of strains according to intended purpose, to expand the pool of potential industrial strains with this collection of 50 genome‐sequenced A. pullulans strains isolated from various habitats across different parts of the world.
Conclusions
Sequencing and analysing the genomes of 50 strains of the polyextremotolerant black yeast A. pullulans has here shown that:
(i) No phylogenetic lineages and no population structure can be detected within these strains isolated from different habitats and from different geographic locations. Instead the worldwide‐occurring A. pullulans appear to have a homogeneous population genetics structure, which can be best explained by good dispersal and a high level of recombination.
(ii) The absence of habitat‐linked strain clustering that would provide a basis for cryptic specialization for specific habitats indicates that A. pullulans is a true generalist that can survive in habitats as different as plant surfaces, glacial ice, hypersaline waters and kitchen surfaces (and many others) without substantial specialization to the individual habitats at the genomic level.
(iii) The decay of LD over short distances provides additional evidence for a high level of recombination within A. pullulans. This should support future genome‐wide association studies with the aim to identify genomic loci that are involved in the many biotechnologically useful traits of A. pullulans. The availability of 50 sequenced and annotated genomes will provide a good basis for such studies.
The great adaptability of A. pullulans is to some extent observed also in other polyextremotolerant fungi, especially in black yeast. These can quickly adapt to novel habitats, such as those created by human activities. Also, their polyextremotolerance and adaptability has been linked to their emerging role in opportunistic infections in mammals (Casadevall and Pirofski, 2007; Onofri et al., 2007; Gostinčar et al., 2010; 2011; 2012; 2015). Further studies will show whether the conclusions of the true generalism of A. pullulans can also be applied to other fungi with ubiquitous distributions and polyextremotolerant ecologies.
Experimental procedures
Culture, medium and growth conditions
Fifty‐four A. pullulans‐like strains collected from various habitats around the world (Table 1) were obtained from the Ex Culture Collection of the Department of Biology, Biotechnical Faculty, University of Ljubljana (Slovenia) and were previously identified based on their internal transcribed spacer sequences. Their cultivation and DNA isolation were performed as described previously (Gostinčar et al., 2018). In short, biomass was grown in the standard chemically defined Yeast Nitrogen Base medium (Qbiogene) with 0.5% (w/v) ammonium sulphate and 2% (w/v) glucose. The pH was adjusted to 7.0 prior to autoclaving. For solid medium, 2% (w/v) agar was added. All of these cultures were grown at 24 °C. Liquid cultures were grown on a rotary shaker at 180 rpm. The cells were harvested in the mid‐exponential growth phase (OD600 = 0.8–1.0) by centrifugation (5000× g for 10 min), and the cell pellets were frozen in liquid nitrogen and kept at −80 °C until DNA isolation.
DNA isolation
The biomass for DNA to be used for sequencing was homogenized using a pestle and mortar, while being kept frozen using liquid nitrogen. Then, 100 mg of homogenate was transferred to 2‐mL microcentrifuge tubes, each of which contained a stainless steel ball. These tubes were placed in holders that were pre‐cooled with liquid nitrogen, and then further homogenized (Retsch Mixer Mill 301; ThermoFisher Scientific) at 20 Hz for 1 min. Then 300 μL of MicroBead solution buffer was added (provided in the UltraClean Microbial DNA isolation kit; see below), and the mixtures were completely thawed on ice. These homogenates were used for DNA extraction (UltraClean Microbial DNA isolation kit; MO BIO Laboratories), according to the manufacturer instructions. Contaminating RNA was removed using RNAse A (ThermoFisher Scientific). The quantity, purity and integrity of the isolated DNA were evaluated using agarose electrophoresis and spectrophotometrically (NanoDrop 2000; ThermoFisher Scientific) and by fluorometry (Qubit; ThermoFisher Scientific).
Genome sequencing
The genome sequencing was performed by SeqMatic, using a Genome Sequencer Illumina NextSeq, with 2× 150‐bp libraries in a multiplexed mode. The resulting output was demultiplexed, the quality was checked (FastQC), and the reads were trimmed for adaptors and quality (removal of bases with Q <20) using the ‘bbduk’ script (https://jgi.doe.gov/data-and-tools/bbtools/).
The sequencing reads, assembly and annotation data have been deposited in Genbank under BioProject PRJNA488010.
Variant calling
Sequencing reads were mapped to the reference A. pullulans genome of strain EXF‐150 (GenBank AYEO00000000.1) (Gostinčar et al., 2014) with ‘bwa mem’, using the default parameters. These mapped reads were sorted with Samtools 1.6 (Li et al., 2009), and duplicates defined with Picard 2.10.2. The density of the reference genome coverage by sequencing reads was calculated using Samtools 1.6 (Li et al., 2009), and visualized in R with ‘ggplot2’ (Wickham, 2009; R Development Core Team, 2017). Variant calling was performed with Genome Analysis Toolkit 3.8 (Alkan et al., 2011), according to the ‘Genome Analysis Toolkit (GATK) Best Practices’, using the ‘hard filtering’ option. Ploidy was set to haploid.
Assembly and annotation
The genomes were assembled using IDBA‐Hybrid 1.1.3 (Peng et al., 2012), with the published A. pullulans genome EXF‐150 (Gostinčar et al., 2014) used as reference to guide the assembly process. The maximum k value selected was 140, the minimum support in each iteration was 2, the similarity for alignment was 0.95, seed kmer was 20, maximum allowed gap in the reference was 100, and the minimum size of contigs was 500.
Annotation of protein‐coding and tRNA genes was performed using MAKER 2.31.8 (Campbell et al., 2014). The fungal subset of the Swissprot database (recovered on 19 July 2017), and the published predicted proteomes of A. pullulans, A. melanogenum, A. subglaciale and A. namibiae (Gostinčar et al., 2014) were used as evidence. Three ab initio gene predictors were used in the MAKER pipeline. Semi‐HMM‐based Nucleic Acid Parser (SNAP) (Korf, 2004) was bootstrap‐trained within MAKER, based on the gene models derived from the alignment of the protein datasets to the genome, as recommended by Campbell et al. (2014). GeneMark‐ES (Lomsadze et al., 2014) was self‐trained (Ter‐Hovhannisyan et al., 2008), and Augustus was used with the training parameters for N. crassa (Stanke and Morgenstern, 2005).
The genome assembly and gene prediction completeness was evaluated with the BUSCO 3 software (Simão et al., 2015), in proteomic mode, using the data set for fungi (Waterhouse et al., 2013). All of the parameters were used as the default values.
The files for submission to GenBank were prepared using the Genome Annotation Generator (GAG) 2.0.1 software (Geib et al., 2018). All of the gene models with a coding region <150 bp or with introns <10 bp were removed.
Variant‐based analysis
The structure of the A. pullulans population(s) was investigated using the Structure 2.3.4 software (Pritchard et al., 2000; Falush et al., 2007), optimized on 10% of randomly sampled loci for maximum population numbers (K) from 1 to 6 (and three runs for each K), and using K 2 for the final analysis with: (i) the full data set; (ii) after removing the most divergent genomes (i.e. 21, 32, 41); and (iii) after additionally removing genomes 12, 13 and 19 due to high similarities to genome 11 (see above). The analysis was run without adding metadata, with the habitat information or with the sampling location data.
Principal component analysis of the SNP data was performed with the ‘glPca’ function from the ‘adgenet’ package (Jombart and Ahmed, 2011). LD was estimated on a dataset of biallelic SNP loci and also on a more stringently filtered dataset with at least 25% frequency of each of the two alleles. For each pair of loci, the normalized coefficient of LD (D′) and the squared correlation coefficient (r 2) were calculated using ‘vcftools’ (Danecek et al., 2011). To investigate LD decay, D′ and r 2 of loci within 2000 nucleotides from each other were plotted as a function of distance (sliding arithmetic means of all D′ or r 2 per each three nucleotide windows were used to reduce noise) using ‘ggplot2’ in R (Wickham, 2009; R Development Core Team, 2017). The LD decay range was determined as the interval outside which all of the arithmetic means of D′ or r 2 were either higher (left interval border) or lower (right interval border) than half of the maximum observed D′ or r 2 means.
Phylogenetic analysis
Gene phylogenetic trees were constructed from the predicted coding sequences of all of the A. pullulans genomes sequenced here, except for 21, 32 and 41, using coding sequences from A. melanogenum, A. subglaciale and A. namibiae as an outgroup. First, all predicted proteins were clustered using the GET_HOMOLOGUES software (Vinuesa and Contreras‐Moreira, 2015) into homologous groups. Clusters containing exactly one protein from each genome and recognized by all three of the clustering algorithms (bidirectional best hits, COGtriangle, OrthoMCL), were used in the consequent analysis. Corresponding coding sequences of proteins from each of the resulting clusters were aligned using MAFFT 7.215, with the ‘‐‐auto’ option and default parameters (Katoh and Toh, 2008). This alignment was optimized using Gblocks 0.91, with the options ‘‐b3=10 ‐b4=3 ‐b5=n’ (Talavera and Castresana, 2007); if this was longer than 200 nucleotides and contained at least 15 nucleotide differences (average per cluster alignment) between the gene pairs, this was used for reconstruction of the phylogeny with PhyML 3.1 (Guindon et al., 2010). The Hasegawa‐Kishino‐Yano 85 (Hasegawa et al., 1985) nucleotide substitution model was used, with the alpha parameter of the gamma distribution of substitution rate categories and the proportion of invariable sites estimated using PhyML. The resulting trees were visualized using DensiTree 2.2.5 (Bouckaert, 2010). A majority rule consensus tree was calculated with the ‘consensus.edges’ function of the package ‘phytools’ in R, using the default parameters (Revell, 2012; R Development Core Team, 2017).
Phylogenies were also estimated for all of the BUSCOs that were identified as complete and single‐copy proteins for all of the genomes investigated. Coding nucleotide sequences were recovered from the annotated genomes and automatically aligned using MAFFT (Katoh and Toh, 2008). These alignments were used for estimation of the phylogenies and a consensus phylogeny, using Gblocks, PhyML and phytools, as described previously.
The phylogenetic network was reconstructed from the SNP data. The dissimilarity distance matrix was calculated using the R package ‘poppr’ (Kamvar et al., 2015), and was used to construct the phylogenetic network with the Neighbor‐Net algorithm, as implemented in the R package ‘phangorn’ (R Development Core Team, 2017; Schliep et al., 2017).
Core genome, GO enrichment
The core genome of 50 here sequenced strains of A. pullulans and the reference strain of the species was estimated with the pipeline GET_HOMOLOGUES 3.0.8 (Vinuesa and Contreras‐Moreira, 2015) as a consensus of bidirectional best hit, COGtriangle and OrthoMCL algorithms using default parameters. Representative sequences of each cluster were annotated using the PANTHER HMM scoring tools 2.1 and the HMM library version 13.1 (Thomas et al., 2003). Statistically significant enrichment of GO‐Slim Biological Process terms was investigated at http://www.pantherdb.org for the lists of core gene clusters (present in all 51 genomes), soft core gene clusters (in at least 48 genomes) and cloud gene clusters (in only 1 or 2 genomes) with a list of all gene clusters used as a reference list. Fisher's exact test and the false discovery rate correction were used. If two or more genes from the same genome were placed in the same gene cluster, they were considered to be the product of gene duplication.
Mating type loci and other genes
Mating genes were identified by BLAST searches against the assembled and annotated A. pullulans genomes and predicted proteomes, using homologues from other dothideomycetous fungi as queries. Annotated genomes were used to identify the flanking genes. The functions of the predicted proteins were inferred by BLAST comparisons with the most similar proteins in the GenBank database.
Sequences of interest from public databases (with GenBank/ Mycocosm accession numbers: adenylation domain of siderophore synthetase from A. melanogenum – GenBank KM272191; pullulan synthase from A. pullulans – Mycocosm 349889; alkali metal cation transporters from Gostinčar et al. (2014)) were used as queries in a stand‐alone BLAST search for homologues in A. pullulans genomes and predicted proteomes. Identified sequences were aligned (together with the query sequences) with MAFFT 7.215, as described above (Katoh and Toh, 2008), and the alignment was used for the reconstruction of phylogeny with PhyML 3.1 (Guindon et al., 2010) as described above for the phylogenetic analysis of core genes, using the Hasegawa‐Kishino‐Yano 85 (Hasegawa et al., 1985) nucleotide substitution model for DNA sequences (siderophore synthases, alkali metal cation transporters) and LG (Le and Gascuel, 2008) amino acid replacement matrix for protein sequences (alkali metal cation transporters).
Supporting information
Supplemental Table S1. Statistics of the sequenced A. pullulans genomes.
Supplemental Table S2. Numbers of genes encoding alkali metal cation transporters in the sequenced A. pullulans genomes.
Supplemental Figure S1. Mapping of the sequencing reads from sequenced genomes 1‐54 to the reference A. pullulans genome EXF‐150. Here, 64 reference contigs longer than 10 kBp are plotted on the horizontal axes. Mapping depth is plotted on the vertical axes, with the maximum plotted value 500×. Possible aneuploidies are in red.
{kind=link}
Acknowledgements
The authors would like to thank Dr. Diego Libkind and Dr. Virginia de Garcia for providing the strains CRUB 1715 / EXF‐8828 and CRUB 1819 / EXF‐8841, Dr. Andrzej Chlebicki for the strain EXF‐6604, Dr. Pierre Amato for the strain EXF‐10751, and Dr. Agapi I. Dougeraki for strains EXF‐9398 and EXF‐9399. The authors acknowledge the financial support from the Slovenian Research Agency to the Infrastructural Centre Mycosmo (MRIC UL), to the programs P1‐0170 and P1‐0207, and to the Postdoctoral Project Z7‐7436 of J. Zajc. The authors would like to thank Chris Berrie for language editing assistance.
References
- Alkan, C. , Coe, B.P. , and Eichler, E.E. (2011) GATK toolkit. Nat Rev Genet 12: 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, J.H. , Spear, R.N. , and Nordheim, E.V. (2002) Population biology of Aureobasidium pullulans on apple leaf surfaces. Can J Microbiol 48: 500–513. [DOI] [PubMed] [Google Scholar]
- Ariño, J. , Ramos, J. , Sychrova, H. , Arino, J. , Ramos, J. , and Sychrova, H. (2010) Alkali metal cation transport and homeostasis in yeasts. Microbiol Mol Biol Rev 74: 95–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert, R.R. (2010) DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26: 1372–1373. [DOI] [PubMed] [Google Scholar]
- Branco, S. , Bi, K. , Liao, H.‐L. , Gladieux, P. , Badouin, H. , Ellison, C.E. , et al (2017) Continental‐level population differentiation and environmental adaptation in the mushroom Suillus brevipes . Mol Ecol 26: 2063–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco, S. , Gladieux, P. , Ellison, C.E. , Kuo, A. , LaButti, K. , Lipzen, A. , et al (2015) Genetic isolation between two recently diverged populations of a symbiotic fungus. Mol Ecol 24: 2747–2758. [DOI] [PubMed] [Google Scholar]
- Branda, E. , Turchetti, B. , Diolaiuti, G. , Pecci, M. , Smiraglia, C. , and Buzzini, P. (2010) Yeast and yeast‐like diversity in the southernmost glacier of Europe (Calderone Glacier, Apennines, Italy). FEMS Microbiol Ecol 72: 354–369. [DOI] [PubMed] [Google Scholar]
- Burt, A. , Carter, D.A. , Koenig, G.L. , White, T.J. , and Taylor, J.W. (1996) Molecular markers reveal cryptic sex in the human pathogen Coccidioides immitis . Proc Natl Acad Sci 93: 770–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butinar, L. , Spencer‐Martins, I. , and Gunde‐Cimerman, N. (2007) Yeasts in high Arctic glaciers: the discovery of a new habitat for eukaryotic microorganisms. Antonie Van Leeuwenhoek 91: 277–289. [DOI] [PubMed] [Google Scholar]
- Campbell, M.S. , Holt, C. , Moore, B. , and Yandell, M. (2014) Genome annotation and curation using MAKER and MAKER‐P. Curr Protoc Bioinforma 2014: 4.11.1–4.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappitelli, F. , and Sorlini, C. (2008) Microorganisms attack synthetic polymers in items representing our cultural heritage. Appl Environ Microbiol 74: 564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreté, L. , Ksiezopolska, E. , Pegueroles, C. , Gómez‐Molero, E. , Saus, E. , Iraola‐Guzmán, S. , et al (2018) Patterns of genomic variation in the opportunistic pathogen Candida glabrata suggest the existence of mating and a secondary association with humans. Curr Biol 28: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall, A. , and Pirofski, L.A. (2007) Accidental virulence, cryptic pathogenesis, Martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 6: 2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, K.C. , Demirci, A. , and Catchmark, J.M. (2011) Pullulan: biosynthesis, production, and applications. Appl Microbiol Biotechnol 92: 29–44. [DOI] [PubMed] [Google Scholar]
- Chi, Z.M. , Wang, F. , Yue, L.X. , Liu, G.L. , Zhang, T. , Chi, Z.M. , et al (2009) Bioproducts from Aureobasidium pullulans, a biotechnologically important yeast. Appl Microbiol Biotechnol 82: 793–804. [DOI] [PubMed] [Google Scholar]
- Cissé, O.H. , Ma, L. , Wei Huang, D. , Khil, P.P. , Dekker, J.P. , Kutty, G. , et al (2018) Comparative population genomics analysis of the mammalian fungal pathogen Pneumocystis . MBio 9: e00381–e00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras‐Moreira, B. , and Vinuesa, P. (2013) GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79: 7696–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , et al (2011) The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoog, G.S. , Guarro, J. , Gene, J. , and Figueras, M.J. (2001) Atlas of clinical fungi 2nd ed. Centraalbureau voor Schimmelcultures / Universitat Rovira i Virgili, Utrecht, The Netherlands / Reus, Spain. [Google Scholar]
- Desjardins, C.A. , Giamberardino, C. , Sykes, S.M. , Yu, C.‐H. , Tenor, J.L. , Chen, Y. , et al (2017) Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans . Genome Res 27: 1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami, M.K. , Hartwig, T. , Letten, A.D. , Banf, M. , and Fukami, T. (2018) Genomic diversity of a nectar yeast clusters into metabolically, but not geographically, distinct lineages. Mol Ecol 27: 2067–2076. [DOI] [PubMed] [Google Scholar]
- Douglass, A.P. , Offei, B. , Braun‐Galleani, S. , Coughlan, A.Y. , Martos, A.A.R. , Ortiz‐Merino, R.A. , et al (2018) Population genomics shows no distinction between pathogenic Candida krusei and environmental Pichia kudriavzevii: One species, four names. PLOS Pathog 14: e1007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, S.‐F. , Han, P.‐J. , Wang, Q.‐M. , Liu, W.‐Q. , Shi, J.‐Y. , Li, K. , et al (2018) The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat Commun 9: 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison, C.E. , Hall, C. , Kowbel, D. , Welch, J. , Brem, R.B. , Glass, N.L. , and Taylor, J.W. (2011) Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc Natl Acad Sci U S A 108: 2831–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , and Pritchard, J.K. (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Francesco, A. , Ugolini, L. , D'Aquino, S. , Pagnotta, E. , and Mari, M. (2017) Biocontrol of Monilinia laxa by Aureobasidium pullulans strains: Insights on competition for nutrients and space. Int J Food Microbiol 248: 32–38. [DOI] [PubMed] [Google Scholar]
- Geib, S.M. , Hall, B. , Derego, T. , Bremer, F.T. , Cannoles, K. , and Sim, S.B. (2018) Genome Annotation Generator: a simple tool for generating and correcting WGS annotation tables for NCBI submission. Gigascience 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser, D.M. , Pitt, J.I. , and Taylor, J.W. (1998) Cryptic speciation and recombination in the aflatoxin‐producing fungus Aspergillus flavus . Proc Natl Acad Sci 95: 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostinčar, C. , Grube, M. , and Gunde‐Cimerman, N. (2011) Evolution of fungal pathogens in domestic environments? Fungal Biol 115: 1008–1018. [DOI] [PubMed] [Google Scholar]
- Gostinčar, C. , Grube, M. , De Hoog, S. , Zalar, P. , and Gunde‐Cimerman, N. (2010) Extremotolerance in fungi: evolution on the edge. FEMS Microbiol Ecol 71: 2–11. [DOI] [PubMed] [Google Scholar]
- Gostinčar, C. , Gunde‐Cimerman, N. , and Grube, M. (2015) Polyextremotolerance as the fungal answer to changing environments In Microbial evolution under extreme conditions, Bakermans C. (ed). Berlin: de Gruyter, pp. 185–208. [Google Scholar]
- Gostinčar, C. , Muggia, L. , Grube, M. , Gostinĉar, C. , Muggia, L. , and Grube, M. (2012) Polyextremotolerant black fungi: oligotrophism, adaptive potential, and a link to lichen symbioses. Front Microbiol 3: 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostinčar, C. , Ohm, R.A. , Kogej, T. , Sonjak, S. , Turk, M. , Zajc, J. , et al (2014) Genome sequencing of four Aureobasidium pullulans varieties: biotechnological potential, stress tolerance, and description of new species. BMC Genomics 15: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostinčar, C. , Stajich, J.E. , Zupančič, J. , Zalar, P. , and Gunde‐Cimerman, N. (2018) Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genomics 19: 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube, M. , Schmid, F. , and Berg, G. (2011) Black fungi and associated bacterial communities in the phyllosphere of grapevine. Fungal Biol 115: 978–986. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.F. , Lefort, V. , Anisimova, M. , Hordijk, W. , and Gascuel, O. (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321. [DOI] [PubMed] [Google Scholar]
- Gunde‐Cimerman, N. , Zalar, P. , de Hoog, S. , and Plemenitaš, A. (2000) Hypersaline waters in salterns ‐ natural ecological niches for halophilic black yeasts. FEMS Microbiol Ecol 32: 235–240. [DOI] [PubMed] [Google Scholar]
- Hasegawa, M. , Kishino, H. , and Yano, T.a. (1985) Dating of the human‐ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174. [DOI] [PubMed] [Google Scholar]
- Johnson, K.B. , and Temple, T.N. (2013) Evaluation of strategies for fire blight control in organic Pome fruit without antibiotics. Plant Dis 97: 402–409. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , and Ahmed, I. (2011) adegenet 1.3‐1: new tools for the analysis of genome‐wide SNP data. Bioinformatics 27: 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarakainen, P. , Rintala, H. , Vepsalainen, A. , Hyvarinen, A. , Nevalainen, A. , and Meklin, T. (2009) Microbial content of house dust samples determined with qPCR. Sci Total Environ 407: 4673–4680. [DOI] [PubMed] [Google Scholar]
- Kamvar, Z.N. , Brooks, J.C. , and Grünwald, N.J. (2015) Novel R tools for analysis of genome‐wide population genetic data with emphasis on clonality. Front Genet 6: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Toh, H. (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9: 286–298. [DOI] [PubMed] [Google Scholar]
- Klein, M.N. , and Kupper, K.C. (2018) Biofilm production by Aureobasidium pullulans improves biocontrol against sour rot in citrus. Food Microbiol 69: 1–10. [DOI] [PubMed] [Google Scholar]
- Koenig, R.L. , Ploetz, R.C. , and Kistler, H.C. (1997) Fusarium oxysporum f. sp. cubense consists of a small number of divergent and globally distributed clonal lineages. Phytopathology 87: 915–923. [DOI] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics 5: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, S.Q. , and Gascuel, O. (2008) An improved general amino acid replacement matrix. Mol Biol Evol. 55: 539–552. [DOI] [PubMed] [Google Scholar]
- Leathers, T.D. (2003) Biotechnological production and applications of pullulan. Appl Microbiol Biotechnol 62: 468–473. [DOI] [PubMed] [Google Scholar]
- Lenassi, M. , Gostinčar, C. , Jackman, S. , Turk, M. , Sadowski, I. , Nislow, C. , et al (2013) Whole genome duplication and enrichment of metal cation transporters revealed by de novo genome sequencing of extremely halotolerant black yeast Hortaea werneckii . PLoS One 8: e71328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , et al (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoBuglio, K.F. , and Taylor, J.W. (2002) Recombination and genetic differentiation in the mycorrhizal fungus Cenococcum geophilum Fr. Mycologia 94: 772–780. [PubMed] [Google Scholar]
- Lomsadze, A. , Burns, P.D. , and Borodovsky, M. (2014) Integration of mapped RNA‐Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42: e119–e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, J.L. , Sychrova, H. , and Ramos, J. (2011) Monovalent cations regulate expression and activity of the Hak1 potassium transporter in Debaryomyces hansenii . Fungal Genet Biol 48: 177–184. [DOI] [PubMed] [Google Scholar]
- Molnárová, J. , Vadkertiová, R. , and Stratilová, E. (2014) Extracellular enzymatic activities and physiological profiles of yeasts colonizing fruit trees. J Basic Microbiol 54: S74–S84. [DOI] [PubMed] [Google Scholar]
- Moore, G.G. (2014) Sex and recombination in aflatoxigenic Aspergilli: global implications. Front Microbiol 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis, B.P.S. , and James, T.Y. (2016) The frequency of sex in fungi. Philos Trans R Soc London B Biol Sci 371: 20150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisiotou, A.A. , Chorianopoulos, N. , Nychas, G.J.E. , and Panagou, E.Z. (2010) Yeast heterogeneity during spontaneous fermentation of black Conservolea olives in different brine solutions. J Appl Microbiol 108: 396–405. [DOI] [PubMed] [Google Scholar]
- Onofri, S. , Seltimann, L. , de Hoog, G.S. , Grube, M. , Barreca, D. , Ruisi, S. , and Zucconi, L. (2007) Evolution and adaptation of fungi at boundaries of life. Adv Sp Res 40: 1657–1664. [Google Scholar]
- Oren, A. , and Gunde‐Cimerman, N. (2012) Fungal life in the dead sea In Biology of Marine Fungi, Raghukumar C. (ed). Berlin, Heidelberg: Springer, pp. 115–132. [DOI] [PubMed] [Google Scholar]
- Otto, S.P. (2009) The Evolutionary Enigma of Sex. Am Nat 174: S1–S14. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Leung, H.C.M. , Yiu, S.M. , and Chin, F.Y.L. (2012) IDBA‐UD: a de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Peter, J. , De Chiara, M. , Friedrich, A. , Yue, J.‐X. , Pflieger, D. , Bergström, A. , et al (2018) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, J.I. , and Hocking, A.D. (1999) Fungi and food spoilage, 2nd ed. Dordrecht, London: Aspen Publishers, Inc. [Google Scholar]
- Plemenitaš, A. , Konte, T. , Gostinčar, C. , Cimerman, N.G. , and Gunde‐Cimerman, N. (2016) Transport Systems in Halophilic Fungi. Adv Exp Med Biol 892: 307–325. [DOI] [PubMed] [Google Scholar]
- Plissonneau, C. , Benevenuto, J. , Mohd‐Assaad, N. , Fouché, S. , Hartmann, F.E. , and Croll, D. (2017) Using population and comparative genomics to understand the genetic basis of effector‐driven fungal pathogen evolution. Front Plant Sci 8, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomraning, K.R. , Smith, K.M. , and Freitag, M. (2011) Bulk segregant analysis followed by high‐throughput sequencing reveals the Neurospora cell cycle gene, ndc‐1, to be allelic with the gene for ornithine decarboxylase, spe‐1. Eukaryot Cell 10: 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasongsuk, S. , Lotrakul, P. , Ali, I. , Bankeeree, W. , and Punnapayak, H. (2018) The current status of Aureobasidium pullulans in biotechnology. Folia Microbiol (Praha) 63: 129–140. [DOI] [PubMed] [Google Scholar]
- Price, N.P.J. , Manitchotpisit, P. , Vermillion, K.E. , Bowman, M.J. , and Leathers, T.D. (2013) Structural characterization of novel extracellular liamocins (mannitol oils) produced by Aureobasidium pullulans strain NRRL 50380. Carbohydr Res 370: 24–32. [DOI] [PubMed] [Google Scholar]
- Pritchard, J.K. , Stephens, M. , and Donnelly, P. (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2017) R: a language and environment for statistical computing.
- Ramos, J. , Arino, J. , and Sychrova, H. (2011) Alkali‐metal‐cation influx and efflux systems in nonconventional yeast species. FEMS Microbiol Lett 317: 1–8. [DOI] [PubMed] [Google Scholar]
- Rauch, M.E. , Graef, H.W. , Rozenzhak, S.M. , Jones, S.E. , Bleckmann, C.A. , Kruger, R.L. , et al (2006) Characterization of microbial contamination in United States Air Force aviation fuel tanks. J Ind Microbiol Biotechnol 33: 29–36. [DOI] [PubMed] [Google Scholar]
- Revell, L.J. (2012) phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3: 217–223. [Google Scholar]
- Samson, R.a. , Houbraken, J. , Thrane, U. , Frisvad, J.C. , and Andersen, B. (2010) Food and Indoor Fungi CBS‐KNAW Fungal Biodiversity Centre. The Netherlands: Utrecht. [Google Scholar]
- Savary, R. , Masclaux, F.G. , Wyss, T. , Droh, G. , Cruz Corella, J. , Machado, A.P. , et al (2018) A population genomics approach shows widespread geographical distribution of cryptic genomic forms of the symbiotic fungus Rhizophagus irregularis . ISME J 12: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep, K. , Potts, A.J. , Morrison, D.A. , and Grimm, G.W. (2017) Intertwining phylogenetic trees and networks. Methods Ecol Evol 8: 1212–1220. [Google Scholar]
- Shabtai, Y. , and Mukmenev, I. (1995) Enhanced production of pigment‐free pullulan by a morphogenetically arrested Aureobasidium pullulans (ATCC 42023) in a two‐stage fermentation with shift from soy bean oil to sucrose. Appl Microbiol Biotechnol 43: 595–603. [Google Scholar]
- Silva, D.N. , Várzea, V. , Paulo, O.S. , and Batista, D. (2018) Population genomic footprints of host adaptation, introgression and recombination in coffee leaf rust. Mol Plant Pathol 19: 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. , and Zdobnov, E.M. (2015) BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Slepecky, R.A. , and Starmer, W.T. (2009) Phenotypic plasticity in fungi: a review with observations on Aureobasidium pullulans . Mycologia 101: 823–832. [DOI] [PubMed] [Google Scholar]
- Spadaro, D. , and Droby, S. (2016) Development of biocontrol products for postharvest diseases of fruit: The importance of elucidating the mechanisms of action of yeast antagonists. Trends Food Sci Technol 47: 39–49. [Google Scholar]
- Stanke, M. , and Morgenstern, B. (2005) AUGUSTUS: a web server for gene prediction in eukaryotes that allows user‐defined constraints. Nucleic Acids Res 33: 465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takesako, K. , Kuroda, H. , Inoue, T. , Haruna, F. , Yoshikawa, Y. , Kato, I. , et al (1993) Biological properties of aureobasidin A, a cyclic depsipeptide antifungal antibiotic. J Antibiot (Tokyo) 46: 1414–1420. [DOI] [PubMed] [Google Scholar]
- Talavera, G. , and Castresana, J. (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56: 564–577. [DOI] [PubMed] [Google Scholar]
- Taylor, J.W. , Hann‐Soden, C. , Branco, S. , Sylvain, I. , and Ellison, C.E. (2015) Clonal reproduction in fungi. Proc Natl Acad Sci 112: 8901–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter‐Hovhannisyan, V. , Lomsadze, A. , Chernoff, Y.O. , and Borodovsky, M. (2008) Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18: 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, P.D. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc, M. , and Ayala, F.J. (2012) Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc Natl Acad Sci 109: 3305–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa, P. , and Contreras‐Moreira, B. (2015) Robust identification of orthologues and paralogues for microbial pan‐genomics using GET_HOMOLOGUES: a case study of pIncA/C plasmids. Methods Mol Biol. 1231: 203–232. [DOI] [PubMed] [Google Scholar]
- Wang, W.L. , Chi, Z.M. , Chi, Z. , Li, J. , and Wang, X.H. (2009) Siderophore production by the marine‐derived Aureobasidium pullulans and its antimicrobial activity. Bioresour Technol 100: 2639–2641. [DOI] [PubMed] [Google Scholar]
- Waterhouse, R.M. , Tegenfeldt, F. , Li, J. , Zdobnov, E.M. , and Kriventseva, E.V. (2013) OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res 41: 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2009) ggplot2. New York, NY: Springer. [Google Scholar]
- Zalar, P. , Gostinčar, C. , de Hoog, G.S. , Uršič, V. , Sudhadham, M. , and Gunde‐Cimerman, N. (2008) Redefinition of Aureobasidium pullulans and its varieties. Stud Mycol 61: 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalar, P. , Novak, M. , De Hoog, G.S. , and Gunde‐Cimerman, N. (2011) Dishwashers ‐ A man‐made ecological niche accommodating human opportunistic fungal pathogens. Fungal Biol 115: 997–1007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Statistics of the sequenced A. pullulans genomes.
Supplemental Table S2. Numbers of genes encoding alkali metal cation transporters in the sequenced A. pullulans genomes.
Supplemental Figure S1. Mapping of the sequencing reads from sequenced genomes 1‐54 to the reference A. pullulans genome EXF‐150. Here, 64 reference contigs longer than 10 kBp are plotted on the horizontal axes. Mapping depth is plotted on the vertical axes, with the maximum plotted value 500×. Possible aneuploidies are in red.