

Abstract
BACKGROUND
Strigolactones (SLs) have a vast number of ecological implications because of the broad spectrum of their biological activities. Unfortunately, the limited availability of SLs restricts their applicability for the benefit of humanity and renders synthesis the only option for their production. However, the structural complexity of SLs impedes their economical synthesis, which is unfeasible on a large scale. Synthesis of SL analogues and mimics with a simpler structure, but with retention of bioactivity, is the solution to this problem.
RESULTS
Here, we present eight new hybrid‐type SL analogues derived from auxin, synthesized via coupling of auxin ester [ethyl 2‐(1H‐indol‐3‐yl)acetate] and of ethyl 2‐phenylacetate with four D‐rings (mono‐, two di‐ and trimethylated). The new hybrid‐type SL analogues were bioassayed to assess the germination activity of seeds of the parasitic weeds Striga hermonthica, Orobanche minor and Phelipanche ramosa using the classical method of counting germinated seeds and a colorimetric method. The bioassays revealed that analogues with a natural monomethylated D‐ring had appreciable to good activity towards the three species and were the most active derivatives. By contrast, derivatives with the trimethylated D‐ring showed no activity. The dimethylated derivatives (2,4‐dimethyl and 3,4‐dimethyl) were slightly active, especially towards P. ramosa.
CONCLUSIONS
New hybrid‐type analogues derived from auxins have been prepared. These analogues may be attractive as potential suicidal germination agents for parasitic weed control because of their ease of preparation and relevant bioactivity. © 2019 The Authors. Pest Management Science published by John Wiley & Sons Ltd on behalf of Society of Chemical Industry.
Keywords: strigolactones, auxins, strigolactone analogues, suicidal germination
Hybrid‐type strigolactone (SL) analogues were prepared from auxin ethyl ester in a two‐step operation. These SL analogues were bioassayed as germination agents for Striga hermonthica, Pelipanche ramosa and Orobanche minor.
1. INTRODUCTION
Strigolactones (SLs) constitute a group of new plant hormones that have received much interest in current plant biology.1, 2, 3, 4, 5, 6 SLs are present in many plants, particularly in root exudates.7 The first SL, strigol, was isolated in 1966 from the root exudate of cotton.8, 9, 10 Interestingly, strigol is an active germination stimulant for seeds of the parasitic weeds Striga, Orobanche and Phelipanche spp.11, 12 SLs invariably contain three annulated rings as the basic scaffold (the ABC ring system) connected to a butenolide (furanone; D‐ring) via an enol ether unit.2, 13 Most members of the SL family have been discovered since 1990. Elucidation of their structures was not easy and, as a consequence, several incorrect assignments appeared in the literature and were later corrected, for example (−)‐orobanchol,14, 15 alectrol14 and solanacol.16 At present, two families of SLs are known: one having stereochemistry as in (+)‐strigol and the other with stereochemistry as in (−)‐orobanchol (Fig. 1).2
Figure 1.
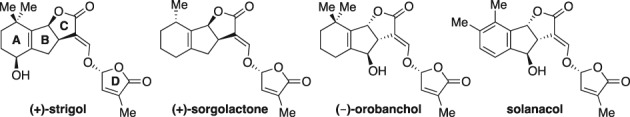
Naturally occurring strigolactones.
New bio‐properties, such as a branching factor for arbuscular mycorrhizal fungi17 and the inhibition of bud outgrowth and shoot branching18, 19, 20, 21 were discovered at the beginning of this century and led to enormous interest in SLs. These findings led to their classification as a new class of plant hormones. Several reviews cover the current state of affairs concerning the chemistry and bio‐properties of SLs.2, 3, 4, 5, 6, 22, 23, 24
The availability of SLs is limited because levels of natural production are minute and their complete synthesis is quite laborious due to their complex structures.25, 26 Synthesis of simple bioactive analogues, having the same bioactiphore as natural SLs, has attracted considerable attention in recent years (Fig. 2). The ‘GR compounds’, such as GR24 and GR7, were the first series of such SL analogues.27 We have recently reported the use of some SL analogues in combating Striga hermonthica infestation in millet in sub‐Saharan Africa.28
Figure 2.
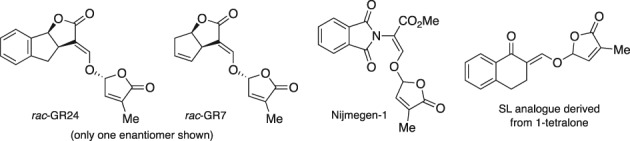
Important strigolactone analogues.
A model containing the features essential for activity was used to construct a large series of new analogues with excellent biological performance (Fig. 3).2, 29 Typical examples are Nijmegen‐130 and analogues derived from simple ketones and keto enols (Fig. 2).31, 32
Figure 3.
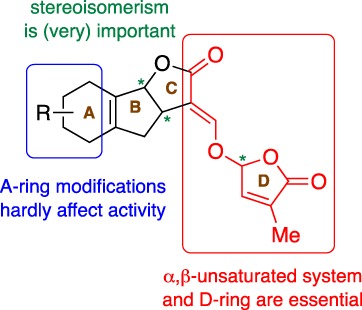
Working model for designing strigolactone analogues.
There is abundant evidence that detachment of the hydroxy D‐ring from SLs is the key step in triggering the germination of seeds of parasitic weeds.5, 33, 34 The part of the SL molecule that remains has, to our knowledge, no further function. Here, however, we construct an SL analogue having an ‘ABC’ scaffold that has a relevant bioactivity after detachment from the D‐ring. We call such compounds hybrid‐type SL analogues. Here, we use auxins, which are well‐known and widely used plant growth regulators, as functional ‘ABC’ scaffolds.35 Further evaluation to examine whether these SL analogues exhibit auxin‐like activity will form part of further research. Recently, Pereira et al.36 described the preparation of hybrid SL mimics derived from gibberellins, and we recently reported the synthesis of hybrid SL mimics from auxins.37 However, hybrid analogues having the same bioactiphore as natural SLs are new.
An additional incentive of the work described here is the preparation of a bioactive SL analogue from a readily available starting material in a simple synthetic operation.
2. MATERIALS AND METHODS
2.1. Synthesis
Reagents were obtained from commercial suppliers and were used without purification, except for ethyl formate which was distilled before use. Standard syringe techniques were used to transfer dry solvents and air‐ or moisture‐sensitive reagents. Reactions were followed, and R F values were obtained using thin‐layer chromatography (TLC) on silica gel‐coated plates (Merck 60 F254) with the indicated solvent mixture. Detection was performed under ultraviolet (UV) light and by charring at ∼ 150 °C after dipping into a solution of either 2% anisaldehyde in ethanol/H2SO4, KMnO4 or ninhydrin. Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded at 298 K on a Varian Inova 400 (400 MHz), Bruker Avance III 400 MHz or Bruker Avance III 500 MHz spectrometer in the solvent indicated. Chemical shifts are given in parts per million (ppm) with respect to tetramethylsilane (0.00 ppm) as the internal standard for 1H NMR and CDCl3 (77.16 ppm) as the internal standard for 13C NMR. Coupling constants are reported as J values in hertz (Hz). High‐resolution mass spectra (HRMS) were recorded with JEOL AccuTOF mass spectrometer. Column or flash chromatography was carried out using ACROS silica gel (0.035–0.070 mm and 60 Å pore diameter).
2.1.1. 5‐Hydroxy‐3‐methylfuran‐2(5H)‐one ( 5 )
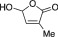 Method 1: Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and a 40% aqueous solution of glyoxal (1.46 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 40% EtOAc in heptane) to yield 5 (630 mg, 65%) as a yellow solid.
Method 1: Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and a 40% aqueous solution of glyoxal (1.46 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 40% EtOAc in heptane) to yield 5 (630 mg, 65%) as a yellow solid.
Method 2: A 40% solution of glyoxal in water (14 mL, 121.0 mmol, 1.4 eq.) and 20 drops of concentrated H2SO4 were added to a solution of methylmalonic acid (10.0 g, 84.7 mmol, 1 eq.) in water (100 mL). The mixture was heated under reflux (100 °C) for 18 h. The solution was then allowed to cool to 20 °C and was saturated with solid NaCl (40 g). The solution was extracted with EtOAc (3 × 60 mL). The combined organic extracts were dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 40% EtOAc in heptane) to yield 5 (4.17 g, 43%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 6.88 (p, J = 1.6 Hz, 1 H), 6.08 (s, 1 H), 3.23 (br s, 1 H), 1.96 (t, J = 1.6 Hz, 3 H).
2.1.2. 5‐Hydroxy‐3,5‐dimethylfuran‐2(5H)‐one ( 6 )
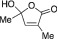 Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and a 40% aqueous solution of pyruvaldehyde (0.8 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 40% EtOAc in heptane) to yield 6 (605 mg, 55%) as a yellow solid. This solid was further purified by recrystallization from DCM/heptane (1:1) to yield 6 as white needles. 1H NMR (400 MHz, CDCl3) δ 6.86 (q, J = 1.7 Hz, 1 H), 3.04 (br s, 1 H), 1.92 (d, J = 1.7 Hz, 3 H), 1.69 (s, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.4, 147.4, 131.8, 104.0, 24.8, 10.4.
Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and a 40% aqueous solution of pyruvaldehyde (0.8 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 40% EtOAc in heptane) to yield 6 (605 mg, 55%) as a yellow solid. This solid was further purified by recrystallization from DCM/heptane (1:1) to yield 6 as white needles. 1H NMR (400 MHz, CDCl3) δ 6.86 (q, J = 1.7 Hz, 1 H), 3.04 (br s, 1 H), 1.92 (d, J = 1.7 Hz, 3 H), 1.69 (s, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.4, 147.4, 131.8, 104.0, 24.8, 10.4.
2.1.3. 5‐Hydroxy‐3,4‐dimethylfuran‐2(5H)‐one ( 7 )
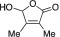 A solution of 2,3‐dimethylmaleic anhydride (1.0 g, 7.9 mmol, 1 eq.) in dry tetrahydrofuran (THF; 25 mL) was stirred and cooled to −15 °C. To this solution, a solution of lithium hydrido[tris(2‐methylpropan‐2‐olato)]aluminate (Li[AlH(OtBu)3]; 2.8 g, 10.9 mmol, 1.4 eq.) in dry THF (15 mL) was added dropwise over 10 min. The mixture was stirred at −15 °C for 1 h, and then at 20 °C for 1 h. The reaction was quenched by adding 2 m aqueous HCl (40 mL), and then extracted with EtOAc (3 × 40 mL). The combined organic extracts were washed with brine, dried over MgSO4, and concentrated in vacuo to yield 7 (560 mg, 55%) as a white solid that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 5.87 (s, 1 H), 3.57 (br s, 1 H), 2.01 (p, J = 1.1 Hz, 3 H), 1.84 (p, J = 1.2 Hz, 3 H).
A solution of 2,3‐dimethylmaleic anhydride (1.0 g, 7.9 mmol, 1 eq.) in dry tetrahydrofuran (THF; 25 mL) was stirred and cooled to −15 °C. To this solution, a solution of lithium hydrido[tris(2‐methylpropan‐2‐olato)]aluminate (Li[AlH(OtBu)3]; 2.8 g, 10.9 mmol, 1.4 eq.) in dry THF (15 mL) was added dropwise over 10 min. The mixture was stirred at −15 °C for 1 h, and then at 20 °C for 1 h. The reaction was quenched by adding 2 m aqueous HCl (40 mL), and then extracted with EtOAc (3 × 40 mL). The combined organic extracts were washed with brine, dried over MgSO4, and concentrated in vacuo to yield 7 (560 mg, 55%) as a white solid that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 5.87 (s, 1 H), 3.57 (br s, 1 H), 2.01 (p, J = 1.1 Hz, 3 H), 1.84 (p, J = 1.2 Hz, 3 H).
2.1.4. 5‐Hydroxy‐3,4,5‐trimethylfuran‐2(5H)‐one ( 8 )
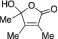 Method 1: Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and butane‐2,3‐dione (1.1 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 30% EtOAc in heptane) to yield 8 (545 mg, 46%) as a yellow solid.
Method 1: Methylmalonic acid (1.0 g, 8.47 mmol, 1 eq.) and phenylboronic acid (1.1 g, 8.89 mmol, 1.05 eq.) were dissolved in water (10 mL) and butane‐2,3‐dione (1.1 mL, 12.7 mmol, 1.5 eq.) was added. The mixture was stirred and heated under reflux (100 °C) for 16 h. The mixture was then allowed to cool to 20 °C, and was saturated with solid NaCl. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 30% EtOAc in heptane) to yield 8 (545 mg, 46%) as a yellow solid.
Method 2: A solution of 2,3‐dimethylmaleic anhydride (1.0 g, 7.9 mmol, 1 eq.) in dry THF (50 mL) was stirred and cooled to −78 °C under a nitrogen atmosphere. A 1.6 m solution of methyllithium in Et2O (5.0 mL, 8.0 mmol, 1 eq.) was added dropwise over 15 min and the resulting mixture was stirred at −78 °C for 15 min. The reaction was quenched by adding a saturated aqueous solution of ammonium chloride (NH4Cl) (20 mL), and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was subjected to flash column chromatography (silica, 30% EtOAc in heptane) to yield 8 (375 mg, 33%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 4.55 (br s, 1 H), 1.97 (q, J = 1.1 Hz, 3 H), 1.78 (q, J = 1.1 Hz, 3 H), 1.62 (s, 3 H).
2.1.4.1. General procedure for the synthesis of chloro furanones ( 9 – 12 )
A solution of thionyl chloride (6 mmol, 1.5 eq.) and one drop of N,N‐dimethylformamide (DMF) in CH2Cl2 (3 mL) was stirred and heated under reflux (40 °C). Hydroxy furanone (5–8, 4 mmol, 1 eq.) was dissolved in CH2Cl2 (3 mL) and then added dropwise to the refluxing solution. The resulting mixture was refluxed for 2 h. After the mixture was cooled to 20 °C, the mixture was diluted with CH2Cl2 (20 mL) and then poured into a cooled (0 °C) solution of saturated aqueous NaHCO3 (25 mL) and stirred until gas evolution had stopped. The aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo to give the crude product as a pale yellow oil. The crude product was distilled using a Kugelrohr apparatus (18 mbar, 150 °C) to give the pure chloro furanone (9–12) as a colourless liquid.
2.1.5. 5‐Chloro‐3‐methylfuran‐2(5H)‐one ( 9 )
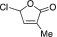 Yield: 349 mg (48%). 1H NMR (400 MHz, CDCl3) δ 7.08 (p, J = 1.6 Hz, 1 H), 6.54 (p, J = 1.5 Hz, 1 H), 2.01 (t, J = 1.6 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.8, 145.9, 131.9, 85.2, 10.5.
Yield: 349 mg (48%). 1H NMR (400 MHz, CDCl3) δ 7.08 (p, J = 1.6 Hz, 1 H), 6.54 (p, J = 1.5 Hz, 1 H), 2.01 (t, J = 1.6 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.8, 145.9, 131.9, 85.2, 10.5.
Method 2: In a flame‐dried flask under nitrogen atmosphere, CCl4 (26.9 g, 16.9 mL, 175.1 mmol, 2.0 eq.) and triphenylphosphane (Ph3P; 34.5 g, 131.5 mmol, 1.5 eq.) were added sequentially to a solution of 5 (10.0 g, 87.6 mmol, 1.0 eq.) in dry THF (300 mL). The reaction was stirred at 60 °C for 3 h. The reaction mixture was then cooled to 23 °C and concentrated in vacuo. The residue was stirred in Et2O/pentane (1:1; 250 mL) until triphenylphosphane oxide (Ph3PO) precipitated. The suspension was filtered; the filtrate was then flushed through a pad of silica gel and concentrated in vacuo to yield 9 (9.55 g, 82%) as a slightly yellow oil.
2.1.6. 5‐Chloro‐3,5‐dimethylfuran‐2(5H)‐one ( 10 )
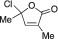 Yield: 572 mg (80%). 1H NMR (500 MHz, CDCl3) δ 7.11 (q, J = 1.7 Hz, 1 H), 2.02 (s, 3 H), 1.98 (d, J = 1.7 Hz, 3 H). 13C NMR (126 MHz, CDCl3) δ 170.5, 150.1, 129.6, 97.9, 29.3, 10.2.
Yield: 572 mg (80%). 1H NMR (500 MHz, CDCl3) δ 7.11 (q, J = 1.7 Hz, 1 H), 2.02 (s, 3 H), 1.98 (d, J = 1.7 Hz, 3 H). 13C NMR (126 MHz, CDCl3) δ 170.5, 150.1, 129.6, 97.9, 29.3, 10.2.
2.1.7. 5‐Chloro‐3,4‐dimethylfuran‐2(5H)‐one ( 11 )
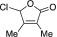 Yield: 590 mg (65%). 1H NMR (400 MHz, CDCl3) δ 6.39–6.35 (m, 1 H), 2.08 (p, J = 1.1 Hz, 3 H), 1.89 (p, J = 1.2 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.1, 156.8, 125.7, 88.2, 11.8, 8.6.
Yield: 590 mg (65%). 1H NMR (400 MHz, CDCl3) δ 6.39–6.35 (m, 1 H), 2.08 (p, J = 1.1 Hz, 3 H), 1.89 (p, J = 1.2 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.1, 156.8, 125.7, 88.2, 11.8, 8.6.
2.1.8. 5‐Chloro‐3,4,5‐trimethylfuran‐2(5H)‐one ( 12 )
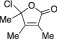 Yield: 328 mg (94%). 1H NMR (400 MHz, CDCl3) δ 2.08 (q, J = 1.1 Hz, 3 H), 1.94 (s, 3 H), 1.86 (q, J = 1.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.4, 160.0, 123.8, 100.4, 28.0, 10.8, 8.6.
Yield: 328 mg (94%). 1H NMR (400 MHz, CDCl3) δ 2.08 (q, J = 1.1 Hz, 3 H), 1.94 (s, 3 H), 1.86 (q, J = 1.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.4, 160.0, 123.8, 100.4, 28.0, 10.8, 8.6.
2.1.9. Ethyl 3‐oxo‐2‐phenylpropanoate ( 16a ) and ethyl (Z)‐3‐hydroxy‐2‐phenylacrylate ( 16b )
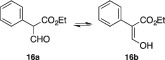 Sodium hydride (2.0 g, 48.7 mmol, 4 eq.; 60% dispersion in mineral oil) was washed with pentane (3 × 10 mL) under a nitrogen atmosphere. Dry THF (40 mL) was added and the mixture was stirred and cooled to 0 °C. Ethyl phenylacetate (2.0 g, 12.2 mmol, 1 eq.) was dissolved in ethyl formate (5 mL) and added dropwise to the stirred solution. The reaction mixture was stirred at 20 °C for 24 h in total. Additional amounts of ethyl formate (2.5 mL per time) were added after 6 and 16 h. The reaction was quenched by adding ethanol (EtOH; 15 mL) slowly, followed by the addition of a 50% (v/v) aqueous solution of acetic acid (AcOH; 20 mL). Diethyl ether was added until the aqueous and organic phases separated. The separated aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo to yield 16a/16b (2.27 g, 97%) as a yellow oil that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 12.12 (d, J = 12.6 Hz, 1 H), 7.30 (d, J = 12.6 Hz, 1 H), 7.37–7.23 (m, 5 H), 4.30 (q, J = 7.1 Hz, 2 H), 1.29 (t, J = 7.1 Hz, 3 H).
Sodium hydride (2.0 g, 48.7 mmol, 4 eq.; 60% dispersion in mineral oil) was washed with pentane (3 × 10 mL) under a nitrogen atmosphere. Dry THF (40 mL) was added and the mixture was stirred and cooled to 0 °C. Ethyl phenylacetate (2.0 g, 12.2 mmol, 1 eq.) was dissolved in ethyl formate (5 mL) and added dropwise to the stirred solution. The reaction mixture was stirred at 20 °C for 24 h in total. Additional amounts of ethyl formate (2.5 mL per time) were added after 6 and 16 h. The reaction was quenched by adding ethanol (EtOH; 15 mL) slowly, followed by the addition of a 50% (v/v) aqueous solution of acetic acid (AcOH; 20 mL). Diethyl ether was added until the aqueous and organic phases separated. The separated aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo to yield 16a/16b (2.27 g, 97%) as a yellow oil that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 12.12 (d, J = 12.6 Hz, 1 H), 7.30 (d, J = 12.6 Hz, 1 H), 7.37–7.23 (m, 5 H), 4.30 (q, J = 7.1 Hz, 2 H), 1.29 (t, J = 7.1 Hz, 3 H).
(Tautomers 16a and 16b are both observed in a 1:12 ratio in the 1H NMR spectrum. The reported signals belong to the major product 16b.)
2.1.10. Ethyl 2‐(1H‐indol‐3‐yl)‐3‐oxopropanoate ( 17a ) and ethyl (Z)‐3‐hydroxy‐2‐(1H‐indol‐3‐yl)acrylate ( 17b )
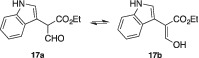 Sodium hydride (1.0 g, 25 mmol, 5 eq.; 60% dispersion in mineral oil) was washed with pentane (3 × 5 mL) under a nitrogen atmosphere. Dry THF (20 mL) was added and the mixture was stirred and cooled to 0 °C. Ethyl 2‐(1H‐indol‐3‐yl)acetate (1.0 g, 4.92 mmol, 1 eq.) was dissolved in ethyl formate (4 mL) and added dropwise to the stirred solution. The reaction mixture was stirred at 20 °C for 36 h in total. Additional amounts of ethyl formate (2 mL per time) were added after 16 and 24 h. The reaction was quenched by adding EtOH (15 mL) slowly, followed by the addition of a 50% (v/v) aqueous solution of AcOH (20 mL). Diethyl ether was added until the aqueous and organic phases separated. The separated aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic extracts were washed with a solution of saturated aqueous NaHCO3, dried over MgSO4 and concentrated in vacuo, to give 17a/17b (1.03 g, 90%) as a grey solid that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 12.13 (d, J = 12.6 Hz, 1 H), 8.12 (br s, 1 H), 7.58–7.54 (m, 1 H), 7.42 (d, J = 12.5 Hz, 1 H), 7.38 (dt, J = 8.1, 1.0 Hz, 1 H), 7.21 (ddd, J = 8.1, 7.1, 1.3 Hz, 1 H), 7.16–7.11 (m, 2 H), 4.29 (q, J = 7.1 Hz, 2 H), 1.26 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 172.3, 162.9, 135.9, 127.2, 123.2, 122.2, 119.8, 119. 8, 111.2, 109.5, 100.6, 60.8, 14.2.
Sodium hydride (1.0 g, 25 mmol, 5 eq.; 60% dispersion in mineral oil) was washed with pentane (3 × 5 mL) under a nitrogen atmosphere. Dry THF (20 mL) was added and the mixture was stirred and cooled to 0 °C. Ethyl 2‐(1H‐indol‐3‐yl)acetate (1.0 g, 4.92 mmol, 1 eq.) was dissolved in ethyl formate (4 mL) and added dropwise to the stirred solution. The reaction mixture was stirred at 20 °C for 36 h in total. Additional amounts of ethyl formate (2 mL per time) were added after 16 and 24 h. The reaction was quenched by adding EtOH (15 mL) slowly, followed by the addition of a 50% (v/v) aqueous solution of AcOH (20 mL). Diethyl ether was added until the aqueous and organic phases separated. The separated aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic extracts were washed with a solution of saturated aqueous NaHCO3, dried over MgSO4 and concentrated in vacuo, to give 17a/17b (1.03 g, 90%) as a grey solid that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 12.13 (d, J = 12.6 Hz, 1 H), 8.12 (br s, 1 H), 7.58–7.54 (m, 1 H), 7.42 (d, J = 12.5 Hz, 1 H), 7.38 (dt, J = 8.1, 1.0 Hz, 1 H), 7.21 (ddd, J = 8.1, 7.1, 1.3 Hz, 1 H), 7.16–7.11 (m, 2 H), 4.29 (q, J = 7.1 Hz, 2 H), 1.26 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 172.3, 162.9, 135.9, 127.2, 123.2, 122.2, 119.8, 119. 8, 111.2, 109.5, 100.6, 60.8, 14.2.
(Tautomers 17a and 17b are both observed in a 1:9 ratio in the 1H NMR spectrum. The reported signals belong to the major product 17b.)
2.1.10.1. General procedure for the synthesis of strigolactone hybrids 18 – 25
A flame‐dried Schlenk flask was loaded with hydroxymethylidene scaffold (16 or 17; 1 mmol, 1 eq.) and potassium carbonate (1.1 eq.), and was dried on a vacuum pump for 2 h. The flask was then filled with nitrogen and cooled on an ice bath (0 °C). Dry DMF (4 mL) was added and the mixture was stirred for 30 min on the ice bath. The stirred mixture was then cooled to −40 °C and a solution of chloro furanone (9–12, 1.2 eq.) in DMF (1 mL) was added dropwise to the stirred solution. The cooling bath was removed and the reaction mixture was stirred at 20 °C for 65 h. The reaction was quenched with water (5 mL), EtOAc (5 mL) was added and the organic phase was washed with ice‐cold brine (4 × 5 mL). The organic phase was separated and the combined washings were back extracted with EtOAc (10 mL). This extract was washed again with ice‐cold brine (4 × 5 mL). The organic extracts were combined, dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (silica, 30% EtOAc in heptane).
2.1.11. Ethyl (E)‐3‐[(4‐methyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐2‐ phenylacrylate ( 18 )
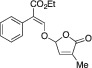 Yellow oil. Yield: 213 mg (71%). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1 H), 7.37–7.23 (m, 5 H), 6.81 (q, J = 1.6 Hz, 1 H), 6.10 (q, J = 1.4 Hz, 1 H), 4.22 (q, J = 7.1 Hz, 2 H), 1.95–1.91 (m, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.6, 166.8, 152.6, 141.5, 135.3, 131.8, 130.1, 127.8, 127.5, 115.8, 100.6, 60.8, 14.3, 10.6. HRMS [ESI+ (m/z)] calcd for (C16H16O5 + Na)+ 311.08954, found 311.08931.
Yellow oil. Yield: 213 mg (71%). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1 H), 7.37–7.23 (m, 5 H), 6.81 (q, J = 1.6 Hz, 1 H), 6.10 (q, J = 1.4 Hz, 1 H), 4.22 (q, J = 7.1 Hz, 2 H), 1.95–1.91 (m, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.6, 166.8, 152.6, 141.5, 135.3, 131.8, 130.1, 127.8, 127.5, 115.8, 100.6, 60.8, 14.3, 10.6. HRMS [ESI+ (m/z)] calcd for (C16H16O5 + Na)+ 311.08954, found 311.08931.
2.1.12. Ethyl (E)‐3‐[(2,4‐dimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐2‐phenylacrylate ( 19 )
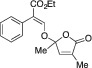 White solid. Yield: 206 mg (70%). 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1 H), 7.38–7.26 (m, 5 H), 6.83 (q, J = 1.7 Hz, 1 H), 4.21 (q, J = 7.1 Hz, 2 H), 2.00 (d, J = 1.7 Hz, 3 H), 1.72 (s, 3 H), 1.26 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.3, 166.9, 148.7, 145.9, 133.8, 132.0, 130.1, 127.7, 127.3, 115.7, 106.3, 60.7, 23.5, 14.3, 10.5. HRMS [ESI+ (m/z)] calcd for (C17H18O5 + Na)+ 325.10519, found 325.10484.
White solid. Yield: 206 mg (70%). 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1 H), 7.38–7.26 (m, 5 H), 6.83 (q, J = 1.7 Hz, 1 H), 4.21 (q, J = 7.1 Hz, 2 H), 2.00 (d, J = 1.7 Hz, 3 H), 1.72 (s, 3 H), 1.26 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.3, 166.9, 148.7, 145.9, 133.8, 132.0, 130.1, 127.7, 127.3, 115.7, 106.3, 60.7, 23.5, 14.3, 10.5. HRMS [ESI+ (m/z)] calcd for (C17H18O5 + Na)+ 325.10519, found 325.10484.
2.1.13. Ethyl (E)‐3‐[(3,4‐dimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐2‐phenylacrylate ( 20 )
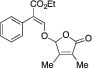 Yellow oil. Yield: 324 mg (86%). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1 H), 7.41–7.18 (m, 5 H), 5.94 (s, 1 H), 4.22 (q, J = 7.1 Hz, 2 H), 1.90 (s, 3 H), 1.81 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.0, 166.8, 152.9, 152.6, 131.8, 130.1, 127.8, 127.8, 127.5, 115.9, 102.1, 60.8, 14.3, 11.4, 8.5. HRMS [ESI+ (m/z)] calcd for (C17H18O5 + Na)+ 325.10519, found 325.10495.
Yellow oil. Yield: 324 mg (86%). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1 H), 7.41–7.18 (m, 5 H), 5.94 (s, 1 H), 4.22 (q, J = 7.1 Hz, 2 H), 1.90 (s, 3 H), 1.81 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 171.0, 166.8, 152.9, 152.6, 131.8, 130.1, 127.8, 127.8, 127.5, 115.9, 102.1, 60.8, 14.3, 11.4, 8.5. HRMS [ESI+ (m/z)] calcd for (C17H18O5 + Na)+ 325.10519, found 325.10495.
2.1.14. Ethyl (E)‐2‐phenyl‐3‐[(2,3,4‐trimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]acrylate ( 21 )
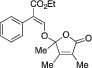 Yellow solid. Yield: 340 mg (86%). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1 H), 7.40–7.29 (m, 5 H), 4.25–4.17 (m, 2 H), 1.91 (s, 3 H), 1.91 (s, 3 H), 1.69 (s, 3 H), 1.28 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 169.4, 165.9, 155.5, 147.5, 131.0, 129.0, 126.6, 126.3, 125.7, 114.8, 106.4, 59.6, 21.2, 13.2, 9.5, 7.5. HRMS [ESI+ (m/z)] calcd for (C18H20O5 + Na)+ 339.12084, found 339.12080.
Yellow solid. Yield: 340 mg (86%). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1 H), 7.40–7.29 (m, 5 H), 4.25–4.17 (m, 2 H), 1.91 (s, 3 H), 1.91 (s, 3 H), 1.69 (s, 3 H), 1.28 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 169.4, 165.9, 155.5, 147.5, 131.0, 129.0, 126.6, 126.3, 125.7, 114.8, 106.4, 59.6, 21.2, 13.2, 9.5, 7.5. HRMS [ESI+ (m/z)] calcd for (C18H20O5 + Na)+ 339.12084, found 339.12080.
2.1.15. Ethyl (E)‐2‐(1H‐indol‐3‐yl)‐3‐[(4‐methyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]acrylate ( 22 )
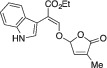 Orange solid. Yield: 121 mg (57%). 1H NMR (400 MHz, CDCl3) δ 8.31 (br s, 1 H), 7.79 (s, 1 H), 7.43–7.39 (m, 1 H), 7.32 (dt, J = 8.1, 1.0 Hz, 1 H), 7.27 (d, J = 2.6 Hz, 1 H), 7.16 (ddd, J = 8.2, 7.6, 1.3 Hz, 1 H), 7.09 (ddd, J = 8.0, 7.0, 1.1 Hz, 1 H), 6.78 (p, J = 1.6 Hz, 1 H), 6.15 (p, J = 1.4 Hz, 1 H), 4.27 (q, J = 7.1 Hz, 2 H), 1.91 (t, J = 1.6 Hz, 3 H), 1.30 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.7, 167.3, 151.7, 141.5, 135.6, 135.2, 126.3, 125.3, 121.9, 120.8, 119.7, 111.2, 109.6, 106.7, 100.5, 60.9, 14.4, 10.6. HRMS [ESI+ (m/z)] calcd for (C18H17NO5 + Na)+ 350.10044, found 350.09998.
Orange solid. Yield: 121 mg (57%). 1H NMR (400 MHz, CDCl3) δ 8.31 (br s, 1 H), 7.79 (s, 1 H), 7.43–7.39 (m, 1 H), 7.32 (dt, J = 8.1, 1.0 Hz, 1 H), 7.27 (d, J = 2.6 Hz, 1 H), 7.16 (ddd, J = 8.2, 7.6, 1.3 Hz, 1 H), 7.09 (ddd, J = 8.0, 7.0, 1.1 Hz, 1 H), 6.78 (p, J = 1.6 Hz, 1 H), 6.15 (p, J = 1.4 Hz, 1 H), 4.27 (q, J = 7.1 Hz, 2 H), 1.91 (t, J = 1.6 Hz, 3 H), 1.30 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.7, 167.3, 151.7, 141.5, 135.6, 135.2, 126.3, 125.3, 121.9, 120.8, 119.7, 111.2, 109.6, 106.7, 100.5, 60.9, 14.4, 10.6. HRMS [ESI+ (m/z)] calcd for (C18H17NO5 + Na)+ 350.10044, found 350.09998.
2.1.16. Ethyl (E)‐3‐[(2,4‐dimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐2‐(1H‐indol‐3‐yl)acrylate ( 23 )
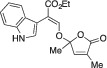 Yellow solid. Yield: 89 mg (46%). 1H NMR (400 MHz, CDCl3) δ 8.26 (br s, 1 H), 7.58 (s, 1 H), 7.46–7.43 (m, 1 H), 7.38 (dt, J = 8.1, 1.0 Hz, 1 H), 7.34 (d, J = 2.6 Hz, 1 H), 7.19 (ddd, J = 8.2, 7.0, 1.3 Hz, 1 H), 7.11 (ddd, J = 8.0, 7.0, 1.1 Hz, 1 H), 6.84 (q, J = 1.7 Hz, 1 H), 4.28–4.21 (m, 2 H), 2.00 (d, J = 1.7 Hz, 3 H), 1.71 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.4, 167.5, 147.9, 146.0, 135.6, 133.9, 126.5, 125.2, 121.9, 121.1, 119.5, 111.1, 109.2, 107.3, 106.3, 60.7, 23.7, 14.3, 10.5. HRMS [ESI+ (m/z)] calcd for (C19H19NO5 + Na)+ 364.11609, found 364.11565.
Yellow solid. Yield: 89 mg (46%). 1H NMR (400 MHz, CDCl3) δ 8.26 (br s, 1 H), 7.58 (s, 1 H), 7.46–7.43 (m, 1 H), 7.38 (dt, J = 8.1, 1.0 Hz, 1 H), 7.34 (d, J = 2.6 Hz, 1 H), 7.19 (ddd, J = 8.2, 7.0, 1.3 Hz, 1 H), 7.11 (ddd, J = 8.0, 7.0, 1.1 Hz, 1 H), 6.84 (q, J = 1.7 Hz, 1 H), 4.28–4.21 (m, 2 H), 2.00 (d, J = 1.7 Hz, 3 H), 1.71 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.4, 167.5, 147.9, 146.0, 135.6, 133.9, 126.5, 125.2, 121.9, 121.1, 119.5, 111.1, 109.2, 107.3, 106.3, 60.7, 23.7, 14.3, 10.5. HRMS [ESI+ (m/z)] calcd for (C19H19NO5 + Na)+ 364.11609, found 364.11565.
2.1.17. Ethyl (E)‐3‐[(3,4‐dimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐2‐(1H‐indol‐3‐yl)acrylate ( 24 )
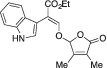 Orange solid. Yield: 64 mg (29%). 1H NMR (400 MHz, CDCl3) δ 8.27 (br s, 1 H), 7.80 (s, 1 H), 7.47–7.43 (m, 1 H), 7.37 (dt, J = 8.1, 0.9 Hz, 1 H), 7.33 (d, J = 2.6 Hz, 1 H), 7.18 (ddd, J = 8.2, 7.1, 1.1 Hz, 1 H), 7.09 (ddd, J = 8.1, 7.1, 1.1 Hz, 1 H), 6.00 (sept, J = 0.9 Hz, 1 H), 4.27 (q, J = 7.1 Hz, 2 H), 1.90 (p, J = 1.0 Hz, 3 H), 1.81 (p, J = 1.2 Hz, 3 H), 1.30 (t, J = 7.1 Hz, 4 H). 13C NMR (101 MHz, CDCl3) δ 171.1, 167.3, 152.8, 151.8, 135.5, 127.8, 126.4, 125.2, 121.9, 121.0, 119.6, 111.1, 109.5, 107.0, 102.2, 60.8, 14.4, 11.6, 8.5. HRMS [ESI+ (m/z)] calcd for (C19H19NO5 + Na)+ 364.11609, found 364.11574.
Orange solid. Yield: 64 mg (29%). 1H NMR (400 MHz, CDCl3) δ 8.27 (br s, 1 H), 7.80 (s, 1 H), 7.47–7.43 (m, 1 H), 7.37 (dt, J = 8.1, 0.9 Hz, 1 H), 7.33 (d, J = 2.6 Hz, 1 H), 7.18 (ddd, J = 8.2, 7.1, 1.1 Hz, 1 H), 7.09 (ddd, J = 8.1, 7.1, 1.1 Hz, 1 H), 6.00 (sept, J = 0.9 Hz, 1 H), 4.27 (q, J = 7.1 Hz, 2 H), 1.90 (p, J = 1.0 Hz, 3 H), 1.81 (p, J = 1.2 Hz, 3 H), 1.30 (t, J = 7.1 Hz, 4 H). 13C NMR (101 MHz, CDCl3) δ 171.1, 167.3, 152.8, 151.8, 135.5, 127.8, 126.4, 125.2, 121.9, 121.0, 119.6, 111.1, 109.5, 107.0, 102.2, 60.8, 14.4, 11.6, 8.5. HRMS [ESI+ (m/z)] calcd for (C19H19NO5 + Na)+ 364.11609, found 364.11574.
2.1.18. Ethyl (E)‐2‐(1H‐indol‐3‐yl)‐3‐[(2,3,4‐trimethyl‐5‐oxo‐2,5‐dihydrofuran‐2‐yl)oxy]‐acrylate ( 25 )
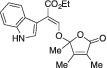 Orange solid. Yield: 74 mg (42%). 1H NMR (400 MHz, CDCl3) δ 8.30 (br s, 1 H), 7.51 (s, 1 H), 7.48–7.44 (m, 1 H), 7.36 (dt, J = 8.1, 1.0 Hz, 1 H), 7.32 (d, J = 2.6 Hz, 1 H), 7.18 (ddd, J = 8.2, 7.0, 1.3 Hz, 1 H), 7.10 (ddd, J = 8.1, 7.0, 1.1 Hz, 1 H), 4.29–4.18 (m, 2 H), 1.87 (q, J = 1.2 Hz, 3 H), 1.83 (q, J = 1.2 Hz, 3 H), 1.67 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.6, 167.5, 156.6, 147.6, 135.6, 126.8, 126.5, 125.2, 121.8, 121.2, 119.5, 111.1, 109.4, 107.4, 107.2, 60.7, 22.4, 14.3, 10.6, 8.6. HRMS [ESI+ (m/z)] calcd for (C20H21NO5 + Na)+ 378.13174, found 378.13106.
Orange solid. Yield: 74 mg (42%). 1H NMR (400 MHz, CDCl3) δ 8.30 (br s, 1 H), 7.51 (s, 1 H), 7.48–7.44 (m, 1 H), 7.36 (dt, J = 8.1, 1.0 Hz, 1 H), 7.32 (d, J = 2.6 Hz, 1 H), 7.18 (ddd, J = 8.2, 7.0, 1.3 Hz, 1 H), 7.10 (ddd, J = 8.1, 7.0, 1.1 Hz, 1 H), 4.29–4.18 (m, 2 H), 1.87 (q, J = 1.2 Hz, 3 H), 1.83 (q, J = 1.2 Hz, 3 H), 1.67 (s, 3 H), 1.27 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, CDCl3) δ 170.6, 167.5, 156.6, 147.6, 135.6, 126.8, 126.5, 125.2, 121.8, 121.2, 119.5, 111.1, 109.4, 107.4, 107.2, 60.7, 22.4, 14.3, 10.6, 8.6. HRMS [ESI+ (m/z)] calcd for (C20H21NO5 + Na)+ 378.13174, found 378.13106.
2.2. Bioassays
Seeds of parasitic weeds were sterilized in a solution of 2% (v/v) sodium hypochorite and 1% (v/v) Triton X‐100 for 6.5 min and washed with MilliQ water. Considering classical germination method,38 glass fibre filter paper discs (10 mm, Grade GF/D, Whatman, GE Healthcare, UK) were placed in a six‐well plate (three per well) and ∼ 25 seeds were spread onto each. MilliQ water was added carefully (1.6 mL/well), plates were sealed and conditioned (i.e. warm stratified) in the dark (for details see Table 1). Consequently, discs were dried on a filter paper for 30 min, placed back into plates and germination was induced with compound solutions (100 μL per disc). Six days later, the assay was evaluated by counting under binocular and calculating percentage of germinated seeds.
Table 1.
Conditioning period and temperature for germination of parasitic weed seeds
| Species | Collection | Temperature (°C) | Conditioning (days) |
|---|---|---|---|
| Striga hermonthica | Sudan, 2007 | 27 | 7 |
| Phelipanche ramosa | St. Martin de Bossenay, France, 2012, host Cannabis sativa | 21 | 7 |
| Orobanche minor | Porto Cedro, Sardinia, 2011, host Asteraceae | 21 | 14 |
For the colorimetric method,39 seeds were sterilized as described above and incubated in 1 mm Hepes, pH 7.5, and 0.1% PPM (Plant Preservative Mixture, Diagnovation Technologies, NY, USA) in 50 mL falcon tubes in the dark. Distribution into a 96‐well plate and stimulation with solutions of tested compounds followed after a conditioning period. MTT solution (5 g/L, 10 μL per well) was added after 4 days of germination. After 24 h, emerged formazan salts were solubilized by 100 μL of lyse buffer (10% Triton X‐100 and 0.04% HCl in propan‐2‐ol). Absorbances at 570 and 690 nm were measured after 24 h and differences (A 570–A 690) calculated.
Dose–response analysis was performed in GraphPad Prism 5.0 and results expressed as EC50 (half‐maximal concentration).
3. RESULTS AND DISCUSSION
3.1. Synthesis
The synthesis of the new hybrid‐type SL analogues from auxins commenced with the synthesis of four hydroxy furanones (5–8; Scheme 1): monomethylated 5, dimethylated 6 and 7, and trimethylated 8. The activity of SL analogues with polymethylated D‐rings is sometimes comparable with the derivatives with the natural monomethylated D‐ring40 and at times, the corresponding SL analogues might be easier (and cheaper on large scale) to synthesize. We developed a new one‐step procedure for the synthesis of hydroxy furanones 5, 6 and 8.41 Methylmalonic acid (1) was condensed with glyoxal (2), pyruvaldehyde (3) and butane‐2,3‐dione (4) with the aid of phenylboronic acid in refluxing water to yield hydroxy furanones 5, 6 and 8 in 65, 55 and 46% yields, respectively. Alternatively, hydroxy furanone 5 was also synthesized by condensation of methylmalonic acid (1) with glyoxal (2) catalysed by sulfuric acid42, 43 and hydroxy furanone 8 was synthesized from dimethylmaleic anhydride and MeLi44 with yields (depending on the scale) comparable with the previous method. Hydroxy furanone 7 was synthesized by reduction of dimethylmaleic anhydride (13) with Li[AlH(OtBu)3] in 55% yield (Scheme 1).44
Scheme 1.
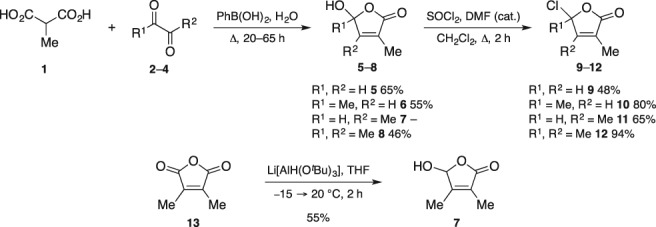
Synthesis of hydroxy furanones 5–8 and chloro furanones 9–12.
The coupling of SL analogues with the D‐ring is commonly achieved with bromo furanones, however, the coupling with the chloro furanones is less capricious. Thus, chloro furanones 9–12 were then synthesized by reaction of hydroxy furanones 5–8 with thionyl chloride in fair to excellent yields (48–94%) depending on the substitution (Scheme 1). The more substituted the reactive carbon is, the higher the yield obtained, because the rate of the competing formation of the side product (D‐ring)2 O is then slowed. Compound 9 was also synthesized in 82% yield using the Appel reaction (CCl4, Ph3P, THF, 60 °C, 3 h).
Finally, the hybrid‐type SL analogues were readily prepared via the regular two‐step sequence utilized at the end of every state‐of‐the‐art total synthesis of SLs: formylation of the scaffolds followed by coupling of the D‐ring. Consequently, the commercially available ethyl ester auxins 14 and 15 were formylated using ethyl formate in the present of a base (NaH) in excellent yields and the resulting enols 16 and 17, respectively, were then coupled with chloro furanones 9–12 using potassium carbonate in DMF to afford hybrid‐type SL analogues 18–25 in poor to very good yields (Scheme 2). In all cases, the yields of the products derived from auxin derivative 14 (18–21) were higher than those of the counterparts of auxin derivative 15 (22–25).
Scheme 2.
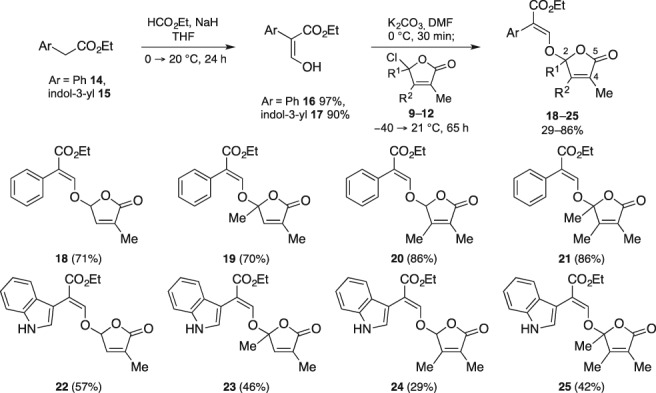
Synthesis of hybrid‐type strigolactone analogues 18–25.
All these syntheses fulfil the criterion of an easy synthetic operation: only two synthetic steps are needed (excluding formation of the always necessary Cl–D‐rings) from cheap commercially available compounds.
3.2. Bioassays
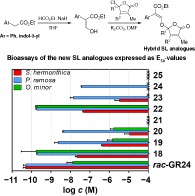
Hybrid‐type SL analogues 18–25 were bioassayed for germination of seeds of the parasitic weeds Striga hermonthica, Phelipanche ramosa and Orobanche minor. The number of germinated seeds was counted in the traditional manner38 and also analysed using a colorimetric method.39 We showed earlier that this colorimetric method gives the same results as the classical seed counting method.37 Some of the compounds showed a remarkably high activity. The compounds were tested in a wide concentration range and dose–response curves were generated in GraphPad Prism 5.0 (Fig. 4).
Figure 4.
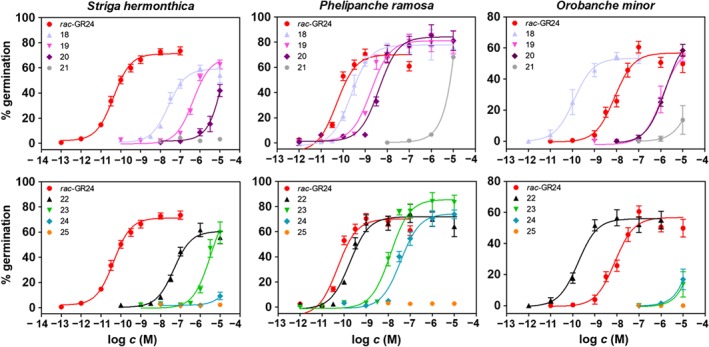
Dose–response curves of hybrid strigolactone analogues tested for germination stimulatory activity of parasitic weed seeds. Data represent averages with standard error (n ≥ 2).
A non‐linear regression was employed to compute EC50 values from dose–response curves. Graphical presentation of the EC50 values is given in Fig. 5.
Figure 5.
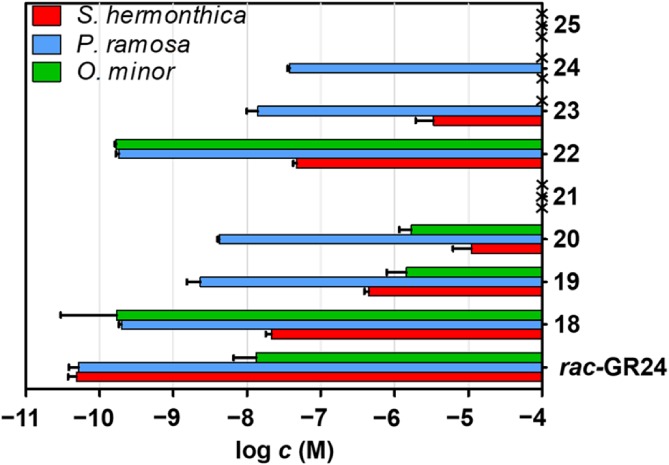
Bioactivity of new strigolactone hybrids expressed as EC50 values. Crosses indicate no stimulatory activity. Data represent averages with standard error (n ≥ 2).
Hybrids 18 and 22, with the monomethylated natural D‐ring, presented similar results and exhibited the best activity towards the three species. Their activities towards P. ramosa were comparable with that of GR24 and those towards O. minor were considerably higher. By contrast, hybrids 21 and 25 (with trimethylated D‐rings) did not present any activity towards any of the three species. Dimethylated hybrid SL analogues derived from ethyl ester auxin 14 (19 and 20) were more active towards the three species than their counterparts derived from auxin derivative 15 (23 and 24). All these compounds were more active towards P. ramosa than to the other two species, especially compounds 23 and 24 that were not active towards O. minor, and hybrid 24 that was not active towards S. hermonthica. Xu et al. recently described a clade of ShHTL receptors from S. hermonthica hyposensitive to light that bind SLs with diverse affinity.45 Some of our new hybrid‐type compounds seem to be species selective, namely 23 and 24. This could be a reason for the different levels of ShHTL receptor expression among species.
We showed that D‐ring modification is important for SL perception. Double methylation of the D‐ring caused a decrease in bioactivity and triple methylation led to total loss of bioactivity. This result is relevant to the previously published mode of action.2, 46 However, it does not correlate with the results obtained on GR24 and Nijmegen‐1 modified at C‐2.40 Possibly, the ethyl ester auxin moiety plays a role in affecting the bioactivity.
Several articles analysing the seed germination inducing activity of SL analogues have been published. Jamil et al. published a remarkable result, where the half maximal concentration of a methyl phenlactonoate (MP1) was 1 nm towards S. hermonthica and the activity on P. ramosa was similar to control.47 Saccharin and cyclic keto–enol analogues were moderately active towards S. hermonthica and comparable with 18 and 22.31, 48 Fluorescent analogues are worth mentioning, although tested on a different parasitic weed (Orobanche aegyptiaca), which showed bioactivity at picomolar levels.49, 50 However, no such high activity was observed for P. ramosa and O. minor compared with our SL analogues 18 and 22, which are outstandingly active. Boyer et al. published 3′‐methyl‐GR24 having an EC50 value one order of magnitude higher than that of GR24 on P. ramosa and comparable with GR24 for O. minor.51 Several hydroxylated analogues of GR24 showed germination‐stimulating activity at 0.1 μm, but results at lower concentrations were not tested.52
The new hybrid molecules 18 and 22 elicited the germination of seeds of parasitic weeds to a significant extent. Considering a suicidal germination approach, they could be agronomically interesting in terms of depleting the parasitic seed bank in soils.24, 53
4. CONCLUSION
In conclusion, new hybrid‐type SL analogues of auxin derivative ethyl 2‐(1H‐indol‐3‐yl)acetate and of ethyl 2‐phenylacetate showed considerable germination activity on parasitic weed seeds. Some of these analogues, namely compounds 18 and 22 (with the monomethylated natural D‐ring), were the most effective germination stimulants, with EC50 values in the nm range. Activity was strongly influenced by the incorporation of methyl groups on the D‐ring, with the trimethylated derivatives presenting no activity. These new analogues are of potential interest for agronomic applications, e.g. for the control of parasitic weeds, because of their ease of preparation and relevant activity.
Supporting information
Appendix S1 Supporting information
ACKNOWLEDGEMENTS
We thank Prof. Philippe Delavault for providing Phelipanche ramosa and Prof. Renata Piwowarczyk for Orobanche minor seeds. This work was supported by grant LO1204 from the National Program of Sustainability I from the Ministry of Education, Youth and Sports of the Czech Republic. A.H. was supported by the Internal Grant Agency of Palacký University Olomouc (IGA_PrF_2018_023). Work of AH and LS was also supported by the ERDF project “Plant as tool for sustainable global deveopment” (no CZ.02.1.01/0.0/0.0/16‐019/0000827)
Contributor Information
Daniel Blanco‐Ania, Email: d.blanco@science.ru.nl.
Binne Zwanenburg, Email: b.zwanenburg@science.ru.nl.
REFERENCES
- 1. Tsuchiya Y and McCourt P, Strigolactones: a new hormone with a past. Curr Opin Plant Biol 12:556–561 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Zwanenburg B and Pospíšil T, Structure and activity of strigolactones: new plant hormones with a rich future. Mol Plant 6:38–62 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Al‐Babili S and Bouwmeester HJ, Strigolactones, a novel carotenoid‐derived plant hormone. Annu Rev Plant Biol 66:161–186 (2015). [DOI] [PubMed] [Google Scholar]
- 4. Screpanti C, Yoneyama K and Bouwmeester HJ, Strigolactones and parasitic weed management 50 years after the discovery of the first natural strigolactone strigol: status and outlook. Pest Manag Sci 72:2013–2015 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Zwanenburg B, Pospíšil T and Ćavar‐Zeljković S, Strigolactones: new plant hormones in action. Planta 243:1311–1326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zwanenburg B and Blanco‐Ania D, Strigolactones: new plant hormones in the spotlight. J Exp Bot 69:2205–2218 (2018). [DOI] [PubMed] [Google Scholar]
- 7. Ćavar S, Zwanenburg B and Tarkowski P, Strigolactones: occurrence, structure, and biological activity in the rhizosphere. Phytochem Rev 14:691–711 (2015). [Google Scholar]
- 8. Cook CE, Whichard LP, Turner B, Wall ME and Egley GH, Germination of witchweed (Striga lutea Lour.): isolation and properties of a potent stimulant. Science 154:1189–1190 (1966). [DOI] [PubMed] [Google Scholar]
- 9. Cook CE, Whichard LP, Wall ME, Egley GH, Coggon P, Luhan PA et al, Germination stimulants. II. The structure of strigol—a potent seed germination stimulant for witchweed (Striga lutea Lour.). J Am Chem Soc 94:6198–6199 (1972). [Google Scholar]
- 10. Brooks DW, Bevinakatti HS and Powell DR, The absolute configuration of (+)‐strigol. J Org Chem 50:3779–3781 (1985). [Google Scholar]
- 11. Siame BA, Weerasuriya Y, Wood K, Ejeta G and Butler LG, Isolation of strigol, a germination stimulant for Striga asiatica, from host plants. J Agric Food Chem 41:1486–1491 (1993). [Google Scholar]
- 12. Yoneyama K, Xie X, Yoneyama K and Takeuchi Y, Strigolactones: structures and biological activities. Pest Manag Sci 65:467–470 (2009). [DOI] [PubMed] [Google Scholar]
- 13. Xie X, Yoneyama K and Yoneyama K, The strigolactone story. Annu Rev Phytopathol 48:93–117 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Ueno K, Nomura S, Muranaka S, Mizutani M, Takikawa H and Sugimoto Y, Ent‐2′‐epi‐Orobanchol and its acetate, as germination stimulants for Striga gesnerioides seeds isolated from cowpea and red clover. J Agric Food Chem 59:10485–10490 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Xie X, Yoneyama K, Kisugi T, Uchida K, Ito S, Akiyama K et al, Confirming stereochemical structures of strigolactones produced by rice and tobacco. Mol Plant 6:153–163 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen VX, Boyer F‐D, Rameau C, Retailleau P, Vors J‐P and Beau J‐M, Stereochemistry, total synthesis, and biological evaluation of the new plant hormone solanacol. Chem Eur J 16:13941–13945 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Akiyama K, Matsuzaki K and Hayashi H, Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 435:824–827 (2005). [DOI] [PubMed] [Google Scholar]
- 18. Gomez‐Roldan V, Fermas S, Brewer PB, Puech‐Pagès V, Dun EA, Pillot J‐P et al, Strigolactone inhibition of shoot branching. Nature 455:189–194 (2008). [DOI] [PubMed] [Google Scholar]
- 19. Umehara M, Hanada A, Yoshida S, Akiyama K, Arite T, Takeda‐Kamiya N et al, Inhibition of shoot branching by new terpenoid plant hormones. Nature 455:195–200 (2008). [DOI] [PubMed] [Google Scholar]
- 20. Umehara M, Hanada A, Magome H, Takeda‐Kamiya N and Yamaguchi S, Contribution of strigolactones to the inhibition of tiller bud outgrowth under phosphate deficiency in rice. Plant Cell Physiol 51:1118–1126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Proust H, Hoffmann B, Xie X, Yoneyama K, Schaefer DG, Yoneyama K et al, Strigolactones regulate protonema branching and act as a quorum sensing‐like signal in the moss Physcomitrella patens . Development 138:1531–1539 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Yoneyama K, Awad AA, Xie X, Yoneyama K and Takeuchi Y, Strigolactones as germination stimulants for root parasitic plants. Plant Cell Physiol 51:1095–1103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zwanenburg B, Ćavar‐Zeljković S and Pospíšil T, Synthesis of strigolactones, a strategic account. Pest Manag Sci 72:15–29 (2016). [DOI] [PubMed] [Google Scholar]
- 24. Zwanenburg B, Mwakaboko AS and Kannan C, Suicidal germination for parasitic weed control. Pest Manag Sci 72:2016–2025 (2016). [DOI] [PubMed] [Google Scholar]
- 25. Reizelman A, Scheren M, Nefkens GHL and Zwanenburg B, Synthesis of all eight stereoisomers of the germination stimulant strigol. Synthesis 1944–1955 (2000). [Google Scholar]
- 26. Bromhead LJ and McErlean CSP, Accessing single enantiomer strigolactones: progress and opportunities. Eur J Org Chem 5712–5723 (2017). [Google Scholar]
- 27. Johnson AW, Gowada G, Hassanali A, Knox J, Monaco S, Razavi Z et al, The preparation of synthetic analogues of strigol. J Chem Soc, Perkin Trans 1:1734–1743 (1981). [Google Scholar]
- 28. Kountche BA, Jamil M, Yonli D, Nikiema MP, Blanco‐Ania D, Asami T et al, Suicidal germination as a control strategy for Striga hermonthica (Benth.) in smallholder farms of sub‐Saharan Africa. Plants People Planet 1:107–118 (2019). [Google Scholar]
- 29. Zwanenburg B, Mwakaboko AS, Reizelman A, Anilkumar G and Sethumadhavan D, Structure and function of natural and synthetic signalling molecules in parasitic weed germination. Pest Manag Sci 65:478–491 (2009). [DOI] [PubMed] [Google Scholar]
- 30. Nefkens GHL, Thuring JWJF, Beenakkers MFM and Zwanenburg B, Synthesis of a phthaloylglycine‐derived strigol analogue and its germination stimulatory activity towards seeds of the parasitic weeds Striga hermonthica and Orobanche crenata . J Agric Food Chem 45:2273–2277 (1997). [Google Scholar]
- 31. Mwakaboko AS and Zwanenburg B, Single step synthesis of strigolactone analogues from cyclic keto enols, germination stimulants for seeds of parasitic weeds. Bioorg Med Chem 19:5006–5011 (2011). [DOI] [PubMed] [Google Scholar]
- 32. Mwakaboko AS and Zwanenburg B, Strigolactone analogs derived from ketones using a working model for germination stimulants as a blueprint. Plant Cell Physiol 52:699–715 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Hamiaux C, Drummond RS, Janssen BJ, Ledger SE, Cooney JM, Newcomb RD et al, DAD2 is an α/β hydrolase to be involved in the perception of the plant branching hormone, strigolactone. Curr Biol 22:2032–2036 (2012). [DOI] [PubMed] [Google Scholar]
- 34. Seto Y, Yasui R, Kameoka H, Tamiru M, Cao M, Terauchi R et al, Strigolactone perception and deactivation by a hydrolase receptor DWARF14. Nat Commun 10:191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Overvoorde P, Fukaki H and Beeckman T, Auxin control of root development. Cold Spring Harb Perspect Biol 2:a001537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pereira RG, Cala A, Fernández‐Aparicio M, Molinillo JMG, Boaventura MAD and Macías FA, Gibberellic and kaurenoic hybrid strigolactone mimics for seed germination of parasitic weeds. Pest Manag Sci 73:2529–2537 (2017). [DOI] [PubMed] [Google Scholar]
- 37. Hýlová A, Pospíšil T, Spíchal L, Mateman JJ, Blanco‐Ania D and Zwanenburg B, New hybrid type strigolactone mimics derived from plant growth regulator auxin. New Biotech 48:76–82 (2019). [DOI] [PubMed] [Google Scholar]
- 38. Mangnus EM, Stommen PLA and Zwanenburg B, A standardized bioassay for evaluation of potential germination stimulants for seeds of parasitic weeds. J Plant Growth Regul 11:91–98 (1992). [Google Scholar]
- 39. Pouvreau J‐B, Gaudin Z, Auger B, Lechat M‐M, Gauthier M, Delavault P et al, A high‐throughput seed germination assay for root parasitic plants. Plant Methods 9:32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mwakaboko AS and Zwanenburg B, Strigolactone analogues with a D‐ring modified at C‐2. Eur J Org Chem 3495–3499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee D, Newman SG and Taylor MS, Boron‐catalyzed direct aldol reactions of pyruvic acids. Org Lett 11:5486–5489 (2009). [DOI] [PubMed] [Google Scholar]
- 42. Malik H, Rutjes FPJT and Zwanenburg B, A new expedient synthesis of 3‐methyl‐2(5H)‐furanone, the common substructure in strigolactones, and its proposed biosynthesis. Synthesis 3271–3273 (2010). [Google Scholar]
- 43. Morris JC and McErlean CSP, Synthesis of highly enantio‐enriched stereoisomers of hydroxy‐GR24. Org Biomol Chem 14:1236–1238 (2016). [DOI] [PubMed] [Google Scholar]
- 44. Surmont R, Verniest G and De Kimpe N, Short synthesis of the seed germination inhibitor 3,4,5‐trimethyl‐2(5H)‐furanone. J Org Chem 75:5750–5753 (2010). [DOI] [PubMed] [Google Scholar]
- 45. Xu Y, Miyakawa T, Nosaki S, Nakamura A, Lyu Y, Nakamura H et al, Structural analysis of HTL and D14 proteins reveals the basis for ligand selectivity in Striga . Nat Commun 9:3947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jamil M, Kountche BA, Haider I, Wang JY, Aldossary F, Zarban RA et al, Methylation at the C‐3 in D‐ring of strigolactone analogs reduces biological activity in root parasitic plants and rice. Front Plant Sci 10:353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jamil M, Kountche BA, Haider I, Guo X, Ntui VO, Jia KP et al, Methyl phenlactonoates are efficient strigolactone analogs with simple structure. J Exp Bot 69:2319–2331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zwanenburg B and Mwakaboko AS, Strigolactone analogues and mimics derived from phthalimide, saccharine, p‐tolylmalondialdehyde, benzoic and salicylic acid as scaffolds. Bioorg Med Chem 19:7394–7400 (2011). [DOI] [PubMed] [Google Scholar]
- 49. Bhattacharya C, Bonfante P, Deagostino A, Kapulnik Y, Larini P, Occhiato EG et al, A new class of conjugated strigolactone analogues with fluorescent properties: synthesis and biological activity. Org Biomol Chem 7:3413–3420 (2009). [DOI] [PubMed] [Google Scholar]
- 50. Prandi C, Occhiato EG, Tabasso S, Bonfante P, Novero M, Scarpi D et al, New potent fluorescent analogues of strigolactones: synthesis and biological activity in parasitic weed germination and fungal branching. Eur J Org Chem 3781–3793 (2011). [Google Scholar]
- 51. Boyer F‐D, de Saint Germain A, Pillot J‐P, Pouvreau J‐B, Chen VX, Ramos S et al, Structure–activity relationship studies of strigolactone‐related molecules for branching inhibition in garden pea: molecule design for shoot branching. Plant Physiol 159:1524–1544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ueno K, Ishiwa S, Nakashima H, Mizutani M, Takikawa H and Sugimoto Y, Regioselective and stereospecific hydroxylation of GR24 by Sorghum bicolor and evaluation of germination inducing activities of hydroxylated GR24 stereoisomers toward seeds of Striga species. Bioorg Med Chem 23:6100–6110 (2015). [DOI] [PubMed] [Google Scholar]
- 53. Rubiales D and Fernández‐Aparicio M, Innovations in parasitic weeds management in legume crops. A review. Agron Sustain Dev 32:433–449 (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information