

Abstract
Risankizumab, a humanized monoclonal antibody that targets interleukin‐23 p19 subunit, was developed for the treatment of psoriasis. This work characterizes risankizumab pharmacokinetics in Japanese and Chinese healthy subjects compared with white healthy subjects and in Japanese patients with moderate to severe plaque psoriasis, generalized pustular psoriasis, or erythrodermic psoriasis. A phase 1, single‐dose study evaluated risankizumab pharmacokinetics and safety/tolerability in healthy white (18 and 300 mg subcutaneous [SC]), Japanese (18, 90, and 300 mg SC and 200, 600, and 1200 mg intravenous [IV]), and Chinese (18, 90, and 300 mg SC) subjects; pharmacokinetic data were analyzed using noncompartmental methods. Risankizumab pharmacokinetic data from phase 2/3 studies in Japanese patients with plaque psoriasis, generalized pustular psoriasis, or erythrodermic psoriasis following multiple SC doses of 75 mg or 150 mg were analyzed using a population pharmacokinetic approach along with data from the phase 1 and global phase 1 to 3 studies. Risankizumab plasma exposures (peak plasma concentration and area under the concentration‐time curve) were approximately dose‐proportional across 18‐ to 300‐mg SC or 200‐ to 1200‐mg IV doses. Risankizumab terminal elimination half‐life (harmonic mean 27–34 days) was comparable across doses and ethnicities. Risankizumab exposures were approximately 20% to 30% higher in Japanese and Chinese healthy subjects compared with white healthy subjects or in Japanese patients compared with non‐Japanese patients. After accounting for body‐weight differences, risankizumab exposures were comparable across ethnicities. Overall, there was no ethnic impact on risankizumab pharmacokinetics, and the small difference in exposure due to body weight has no clinical relevance.
Keywords: erythrodermic psoriasis, ethnicity, generalized pustular psoriasis, Japanese patients, plaque psoriasis, risankizumab
Psoriasis is a common, chronic, autoimmune disorder of the dermis and epidermis with clinical manifestations including prominent, often painful, skin lesions that negatively impact patient quality of life. Psoriatic patients are also at an increased risk for other comorbidities including psoriatic arthritis, uveitis, and cardiometabolic diseases.1 Generalized pustular psoriasis (GPP) is a rare and severe form of psoriasis that presents with flares of widespread sterile pustules on a background of red and tender skin and in rare cases can be fatal. About 10% of subjects with GPP have a preceding history of psoriasis.2 Erythrodermic psoriasis (EP) is another rare and severe variant of psoriasis that is difficult to treat and can be fatal. It has an estimated prevalence among psoriatic patients ranging from 1% to 2.25%.3 It is marked by widespread involvement (>75% of the body surface area) with redness and scaling and can be accompanied by systemic manifestations such as fever, chills, or malaise.3
Interleukin‐23 (IL‐23) is a key regulator of multiple effector cytokines that has been demonstrated to play a crucial role in the pathogenesis of several autoimmune diseases, including psoriasis. T helper 17 cells are a major source of proinflammatory cytokines, including IL‐17A,4 that can promote chronic tissue inflammation via IL‐23 stimulation. Risankizumab is a humanized IgG1 monoclonal antibody that prevents IL‐23 from binding to its receptor by selectively inhibiting the p19 subunit of IL‐23,5 thereby blocking IL‐23–dependent cell signaling and the release of proinflammatory cytokines.
In 4 phase 3 clinical trials (UltIMMa‐1 [NCT02684370], UltIMMa‐2 [NCT02684357], IMMvent [NCT02694523], and IMMhance [NCT02672852]) conducted globally, patients with moderate to severe plaque psoriasis who were treated with a 150‐mg subcutaneous (SC) dose of risankizumab administered at weeks 0, 4, and every 12 weeks (q12w) thereafter, demonstrated rapid and durable skin clearance with approximately 84% to 88% of patients with clear or almost clear skin after 16 weeks of treatment with further improvement in efficacy responses beyond 16 weeks following continued treatment with risankizumab.6, 7, 8 Risankizumab at doses of 75 mg SC and 150 mg SC have also been evaluated in Japanese patients with moderate to severe plaque psoriasis, GPP, or EP in phase 2/3 (phases 2 and 3) clinical trials; efficacy in Japanese moderate to severe plaque psoriasis patients was consistent with global phase 3 trials, with 93% of patients demonstrating clear or almost clear skin after 16 weeks of treatment with 150‐mg SC doses of risankizumab and further improvement in efficacy following continued treatment.9
This work characterized the pharmacokinetics and safety/tolerability of risankizumab following administration of single SC or intravenous (IV) doses in healthy white, Japanese, and Chinese subjects in a phase 1 study as well as the pharmacokinetics of risankizumab following administration of multiple SC doses in Japanese patients with moderate to severe plaque psoriasis, GPP, or EP in phase 2/3 trials. This article also describes the impact of the relevant patient‐specific covariates on risankizumab systemic exposures that were evaluated to inform dosing recommendations and the comparability of risankizumab exposures across Japanese patients with plaque psoriasis, GPP, or EP as well as between Japanese and non‐Japanese patients with psoriasis enrolled across all global and Japanese phase 2/3 clinical trials.
Methods
Study Designs
The studies were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocol and informed consent forms were approved by the institutional review boards (Alpha IRB, San Clemente, California; Institutional Review Board for Human Research, Inje University Busan Paik Hospital, Busan, Republic of Korea; Hakata Clinic Institutional Review Board, Fukuoka, Japan), and participants provided written informed consent before any study‐related procedures were performed.
Study 1, the phase 1 study (N = 89) in healthy white, Japanese, and Chinese subjects, was conducted according to a randomized, double‐blind, placebo‐controlled design. This was a 2‐part sequential‐dose study with 8 subjects randomized to risankizumab or placebo (6:2 ratio) at each dose level. In part 1, healthy white subjects (18 and 300 mg), Japanese subjects (18, 90, and 300 mg), and Chinese subjects (18, 90, and 300 mg) received a single SC dose of either risankizumab or matching placebo. In part 2, healthy Japanese subjects received single doses as an IV infusion over 2 hours of either risankizumab (200 mg, 600 mg, or 1200 mg) or placebo. In both parts, dose escalation proceeded after evaluation of the safety, tolerability, and pharmacokinetic data from the preceding dose level. Subjects were confined to the study site beginning 1 day before the study drug administration until day 8. Subjects returned to the study site at the specified time interval for continued safety assessments and pharmacokinetic sampling. The study was conducted at Inje University Busan Paik Hospital Clinical Trial Center in Busan, Korea for the white and Chinese subjects and at Sumida Hospital, LTA Medical Corp, Tokyo, Japan for the Japanese subjects.
Study 2, the phase 2/3 study in Japanese patients (N = 167) with severe chronic plaque psoriasis, was a randomized, double‐blind, double‐dummy, placebo‐controlled, parallel design study that evaluated 2 different dosing regimens of risankizumab. Eligible subjects were randomized in a ratio of 2:2:1:1 to 1 of 4 treatment arms (75 mg SC or 150 mg SC risankizumab at weeks 0, 4, and q12w; or placebo for 75‐mg and 150‐mg doses at weeks 0 and 4). After the 16‐week treatment period, all subjects who initially had been randomized to placebo began receiving either 75 or 150 mg risankizumab q12w. All subjects continued to receive treatment through week 40 and were followed through week 52. The study was conducted at 38 clinical trial sites in Japan.
Study 3, the phase 3 study in Japanese patients with GPP (N = 8) or EP (N = 9), was a randomized, open‐label study that compared 2 different dosing regimens of risankizumab (75 mg SC and 150 mg SC administered at weeks 0, 4, and q12w thereafter). Eligible subjects were randomized in a ratio of 1:1 to the 75‐mg or 150‐mg risankizumab treatment groups. The subjects received treatment through week 172 in the study, and the data until interim analysis at week 52 were summarized. The study is being conducted in Japan at 9 clinical trial sites.
Study Participants
Study 1 included healthy male subjects of white, Japanese, or Chinese ethnicity aged 21 to 45 years (inclusive) with body mass index 18.5 to 25 kg/m2 (inclusive) for Chinese and Japanese subjects and 20.0 to 29.1 kg/m2 (inclusive) for white subjects.
Study 2 included adults aged ≥20 years with stable moderate to severe chronic plaque psoriasis of ≥6 months’ duration with or without psoriatic arthritis, body surface area (BSA) involvement ≥10%; Psoriasis Area and Severity Index (PASI) ≥12; Static Physician Global Assessment (sPGA) ≥3; and candidates for systemic therapy or phototherapy.
Study 3 included adults aged ≥20 years with a clinical diagnosis of GPP or EP for >60 days who were candidates for systemic therapy or phototherapy. Subjects with GPP were to have ≥10% of the BSA involved with erythema with pustules and a Japanese Dermatological Association total score <14. Subjects with EP were to have ≥80% BSA involvement for inflammatory erythema.
Pharmacokinetic Sampling and Bioanalytical Methods
In study 1, serial blood samples for assessment of risankizumab pharmacokinetics, antidrug antibody (ADA), and neutralizing antibody (NAb) against risankizumab were collected for up to 137 days after a single dose of risankizumab. Pharmacokinetic samples following SC dosing were collected predose and on days 2, 3, 4, 8, 15, 29, 57, 85, and 137 after dosing; following IV dosing, samples were collected predose and 20 minutes, 1.5, 2, 4, 8, and 12 hours on day 1, and on days 2, 3, 4, 8, 15, 29, 57, 85, and 137 after dosing. ADA and NAb samples following both SC and IV doses were collected predose and on days 8, 29, 85, and 137. In studies 2 and 3 in patients, sparse sampling, including predose trough sampling at weeks 4, 16, 28, 40 (only for plaque psoriasis), and 52 was performed for assessment of risankizumab pharmacokinetics, ADA, and NAb. Risankizumab plasma concentrations and ADA were measured as previously described10, 11 and are briefly summarized here.
Plasma concentrations of risankizumab were determined using a validated enzyme‐linked immunosorbent assay. The assay was validated over the nominal risankizumab concentration range of 5 to 100 ng/mL with a lower limit of quantitation of 5.00 ng/mL. The assay had interrun precision (percentage coefficient of variation [%CV]) ≤7.2%, and the bias was –2.8% to 3.61% across studies. Plasma samples above the upper limit of quantitation were diluted and reassayed.
The presence of ADA was determined using a validated electrochemiluminescent immunoassay. ADA titers were determined for all confirmed ADA‐positive samples. Samples that were confirmed positive were also characterized using a validated NAb assay. Assay details are available from Suleiman et al.10 The ADA assay had a sensitivity of 0.119 ng/mL, and drug tolerance allowed detection of 0.7 ng/mL of a positive control in the presence of 5 µg/mL of risankizumab.
Pharmacokinetic and Safety Analyses in Healthy Subjects (Study 1)
Values for the pharmacokinetic parameters of risankizumab in the phase 1 study were estimated using noncompartmental methods with Phoenix WinNonlin Version 6.3 (Pharsight Corporation, Mountain View, California). The maximum observed plasma concentration (Cmax) and the time to Cmax (Tmax) were determined directly from the plasma concentration‐time data. Calculated parameters included the terminal‐phase elimination half‐life (t½) and the area under the plasma concentration‐time curve (AUC) from time 0 to the last measurable concentration (AUCt) or infinite time (AUC∞). Dose‐normalized values of pharmacokinetic parameters (Cmax and AUC∞) were calculated by dividing each of the selected parameters by the administered dose.
Dose proportionality of the pharmacokinetic parameters (Cmax and AUC∞) was assessed using a power model for SC administration and IV administration separately for each ethnicity, using SAS software Version 9.4 (SAS Institute Inc, Cary, North Carolina).
An analysis of covariance (ANCOVA) was performed for the natural logarithms of Cmax and AUC∞ for SC administration in Japanese and Chinese subjects relative to white subjects. Ethnicity, dose, and body weight were included as the covariates. A separate set of analyses was performed without body weight as a covariate. Within the ANCOVA framework, the ratios and 90%CIs of the ratios of risankizumab Cmax and AUC∞ in Japanese and Chinese ethnicity relative to white ethnicity were determined.
Safety was evaluated throughout the study based on assessments of adverse events, vital sign values, physical examinations, laboratory values, and 12‐lead electrocardiograms.
Pharmacokinetic Analyses in Psoriasis Patients (Study 2 and Study 3)
Risankizumab plasma concentrations that were observed at the end of the dosing interval (Ctrough) and at weeks 4, 16, 28, 40, and 52 were summarized by dose levels. In addition, risankizumab plasma concentration data from studies 1, 2, and 3 were combined with data from plaque psoriasis patients enrolled in global clinical trials—a phase 1, a phase 2, and 4 phase 3 trials—and analyzed using a population pharmacokinetic modeling approach using NONMEM version 7.4.1 (ICON Development Solutions, Ellicott City, Maryland). Two of the global phase 3 trials (UltIMMa‐1 and IMMhance) also included Japanese sites. This approach allowed a combined assessment of risankizumab pharmacokinetics across all Japanese patients enrolled in Japan and global trials.
A population pharmacokinetic model of risankizumab has been developed previously using global phase 1, phase 2, and phase 3 trials as well as study 1 in healthy volunteers and moderate to severe plaque psoriasis patients (N = 1899).12 Briefly, a 2‐compartment model with first‐order absorption and elimination best described the pharmacokinetics of risankizumab in healthy subjects and patients with psoriasis in these studies. In the covariate analysis, body weight, baseline serum albumin, baseline serum creatinine, and baseline high‐sensitivity C‐reactive protein (hs‐CRP) and ADA titers ≥128 showed statistically significant relationships with risankizumab clearance. Among these covariates, baseline serum albumin, baseline serum creatinine, and baseline hs‐CRP resulted in no impact on risankizumab steady‐state exposures (Cmax and AUC) because risankizumab exposures were predicted to be within the default 80% to 125% equivalence boundaries for patients with extremes of covariate values (less than 25th percentile or greater than 75th percentile) compared to middle 50% of patients for the covariate distribution in phase 3 clinical trials. Patients with body weight >100 kg or those with ADA titers above 128 had approximately 30% decrease in risankizumab exposures compared with patients with body weight ≤100 kg or those who were ADA‐negative or ADA‐positive but with titers ≤128, respectively. The details of the analysis and parameter estimates for the model were published previously.12
In the current analysis the previously developed population pharmacokinetic model of risankizumab was first evaluated for its appropriateness for describing the risankizumab plasma concentration data for the recent studies conducted in Japan (study 2 and study 3). This was performed using a simulation‐based approach (visual predictive check [VPC]). By use of the parameter estimates from the previously developed population pharmacokinetic model and the subject‐specific information in Japan study 2 and study 3, 1000 replicates of the plasma concentration data for the Japan‐only studies were simulated based on the actual dosing and sampling times. Median and 5th and 95th percentiles of the simulated data representing the 90% prediction intervals along with the 95%CIs of median, 5th, and 95th percentiles were calculated by binning the simulated plasma concentration data on the basis of protocol‐defined times for each risankizumab dose level and compared graphically with the observed data (median and 5th and 95th percentiles of the risankizumab observed plasma concentration data) from Japan‐only studies. After confirming that the population pharmacokinetic model adequately described the pharmacokinetic data from the Japan studies, the parameter estimates of the pharmacokinetic model were updated by fitting the model to a pooled data set including data from global studies and the 2 Japan studies (study 2 and study 3; the data set already included study 1).
With the updated population pharmacokinetic model, risankizumab steady‐state plasma exposures (Cmax, AUC, and Ctrough) over the dosing interval in subjects with moderate to severe plaque psoriasis, GPP, or EP following administration of 150 mg SC at weeks 0, 4, and q12w thereafter across all Japanese subjects (Japanese subjects enrolled in global phase 3 studies, as well as in Japan studies 2 and 3) were calculated based on the empirical Bayesian individual pharmacokinetic parameter estimates. The plasma concentration‐time course was simulated using the actual dosing history and by adding dummy observation time records to the data set for daily risankizumab concentration sampling.
To evaluate the clinical relevance of the statistically significant covariates based on the updated population pharmacokinetic model, simulations were carried out to compare risankizumab steady‐state Cmax and AUC over a dosing interval (AUCtau) for the risankizumab dosing regimen of 150 mg SC at weeks 0 and 4 and q12w thereafter in a subset of patients, the test group, relative to the reference group. For body weight, patients weighing ≤100 kg or ≤90 kg were considered the reference group, and patients weighing >100 kg or >90 kg were considered the test group. The 100‐kg and 90‐kg body‐weight cutoffs were the same as utilized in efficacy subgroup analyses of risankizumab in global phase 3 clinical trials and phase 2/3 clinical trial in Japan, respectively. It is noteworthy that 100 kg is also the body‐weight cutoff that determines the dose of ustekinumab, which was an active comparator in 2 of risankizumab's global phase 3 trials. In the overall analysis data set, 15% and 8% of all Asian subjects as well as 50% and 32% of all white subjects plus others (including all categories except Asians) were above 90‐kg and 100‐kg cutoff values, respectively. For other continuous covariates, subjects within the 25th to 75th percentiles of the covariate distribution in the data set served as the reference group, and subjects below the 25th percentile or above the 75th percentile were 2 separate test groups. For categorical covariates, subjects with most frequent category were considered the reference group, and the other categories were the test groups.
Simulations for each of the identified covariates of interest were carried out separately, whereas other covariates were fixed at their reference values. To include parameter uncertainty in simulations, the fixed‐effect parameters for each replicate were sampled from the variance‐covariance matrix of the final pharmacokinetic model parameters, and the interindividual random effects distributions determined the individual subject pharmacokinetic parameters within a replicate. Cmax and AUCtau values over the week‐40 to week‐52 interval were calculated, and within each simulation replicate, the ratios of median Cmax and AUCtau values for each test group relative to the median Cmax and AUCtau of the reference group (normalized exposure ratio) were calculated. This process was repeated 200 times, and the median of the normalized exposure ratios across the 200 replicates and the nonparametric 95%CIs (2.5th and 97.5th percentiles of the ratios) were calculated and summarized.
The evaluation of the updated population pharmacokinetic model was performed using goodness‐of‐fit plots, VPCs with 1000 simulated replicates, and bootstrap analyses using 1000 replicate data sets as described previously.12 For the VPCs, the median and 5th and 95th percentiles of the simulated data representing the 90% prediction intervals along with the 95%CIs of medians and 5th and 95th percentiles were calculated and compared graphically with the observed data. For the bootstrap analysis, the medians and corresponding nonparametric 95%CIs (2.5th to 97.5th percentiles) were constructed for each parameter based on the successfully converging runs and compared to the parameter estimates of the final model.
Results
Participants
Participants in study 1 had an average age of approximately 28 years, with average weights for Japanese, Chinese, and white subjects of approximately 62, 68, and 79 kg, respectively. A demographic summary on participants from Japan study 2 and study 3, and all Japanese patients across Japan, and global phase 3 studies is presented in Table 1.
Table 1.
Demographic and Baseline Characteristics of Patients With Moderate to Severe Plaque Psoriasis, GPP, or EP Included in the Risankizumab Population Pharmacokinetic Analyses
| Characteristic | Japan Study 2 | Japan Study 3 | Japanese Subjects in Global Phase 3 Studies (UltIMMa‐1, and IMMhance) | All Japanese Subjects in Phase 2/3 Studies | All Subjects in Phase 1 to 3 Studies Included in the Data Set | |
|---|---|---|---|---|---|---|
| N | 167 | 17 | 52 | 236 | 2095 | |
| Age (y) | Mean (SD) | 51.6 (11.7) | 48.4 (20.2) | 49.2 (12.0) | 50.8 (12.6) | 47.2 (13.8) |
| Median | 51 | 51 | 51 | 51 | 48 | |
| Min‐Max | 23–80 | 21–84 | 22–73 | 21–84 | 18–85 | |
| Body weight (kg) | Mean (SD) | 74.3 (17.1) | 71.8 (17.1) | 71.9 (16.3) | 73.6 (16.9) | 88.3 (22.6) |
| Median | 71.5 | 71.0 | 70.4 | 70.8 | 85.5 | |
| Min‐Max | 38.5–121 | 48.0–106 | 46.9–125 | 38.5–125 | 38.5–193 | |
| Body mass index (kg/m2) | Mean (SD) | 26.5 (5.30) | 25.6 (5.44) | 25.7 (5.35) | 26.3 (5.31) | 30.0 (7.10) |
| Median | 25.3 | 25.1 | 24.4 | 25.1 | 29.0 | |
| Min‐Max | 15.8–43.0 | 17.3–36.6 | 18.0–45.5 | 15.8–45.5 | 15.0–92.7 | |
| Sex | Male, N (%) | 141 (84%) | 14 (82%) | 42 (81%) | 197 (83%) | 1505 (72%) |
| Female, N (%) | 26 (16%) | 3 (18%) | 10 (19%) | 39 (17%) | 590 (28%) | |
| Race | White and others (including all categories except Asians), N (%) | … | … | … | … | 1578 (75%) |
| Asian, N (%) | 167 (100%) | 17 (100%) | 52 (100%) | 236 (100%) | 517 (25%) | |
| Psoriasis disease status | Plaque psoriasis, N (%) | 167 (100%) | … | 52 (100%) | 219 (93%) | 2011 (96%) |
| Generalized pustular psoriasis, N (%) | … | 8 (47%) | … | 8 (3%) | 8 (0.38%) | |
| Erythrodermic psoriasis, N (%) | … | 9 (53%) | … | 9 (4%) | 9 (0.43%) | |
| Healthy, N (%) | … | … | … | … | 67 (3.2%) | |
| C‐reactive protein (mg/L) | Mean (SD) | 6.9 (11.4) | 7.5 (12.4) | 6.6 (11.3) | 6.9 (11.4) | 6.0 (10.2) |
| Median | 2.3 | 1.4 | 2.7 | 2.3 | 2.8 | |
| Min‐Max | 0.2–61.3 | 0.2–42.4 | 0.2–63.1 | 0.2–63.1 | 0.1–131.0 | |
| Baseline PASI score | Mean (SD) | 25.7 (10.2) | 35.7 (21.2) | 25.6 (11.3) | 26.4 (11.8) | 20.6 (8.35)a |
| Median | 22.8 | 32.3 | 23.7 | 23.0 | 18.0a | |
| Min‐Max | 12.0–59.0 | 7.2–69.6 | 12.2–63.4 | 7.2–69.6 | 7.2–69.6a | |
| Serum albumin (g/L) | Mean (SD) | 43.7 (3.12) | 43.4 (3.18) | 44.6 (2.97) | 43.9 (3.10) | 44.2 (2.99)b |
| Median | 44.0 | 44.0 | 45.0 | 44.0 | 44.0b | |
| Min‐Max | 33.0–54.0 | 36.0–50.0 | 39.0–52.0 | 33.0–54.0 | 33.0–58.0b | |
| Serum creatinine (µmol/L) | Mean (SD) | 73.0 (18.3) | 69.1 (17.5) | 74.5 (15.6) | 73.0 (17.6) | 76.4 (16.4) |
| Median | 70.7 | 70.7 | 75.0 | 70.7 | 75.0 | |
| Min‐Max | 44.2–212 | 44.2–106 | 44.2–124 | 44.2–212 | 35.4–212 | |
| Creatinine clearance (mL/min) | Mean (SD) | 112 (36.3) | 118 (40.8) | 108 (36.6) | 112 (36.6) | 131 (46.43) |
| Median | 106 | 115 | 107 | 107 | 122 | |
| Min‐Max | 50.2–224 | 54.0–195 | 52.1–251 | 50.2–251 | 33.1–404 | |
| ADA treatment‐emergent status | Yes, N(%)c | 39 (23%) | 4 (24%) | 15 (29%) | 58 (24%) | 468 (22%) |
| ADA titer ≥ 128 | Nd | 1 | … | … | 1 | 33 |
| NAb treatment‐emergent status | Yes, N (%) | 27 (16%) | 0 (0%) | 3 (6%) | 28 (12%) | 263 (13%) |
ADA indicates antidrug antibodies to risankizumab; Max, maximum; Min, minimum; N, number of subjects; NAb, neutralizing antibodies to risankizumab; PASI, Psoriasis Area and Severity Index.
N = 2028, 67 healthy subjects excluded.
N = 2064.
The percentage value was calculated based on the number of subjects included in the data set for each study.
Summary calculated only among those subjects with an ADA titer ≥128.
Pharmacokinetics
Single Dose in Healthy Subjects
A total of 67 healthy subjects received risankizumab, and 22 healthy subjects received placebo in study 1; 1 Chinese subject (90 mg SC) withdrew from the study on day 8 and was excluded from pharmacokinetic analysis and replaced with another subject to have a total of 6 subjects for the dose level. The mean plasma concentration‐time profiles for risankizumab following single SC doses of 18 mg, 90 mg, and 300 mg for each ethnicity and following single IV doses of 200 mg, 600 mg, and 1200 mg in Japanese subjects are presented in Figure 1. A summary of the pharmacokinetic parameters of risankizumab following single SC and IV doses for each ethnicity is shown in Table 2 and Table 3, respectively.
Figure 1.
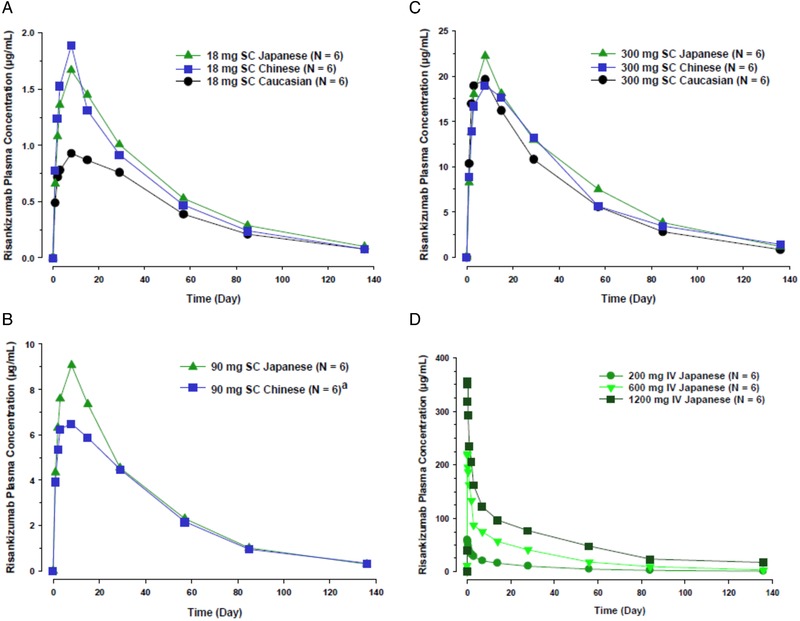
Risankizumab mean plasma concentration‐time profile following single subcutaneous (SC) doses in panels A, B, and C or intravenous (IV) doses in panel D in healthy subjects.
Table 2.
Pharmacokinetic Parameters of Risankizumab Following Single Doses of 18 mg, 90 mg, 300 mg SC in Healthy Subjects
| Risankizumab Dose | Pharmacokinetic Parameters | Units | Japanese (N = 6) | Chinese (N = 6) | White (N = 6) |
|---|---|---|---|---|---|
| 18 mg SC | Cmax | µg/mL | 1.70 (30) | 1.91 (29) | 1.04 (21) |
| Tmax a | d | 7.0 (7.0–14.0) | 7.0 (3.0–7.0) | 7.0 (3.0–28.1) | |
| t½ b | d | 32.5 (4.95) | 28.5 (3.78) | 30.5 (8.93) | |
| AUC∞ | µg·d/mL | 84.7 (17) | 77.3 (22) | 59.3 (18) | |
| 90 mg SC | Cmax | µg/mL | 9.08 (7) | 6.81 (27) | |
| Tmax a | d | 7.0 (7.0–7.0) | 5.0 (3.0–7.0) | ||
| t½ b | d | 26.9 (1.54) | 26.7 (3.73) | ||
| AUC∞ | µg·d/mL | 377 (6) | 336 (22) | ||
| 300 mg SC | Cmax | µg/mL | 22.3 (41) | 19.5 (26) | 20.4 (39) |
| Tmax a | d | 7.0 (7.0–14.0) | 7.0 (3.0–14.0) | 7.0 (3.0–7.0) | |
| t½ b | d | 29.7 (4.80) | 33.8 (7.15) | 28.7 (3.77) | |
| AUC∞ | µg·d/mL | 1100 (37) | 1030 (28) | 915 (33) |
AUC∞ indicates area under the plasma concentration‐time curve from time 0 to infinity; Cmax, maximum plasma concentration; SC, subcutaneous; t½, elimination half‐life; Tmax, time to maximum plasma concentration.
Data are presented as mean (%CV), unless noted otherwise.
Median (minimum through maximum).
Harmonic mean (pseudo‐SD); evaluations of t½ were based on statistical tests for β.
Table 3.
Mean Pharmacokinetic Parameters of Risankizumab Following Single Doses of 200 mg, 600 mg, and 1200 mg IV in Healthy Japanese Subjects
| Pharmacokinetic Parameters | Units | Risankizumab 200 mg IV (N = 6) | Risankizumab 600 mg IV (N = 6) | Risankizumab 1200 mg IV (N = 6) |
|---|---|---|---|---|
| Cmax | µg/mL | 60.1 (14) | 225 (8) | 363 (15) |
| Tmax a | d | 0.1 (0.1–0.2) | 0.1 (0.1–0.2) | 0.1 (0.1–0.3) |
| t½ b | d | 31.2 (7.38) | 30.7 (2.56) | 32.7 (14.2) |
| AUC∞ | µg·d/mL | 998 (12) | 3620 (9) | 7020 (28) |
AUC∞ indicates area under the plasma concentration‐time curve from time 0 to infinity; Cmax, maximum plasma concentration; IV, intravenous; t½, elimination half‐life; Tmax, time to maximum plasma concentration.
Median (minimum through maximum).
Harmonic mean (pseudo‐SD); evaluations of t½ were based on statistical tests for β.
Following administration of single SC doses of risankizumab 18 mg to 300 mg across ethnicities, risankizumab Tmax was 5 to 7 days after dosing. Following administration of single SC or IV doses of risankizumab the harmonic mean t½ ranged from approximately 27 to 34 days across doses and ethnicities. Risankizumab Tmax (for SC doses) and t½ (for SC or IV doses) were comparable across doses and ethnicities. Risankizumab exposure (AUC) was 31% higher in Japanese subjects compared with white subjects and 22% higher in Chinese subjects compared with white subjects (Table 4). After accounting for differences in body weight, risankizumab exposures were comparable (less than 5% difference) across the ethnic groups suggesting that the observed small exposure differences were driven by body‐weight differences across these ethnicities (Table 4).
Table 4.
Ratios of Risankizumab Cmax and AUC in Healthy Japanese and Chinese Subjects Relative to White Subjects
| Regimens | Exposure Ratiosa (Not Accounting for Body Weight) | Exposure Ratiosa (Adjusted for Body Weight) | |||
|---|---|---|---|---|---|
| Test vs Reference | Pharmacokinetic Parameter | Point Estimate | 90%CI | Point Estimate | 90%CI |
| Japanese vs white | Cmax | 1.32 | 1.03–1.69 | 0.84 | 0.61–1.14 |
| AUC∞ | 1.31 | 1.10–1.56 | 1.02 | 0.81–1.30 | |
| Chinese vs white | Cmax | 1.35 | 1.06–1.70 | 1.08 | 0.86–1.35 |
| AUC∞ | 1.22 | 1.03–1.45 | 1.05 | 0.88–1.25 | |
AUC∞ indicates area under the plasma concentration‐time curve from time 0 to infinity; Cmax, maximum plasma concentration.
Ratio was calculated by exponentiation of the difference between the logarithms of the least‐squares means calculated based on 1‐way ANOVA.
The mean ± SD dose‐normalized Cmax and AUC∞ values following SC or IV doses of risankizumab are presented in Supplementary Figure 1. These data show approximate dose proportionality across the evaluated dose range for all ethnicities. Based on statistical analyses, risankizumab exposures (Cmax and AUC) did not deviate significantly from dose proportionality for the 18‐mg and 300‐mg SC doses for white subjects and for 200‐mg to 1200‐mg IV doses for Japanese subjects (P > .05). For Japanese subjects across the 18‐mg to 300‐mg SC doses, there was no significant deviation from dose proportionality for Cmax (P > .05), but AUC showed a slightly less than dose‐proportional increase, with the slope estimate close to 1 (0.9, 95%CI 0.81–0.993). For Chinese subjects there was no significant deviation from dose proportionality for AUC (P > .05), although the Cmax showed a slightly less than dose‐proportional increase, with the slope estimate close to 1 (0.83, 95%CI 0.731–0.932). Overall these data indicate that deviation from dose proportionality is minimal and likely driven by small sample size per dose level for these analyses.
All subjects were ADA negative at baseline, except for 1 Chinese subject who had received placebo treatment. Following administration of single SC or IV doses of risankizumab, treatment‐emergent ADA incidence (0% to 11%) was low and comparable across ethnicities. No dose‐dependent trend in ADA incidence was noted. All the ADA positive subjects had low titers (≤16). None of the subjects was positive for neutralizing antibody against risankizumab. Risankizumab exposures in ADA‐positive subjects were comparable to ADA‐negative subjects indicating lack of impact by the presence of ADA.
Risankizumab was well tolerated with no dose‐limiting toxicities (ie, the maximum tolerated dose was not reached). No serious adverse events or clinically significant vital signs or laboratory measurements were observed during the course of the study.
Plaque Psoriasis, GPP, or EP Patients
Risankizumab plasma Ctrough values in Japanese patients with plaque psoriasis, GPP, or EP are shown in Supplementary Table 1. Risankizumab plasma Ctrough values at week 4 were higher than those at week 16 or week 52 primarily because of the shorter interdose interval at week 4 (4 weeks) compared with week 52 (12 weeks). Risankizumab steady state in patients was achieved by approximately 16 weeks after initiation of dosing. Risankizumab plasma Ctrough values were approximately dose proportional across the 75 mg and 150 mg SC doses and generally comparable between Japanese subjects with moderate to severe plaque psoriasis and those with GPP or EP.
Population Pharmacokinetics
The risankizumab population pharmacokinetic model previously developed using global phase 1, 2, and 3 trials described the central tendency and variability in the observed risankizumab plasma concentration data for both 75‐mg and 150‐mg SC doses from Japan studies very well, confirming the adequacy of the global population pharmacokinetic model for Japanese subjects (Supplementary Figure 2). Table 5 shows the estimated pharmacokinetic parameter values and their associated variability for the updated pharmacokinetic model based on the combined data set including a phase 1 trial in healthy volunteers, global phase 1, 2, and 3 trials in patients with plaque psoriasis, and Japan phase 2/3 trials (studies 2 and 3) in patients with plaque psoriasis, GPP, or EP. Based on the population pharmacokinetic analyses, risankizumab plasma clearance, the steady‐state volume of distribution, and terminal‐phase elimination half‐life were estimated to be approximately 0.24 L/day, 9.12 L, and 28 days, respectively, for a typical 70‐kg patient with psoriasis, GPP, or EP (70 kg selected here because it approximates the median body weight in Japanese psoriasis patients). The interindividual variability (%CV) for risankizumab clearance, volume of distribution, and absorption rate constant were 24%, 34%, and 63%, respectively.
Table 5.
Fixed and Random Effects Parameter Estimates for the Risankizumab Population Pharmacokinetic Model Based on Global and Japan Studies
| Parameter | Population Estimate (%RSE)a , b | Bootstrap 95%CI |
|---|---|---|
| Clearance (CL; L/d) | 0.244 (1.8) | 0.218 to 0.265 |
| Central volume of distribution (Vc; L) | 4.87 (3.8) | 4.04 to 5.65 |
| Absorption rate constant (Ka; d‐1) | 0.230 (4.8) | 0.180 to 0.302 |
| Intercompartmental clearance (Q; L/d) | 0.648 (3.7) | 0.529 to 0.784 |
| Peripheral volume of distribution (Vp; L) | 4.25 (2.0) | 3.87 to 4.66 |
| Absolute SC bioavailability (F)c , d | 0.89 (7.2) | 0.796 to 0.970 |
| Exponent for effect of body weight on risankizumab clearance (CL) | 0.935 (3.1) | 0.871 to 0.996 |
| Exponent for effect of body weight on risankizumab central volume of distribution (Vc) | 1.18 (6.9) | 0.976 to 1.36 |
| Exponent for effect of serum albumin on risankizumab clearance (CL) | –0.717 (10.2) | –0.899 to –0.535 |
| Exponent for effect of serum creatinine on risankizumab clearance (CL) | –0.242 (10.1) | –0.294 to –0.189 |
| Exponent for effect of C‐reactive protein on risankizumab clearance (CL) | 0.044 (10.1) | 0.034 to 0.053 |
| Exponent for effect of body weight on risankizumab peripheral volume of distribution (Vp) | 0.360 (12.3) | 0.220 to 0.499 |
| Proportional increase in CL for ADA titer ≥128 | 0.431 (5.0) | 0.292 to 0.757 |
| Variance of interindividual variability in CL, exponential error model | 0.055 | 0.039 to 0.067 |
| Variance of interindividual variability in Vc, exponential error model | 0.111 | 0.053 to 0.154 |
| Variance of interindividual variability in Ka, exponential error model | 0.336 | 0.140 to 0.575 |
| Variance of interindividual variability in Fe; additive error model in logit domain | 0.489 | 0.270 to 0.886 |
| Covariance between IIV CL and IIV Vc, % correlation | 0.031 | ‐0.0004 to 0.051 |
| Variance of proportional residual error | 0.036 | 0.033 to 0.038 |
ADA indicates antidrug antibodies; CL, clearance; F, bioavailability; IIV, interindividual variability; Ka, first‐order absorption rate constant; Q, intercompartmental clearance; SC, subcutaneous; Vc, central volume of distribution; Vp, peripheral volume of distribution.
% Relative standard error (%RSE) was estimated as the standard error of the estimate divided by the population estimate multiplied by 100.
%RSE and 95%CIs are relevant for the updated refined population pharmacokinetic model.
Bioavailability for SC dosing in global phase 3 or Japan phase 2/3 studies.
Estimate was back‐transformed from the logit scale (estimate on the logit scale was 2.09).
The estimates are provided in the logit domain.
Following administration of 75‐mg SC or 150‐mg SC doses of risankizumab at week 0, 4 and q12w thereafter in Japanese patients with moderate to severe plaque psoriasis, GPP, or EP, the incidence (treatment emergent) of ADA to risankizumab was 22.4% (15/67) and 31% (31/100), respectively, and the incidence of NAb was 14.9% (10/67) and 12% (12/100), respectively, over the 52‐week treatment duration. Among ADA‐evaluable Japanese subjects in phase 2/3 studies (N = 235), only 1 subject (∼0.4%) developed ADA titer ≥128. Subjects with ADA titer above 128 were estimated to have increased risankizumab clearance by 43% corresponding to an AUC decrease by 30%, on average (Figure 2).
Figure 2.
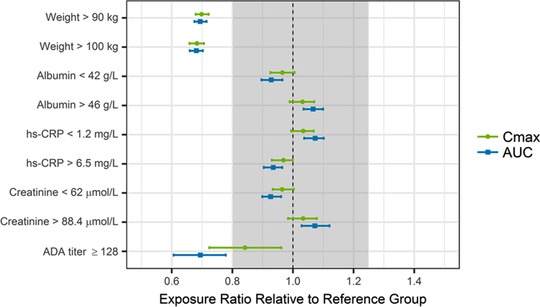
Impact of covariates on risankizumab steady‐state exposures. ADA indicates antidrug antibody; AUC, area under the plasma concentration‐time curve in dosing interval at steady state; Cmax, maximum plasma concentration at steady state; hs‐CRP, high‐sensitivity C‐reactive protein. Points and squares represent median values, and error bars represent 95%CIs of the normalized exposure ratios across 200 simulation replicates. The vertical black dashed line shows an exposure ratio of 1 relative to the reference group, and the shaded area represents the 0.8 to 1.25 default equivalence boundaries.
Based on the simulations, patients with body weight >90 or >100 kg were predicted to have ∼30% lower exposures (Cmax and AUC at steady state), on average, compared to patients weighing ≤100 kg or ≤90 kg following administration of risankizumab (Figure 2). None of the other statistically significant covariates, including baseline hs‐CRP, serum albumin, and serum creatinine, showed a meaningful impact on risankizumab exposures as risankizumab exposures in patients within upper or lower 25th percentiles of covariate distribution were estimated to be well within the default equivalence boundaries of 0.8 to 1.25 relative to the patients in the middle 50% of the covariate distribution for baseline hs‐CRP, serum albumin, or serum creatinine.
In this analysis risankizumab steady‐state exposures (Cmax, AUCtau, and Ctrough) in Japanese subjects with moderate to severe plaque psoriasis were comparable to those with GPP or EP. Consistent with the pharmacokinetic analysis based on study 1, risankizumab steady‐state exposures following administration of 150 mg SC in Japanese patients with psoriasis (moderate to severe plaque psoriasis, GPP, or EP) were approximately 17% higher than those in non‐Japanese patients with psoriasis (with moderate to severe plaque psoriasis). Risankizumab mean (SD) steady‐state Cmax, AUCtau, and Ctrough values in Japanese patients with psoriasis were approximately 14.5 (3.4) µg/mL, 560 (190) µg·day/mL, and 2.2 (1.3) µg/mL, respectively, and those in non‐Japanese patients with psoriasis were 12.4 (3.3) µg/mL, 490 (180) µg·day/mL, and 2.0 (1.2) µg/mL, respectively.
The population pharmacokinetic model adequately described the data as demonstrated with the goodness‐of‐fit plots (Supplementary Figure 3). These plots based on population and individual predictions showed lack of any bias when plotted against observed data, and with no obvious trends in plots of conditional weighted residuals versus population predictions and time. In the bootstrap analyses, out of 1000 replicates, 992 runs converged successfully. Based on the successful runs, the model parameters were precisely estimated as indicated by the 95%CIs, with negligible deviation from the original data set estimates (Table 5). Furthermore, VPCs for risankizumab plasma concentration data demonstrated that the model was able to describe the central tendency and variability in the observed data well (Figure 3).
Figure 3.
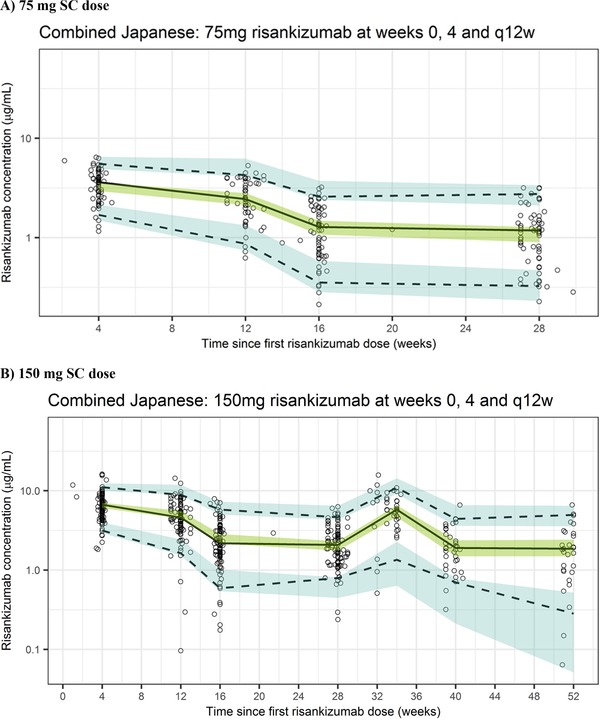
Visual predictive checks across phase 2/3 studies in Japanese patients with psoriasis who received risankizumab 75 mg SC (A) or 150 mg SC (B) at weeks 0 and 4 and every 12 weeks (q12w) thereafter using the updated population pharmacokinetic model. The black circles represent observed data; the lines represent observed median (solid black) and observed 5%/95% percentiles (dashed); the shaded regions represent the 95%CIs for the simulated median (green) and simulated 5%/95% percentiles (blue).
Discussion
Risankizumab is an anti‐IL‐23 antibody being developed for the treatment of moderate to severe plaque psoriasis and other inflammatory diseases. In this article pharmacokinetic results of risankizumab from a phase 1 trial in healthy Asian (Japanese or Chinese) subjects were presented and compared with healthy white subjects. Additionally, pharmacokinetic assessment from Japanese patients with moderate to severe plaque psoriasis, GPP, or EP are presented and compared with non‐Japanese patients. The analyses summarized here provide a robust assessment of pharmacokinetics of risankizumab in Japanese and Chinese populations.
Risankizumab displayed linear pharmacokinetics across doses evaluated in phase 1, 2, and 3 clinical trials (0.01 mg/kg to 5 mg/kg IV, 200 mg to 1200 mg IV, 0.25 mg/kg to 1 mg/kg SC, and 18 mg to 300 mg SC) with no apparent target‐mediated drug disposition across the evaluated dose range. This is consistent with pharmacokinetics of other monoclonal antibodies directed against soluble targets, which are primarily eliminated by Fc receptor‐mediated nonspecific clearance.13, 14 The pharmacokinetic profile of risankizumab following administration of single SC doses of 18–300 mg to healthy white, Japanese, and Chinese subjects in the phase 1 trial showed that risankizumab pharmacokinetics was consistent across ethnicities. Similarly, pharmacokinetic data following administration of repeated doses of 75–150 mg SC in patients showed that risankizumab pharmacokinetics was consistent across Japanese patients with different forms of psoriasis (plaque psoriasis, GPP, or EP) as well as across Japanese and non‐Japanese patients with plaque psoriasis.
A population pharmacokinetic model for risankizumab was developed previously for patients with moderate to severe plaque psoriasis, which was able to adequately describe risankizumab plasma concentration data from the Japan phase 2/3 trials (studies 2 and 3).12 Therefore, the previously developed population pharmacokinetic model was applied to the pooled data set including global clinical trials of risankizumab and Japan phase 2/3 trials (studies 2 and 3) to characterize risankizumab pharmacokinetics across the entire data set. Risankizumab pharmacokinetic parameters based on population pharmacokinetic analyses of the combined dataset of global and Japan trials (studies 2 and 3) were consistent (<5% relative difference) with the values obtained based on the data set comprising global trials alone (excluding Japan studies 2 and 3), confirming that the risankizumab plasma concentration data in Japanese patients is consistent with that in the global population.
Consistent with the pharmacokinetics of several other monoclonal antibodies, such as golimumab, ustekinumab, or cetuximab, risankizumab plasma exposures were numerically higher in the Asian population compared with non‐Asians.15 Risankizumab plasma exposures were up to 30% higher in Japanese and Chinese healthy subjects compared with whites and approximately 17% higher in Japanese patients with psoriasis compared with non‐Japanese patients with psoriasis. In the population pharmacokinetic analyses of risankizumab, body weight, but not race, showed a significant correlation with risankizumab clearance. The comparable body‐weight–adjusted exposures of risankizumab in healthy subjects and lack of correlation of race with risankizumab clearance in population pharmacokinetic analyses indicate that the observed differences in risankizumab exposures between Asian and white subjects are primarily driven by the differences in body weight and not due to race or ethnic differences. This is supported by the absence of ethnic differences in Fc receptor‐mediated nonspecific clearance for monoclonal antibodies and consistent with other published literature summarizing the pharmacokinetic comparison of different monoclonal antibodies between Japanese and non‐Japanese populations,15 especially for drugs that have soluble targets similar to risankizumab. The small difference in exposure (less than 20%) between Japanese and non‐Japanese patients is clinically not relevant and does not warrant any dose adjustment due to lack of any impact on safety based on observed data in Japan phase 2/3 studies as well as based on exposure‐response analyses for safety events using data from global phase 2 and 3 trials of risankizumab in patients with psoriasis.16
Because of the correlation of body weight with clearance, patients with body weights >100 kg or >90 kg were estimated to have 30% lower exposures compared with those weighing ≤100 kg or ≤90 kg. Exposure‐response analyses indicated that the clinical regimen of risankizumab 150 mg SC at week 0, week 4, and q12w thereafter achieved the plateau of efficacy (PASI 90 and sPGA 0/1) across the entire body‐weight range evaluated in subjects with psoriasis. Detailed reports of these analyses are forthcoming.
Immunogenicity of biologics can potentially affect the elimination clearance and consequently the systemic exposure. We have demonstrated previously that antibodies to risankizumab do not impact risankizumab clearance except in subjects with high titers ≥128, who represent a very small fraction (∼1.5%) of the subjects evaluated in global phase 2 or 3 psoriasis trials. In subjects with ADA titer ≥128, risankizumab clearance was estimated to increase by 43% (corresponding to AUC decrease by 30%), on average.12 Among ADA‐evaluable Japanese subjects in phase 2/3 studies (N = 235), only 1 subject developed ADA titer ≥128. Consistent with the lack of impact of 30% lower exposure in heavier patients on efficacy, the estimated average effect of ADA titer ≥128 is not clinically relevant. Similarly, none of the other covariates identified to be statistically correlated with risankizumab clearance (baseline serum creatinine, baseline albumin, and baseline hs‐CRP) in the population pharmacokinetic analyses of risankizumab is expected to be clinically relevant based on the lack of meaningful impact on risankizumab exposures as has been previously demonstrated.12
Conclusions
Risankizumab pharmacokinetics was comparable among healthy white, Japanese, and Chinese subjects, between Japanese patients with plaque psoriasis and those with GPP or EP, and also between Japanese and non‐Japanese patients with psoriasis, indicating a lack of ethnic impact on risankizumab pharmacokinetics. Compared with white subjects, Asian subjects tend to have slightly higher plasma exposures due to their lower body weight on average. These small differences in exposure have no clinical relevance and do not warrant any dose adjustment.
Conflicts of Interest
Doerthe Eckert, Ahmed Suleiman, Yinuo Pang, Ling Cheng, and Ahmed A. Othman are employees of AbbVie and may hold AbbVie stock or stock options. Amit Khatri and Rajneet Oberoi are former employees of AbbVie and may hold AbbVie stock or stock options.
Supporting information
Supplementary Figure 1. Risankizumab dose‐normalized Cmax and AUC∞ values (arithmetic mean ± SD) vs dose following single subcutaneous (SC) or intravenous (IV) doses in healthy subjects A, 18 mg, 90 mg, and 300 mg risankizumab SC. B, Risankizumab 200 mg, 600 mg, and 1200 mg IV.
Supplementary Figure 2. Observed and simulated risankizumab plasma concentration data in Japanese subjects who received 75 mg SC or 150 mg SC doses using the previously developed population pharmacokinetic model based on global phase 1, 2, and 3 studies.
Supplementary Figure 3. Goodness‐of‐fit plots for the updated risankizumab population pharmacokinetic model.
Supplementary Table 1. Risankizumab Plasma Ctrough (µg/mL) in Japanese Patients With Moderate to Severe Plaque Psoriasis, Generalized Pustular Psoriasis, or Erythrodermic Psoriasis
Financial Disclosures
This work was sponsored by Boehringer Ingelheim and AbbVie. Boehringer Ingelheim contributed to the study designs and data collection, and AbbVie contributed to the analysis and interpretation of data and the writing, review, and approval of the publication.
Acknowledgments
The authors thank Wesley Wayman, PhD, an employee of AbbVie, and Mia DeFino, MS, ELS, a freelance medical writer under contract with AbbVie, for medical writing support.
Data‐Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets) as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan and execution of a Data‐Sharing Agreement. Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Dr Ahmed A. Othman is a Fellow of the American College of Clinical Pharmacology.
Clinical Trial numbers for included studies: NCT02596217, NCT03000075, NCT03022045.
References
- 1. Conrad C, Gilliet M. Psoriasis: from pathogenesis to targeted therapies. Clin Rev Allergy Immunol. 2018;54(1):102‐113. [DOI] [PubMed] [Google Scholar]
- 2. Hoegler KM, John AM, Handler MZ, Schwartz RA. Generalized pustular psoriasis: a review and update on treatment. J Eur Acad Dermatol Venereol. 2018;32(10):1645‐1651. [DOI] [PubMed] [Google Scholar]
- 3. Singh RK, Lee KM, Ucmak D, et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:93‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh S, Kroe‐Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high‐affinity humanized anti‐IL23A antibody. mAbs. 2015;7(4):778‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. AbbVie . Press release: Risankizumab Meets All Co‐Primary and Ranked Secondary Endpoints, Achieving Significantly Greater Efficacy Versus Standard Biologic Therapies in Three Pivotal Phase 3 Psoriasis Studies. https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm. Accessed June 4, 2018.
- 7. AbbVie . Press release: Risankizumab Meets All Primary Endpoints Reporting Positive Results in Fourth Pivotal Phase 3 Psoriasis Study. https://news.abbvie.com/news/risankizumab-meets-all-primary-endpoints-reporting-positive-results-in-fourth-pivotal-phase-3-psoriasis-study.htm. Accessed June 4, 2018.
- 8. Gordon K, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab: results from two double‐blind, randomised, placebo‐ and ustekinumab‐controlled, phase 3 trials in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2). Lancet. 2018;392(10148):650‐661. [DOI] [PubMed] [Google Scholar]
- 9. Ohtsuki M, Fujita H, Watanabe M, et al. Efficacy and safety of risankizumab in Japanese patients with moderate to severe plaque psoriasis: results from the SustaIMM phase 2/3 trial. J Dermatol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suleiman AA, Khatri A, Minocha M, Othman AA. Population pharmacokinetics of the interleukin‐23 inhibitor risankizumab in subjects with psoriasis and Crohn's disease: analyses of phase I and II trials. Clin Pharmacokinet. 2019;58(3):375‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate‐to‐severe plaque psoriasis. N Engl J Med. 2017;376(16):1551‐1560. [DOI] [PubMed] [Google Scholar]
- 12. Suleiman AA, Minocha M, Khatri A, Pang Y, Othman AA. Population pharmacokinetics of risankizumab in healthy volunteers and subjects with moderate to severe plaque psoriasis: integrated analyses of phase I‐III clinical trials. Clin Pharmacokinet. 2019. May 4. 10.1007/s40262-019-00759-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24(1):23‐39. [DOI] [PubMed] [Google Scholar]
- 14. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493‐507. [DOI] [PubMed] [Google Scholar]
- 15. Chiba K, Yoshitsugu H, Kyosaka Y, et al. A comprehensive review of the pharmacokinetics of approved therapeutic monoclonal antibodies in Japan: are Japanese phase I studies still needed? J Clin Pharmacol. 2014;54(5):483‐494. [DOI] [PubMed] [Google Scholar]
- 16. Khatri A, Suleiman, AA , Polepally, AR , Othman, AA . Exposure‐ response relationships for efficacy and safety of risankizumab in patients with moderate to severe plaque psoriasis: integrated analyses of phase 2 and 3 clinical trials. American Academy of Dermatology Annual Meeting, March 1‐5, 2019; Washington, DC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Risankizumab dose‐normalized Cmax and AUC∞ values (arithmetic mean ± SD) vs dose following single subcutaneous (SC) or intravenous (IV) doses in healthy subjects A, 18 mg, 90 mg, and 300 mg risankizumab SC. B, Risankizumab 200 mg, 600 mg, and 1200 mg IV.
Supplementary Figure 2. Observed and simulated risankizumab plasma concentration data in Japanese subjects who received 75 mg SC or 150 mg SC doses using the previously developed population pharmacokinetic model based on global phase 1, 2, and 3 studies.
Supplementary Figure 3. Goodness‐of‐fit plots for the updated risankizumab population pharmacokinetic model.
Supplementary Table 1. Risankizumab Plasma Ctrough (µg/mL) in Japanese Patients With Moderate to Severe Plaque Psoriasis, Generalized Pustular Psoriasis, or Erythrodermic Psoriasis