

Abstract
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disorder caused by opportunistic infection of JC polyomavirus (JCV). Today, increased attention has been focused on PML development in multiple sclerosis (MS) patients under disease‐modifying therapies (DMT). Although in the acquired immunodeficiency syndrome (AIDS) era, PML was thought to be a rapidly progressive disease with poor prognosis, drug‐associated PML is relatively slow in progress, and a favorable outcome may be expected with early diagnosis. However, early PML diagnosis on magnetic resonance imaging (MRI) is frequently difficult, and JCV DNA copy number in cerebrospinal fluid (CSF) is usually low. To facilitate early PML diagnosis on MRI, the pre‐mortem images were compared with neuropathology of the post‐mortem brain, and underlying pathology corresponding to the MRI findings was evaluated. As a result, PML lesions of the autopsied brain were divided into three parts, based on the disease extension patterns: (A) Progressive white matter lesion in the right frontoparietal lobe including the precentral gyrus. Huge demyelinated lesions were formed with fusions of numerous small lesions. (B) Central lesion including deep gray matters, such as the putamen and thalamus. The left thalamic lesion was contiguous with the pontine tegmentum. (C) Infratentorial lesion of brainstem and cerebellum. Demyelination in the pontine basilar region and in cerebellar white matter was contiguous via middle cerebellar peduncles (MCPs). In addition, (D) satellite lesions were scattered all over the brain. These observations indicate that PML lesions likely evolve with three steps in a tract‐dependent manner: (1) initiation; (2) extension/expansion of demyelinating lesions; and (3) fusion. Understanding of the PML disease evolution patterns would enable confident early diagnosis on MRI, which is essential for favorable prognosis with good functional outcome.
Keywords: disease‐modifying therapy (DMT), JC virus (JCV), magnetic resonance imaging (MRI), multiple sclerosis (MS), progressive multifocal leukoencephalopathy (PML)
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disorder of the central nervous system (CNS) caused by opportunistic infection with JC polyomavirus (JCV). JCV reactivates in patients with immunosuppressive states, and since the 1980s, a number of PML cases have been reported, mainly as complications of acquired immunodeficiency syndrome (AIDS). Advanced PML lesions in patients with AIDS typically present as huge hyperintense signals on T2‐weighted/fluid‐attenuated inversion recovery (FLAIR) images, which spread in the white matter of the frontoparietal lobes.1, 2 In the AIDS era, it was thought that PML white matter lesions rapidly progress, and prognosis is poor (classical PML).
Recently, increased attention has been focused on PML development in patients with multiple sclerosis (MS) who are under disease‐modifying therapies (DMT).3 Natalizumab (NTZ) is a monoclonal antibody against the integrin alpha‐4 subunit CD49a and is highly effective on relapsing and remitting MS. However, the higher risk of PML development has been a serious concern. As of 30 November, 2018, the global overall incidence of PML in NTZ‐treated patients is: 4.15 per 1000 patients (95% CI 3.87 to 4.44 per 1000 patients), and as of 5 December, 2018 there have been 804 confirmed PML cases. (Biogen, data on file, https://tys.ms-supportnavi.com/ja-jp/home/risk/risk01.html). Fingolimod (FTY720), which is a first‐in‐class orally bioavailable compound for the treatment of MS, is also at issue. So far, 15 patients with MS have developed FTY‐associated PML (FTY‐PML) worldwide, and four of these patients were Asian.4, 5 The incidence of FTY‐PML is 0.082 per 1000 worldwide (Novartis, data on file). Early diagnosis, preferably in an asymptomatic stage is essential (asymptomatic PML). Small asymptomatic PML lesions slowly progress, relating to favorable prognosis with a better functional outcome.6 However, JCV DNA copy numbers measured by quantitative polymerase chain reaction (PCR) in cerebrospinal fluid (CSF) are usually low or under the detection level,7, 8 which leads to difficulty in diagnosis.
On magnetic resonance imaging (MRI), early signs of NTZ‐PML have been described as punctate pattern, star‐like or Milky Way appearance. There is also cortical gray matter involvement and juxtacortical/subcortical white matter lesions.9, 10, 11, 12, 13, 14, 15, 16 However, most MRI findings have been acquired using 3T, and even 7T MRI, and it is thus not known whether the above signs are specific to NTZ‐PML or ubiquitously seen in patients with PML in the early stages. Moreover, the underlying PML pathology corresponding to the newly described MRI findings is not yet fully elucidated, and it is thus difficult to differentiate early PML lesions from those associated with MS. Therefore, in this study, an autopsied PML brain without MS lesions was examined and compared to the pre‐mortem MR images. The patient died from arrhythmia, and most of the PML lesions were still developing at the time of death. Pathologically proven PML disease evolution patterns in the absence of MS lesions may contribute to diagnostic assurance for early PML detection on MRI. The aim of this study is to elucidate pathology of PML evolution patterns and to facilitate early diagnosis on MRI.
CLINICAL SUMMARY
A 52‐year‐old man with a history of severe chronic heart failure due to myocardial infarction, diabetes mellitus, hypertension, dyslipidemia, and chronic kidney disease presented with a 170‐day clinical course, as summarized in Figure 1A. Due to diagnostic uncertainty, MRI was performed 10 times during the 170‐day clinical course. Contrast administration was not performed due to renal dysfunction. Representative MR images are presented in Figure 1B.
Figure 1.
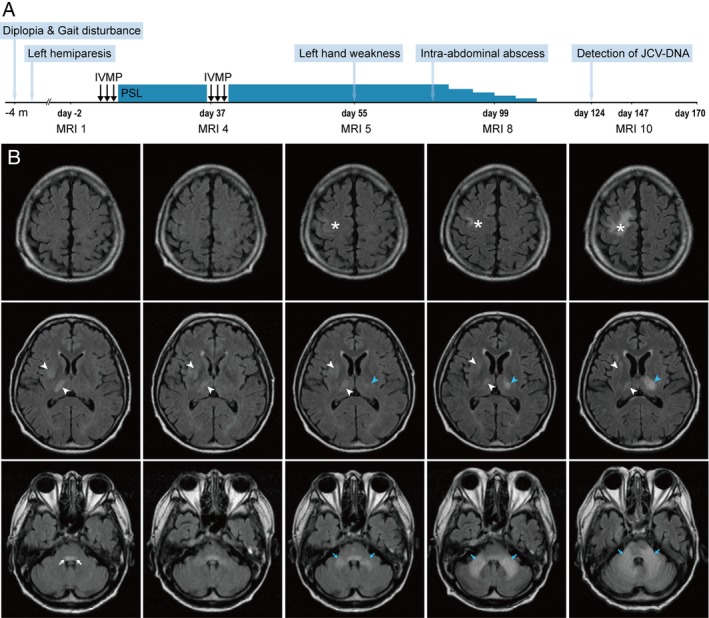
Clinical course and FLAIR images. (A) Clinical course. A 52‐year‐old man first developed diplopia, gait disturbance, and left hemiparesis. After hospital admission, the patient was treated with intravenous methylprednisolone (IVMP) and prednisolone (PSL) under the provisional diagnosis of malignant lymphoma. On day 124, JCV DNA was detected in CSF, leading to the diagnosis of PML. The patient died of arrhythmia on day 170. (B) FLAIR images. During the clinical course, MRI were acquired 10 times. Representative FLAIR images (MRI 1, 4, 5, 8, and 10) acquired respectively on days ‐2, 37, 55, 99, and 147, are presented here. On day ‐2 (MRI 1), FLAIR hyperintense lesions in the right thalamus and right putamen (white arrowheads) were observed but these lesions did not grow. A hyperintense lesion was also seen in the pontine tegmentum (white arrows). The patient gradually developed weakness of the left hand. A new lesion appeared in the right precentral gyrus on day 55 (MRI 5) and grew larger until the patient's death (asterisks). Lesions of the left thalamus (blue arrowheads) and bilateral MCPs (blue arrows) also emerged and progressed, although lesions of the right thalamus and putamen did not change after admission.
The patient developed diplopia and gait disturbance 4 months before admission. Dysesthesia around the left corner of the mouth and mild weakness of the left upper and lower limbs followed. Before hospital admission, hyperintense signals on T2‐weighted/fluid‐attenuated inversion recovery (FLAIR) images appeared in the right putamen, right thalamus, and pontine tegmentum (Fig. 1B, white arrowheads and white arrows). On admission, neurological examination revealed left abducens nerve palsy, mild left hemiparesis, and truncal cerebellar ataxia. The results of screening for human immunodeficiency virus, autoimmune disease, and vitamin deficiency were negative. Although the potential diagnoses included malignant lymphoma, cytology of CSF and a random skin biopsy did not suggest the presence of any malignancy. His chronic heart failure was so severe that we could not perform brain biopsy. During hospitalization, the T2/FLAIR hyperintense lesions in the pontine tegmentum gradually enlarged (Fig. 1B, white arrows), although those in the right putamen and thalamus were rather stable (Fig. 1B, white arrowheads). On day 9, corticosteroid therapy was initiated under the provisional diagnosis of malignant lymphoma, but resulted in no remarkable response.
On day 55, the weakness in the left hand was exacerbated and MRI revealed three new lesions on T2‐weighted/FLAIR images. Cortical signals appeared in the precentral gyrus of the right frontal lobe and later extended to the deep white matter (Fig. 1B, asterisks). In the posterior fossa, the signals of bilateral middle cerebellar peduncles (MCPs) emerged, and extended to both the pontine basilar region and the cerebellar hemisphere (Fig. 1B, blue arrows). In the left thalamus, new hyperintense signals developed and slowly grew (Fig. 1B, blue arrowheads). On day 78, an intra‐abdominal abscess was discovered as a complication and corticosteroid therapy was gradually tapered off. The neurological manifestations worsened at this time. On day 124, JCV DNA was detected with high titer (1.0 × 104 copies/mL) in the CSF, and the clinical diagnosis of PML was determined. The patient died of arrhythmia on day 170.
PATHOLOGICAL FINDINGS
Neuropathological findings from the post‐mortem brain are summarized in Table 1. There were three major lesions of different extension patterns, with different disease activities: (A) progressive white matter lesion in the right frontoparietal lobe; (B) central lesion including deep gray matter, thalamus and putamen, associated with the pontine tegmentum; and (C) infratentorial lesion of brainstem and cerebellar white matters, associated via the MCPs. In addition, (D) satellite lesions were scattered in the corticomedullary borders and in the deep white matter all over the brain. Although some autopsied PML cases have presented fatal inflammation called PML‐immune reconstitution inflammatory syndrome (IRIS),17, 18 inflammatory reactions were not apparent in this case.
Table 1.
Demyelinating/demyelinated lesions in an autopsied progressive multifocal leukoencephalopathy (PML) brain
|
Progressive white matter lesion in the right frontoparietal lobe: Typical PML lesion due to “extension, expansion, and fusion” of demyelinating lesions
Histopathological sections of the right cerebral hemisphere of the post‐mortem brain displayed a large demyelinated lesion occupying the frontal and parietal lobes, including the precentral gyrus. Klüver‐Barrera (KB) staining revealed that well‐demarcated oval demyelinating lesions approximately 2–3 mm in size were fused to one another to form a huge white matter lesion (Fig. 2A). This finding corresponded well with the last FLAIR images obtained on day 147 (Fig. 2B). In both the post‐mortem brain and the pre‐mortem MR images, demyelination or degeneration was more advanced in the deeper white matter, whereas cortical and subcortical areas of the precentral gyrus were rather mildly affected.
Figure 2.
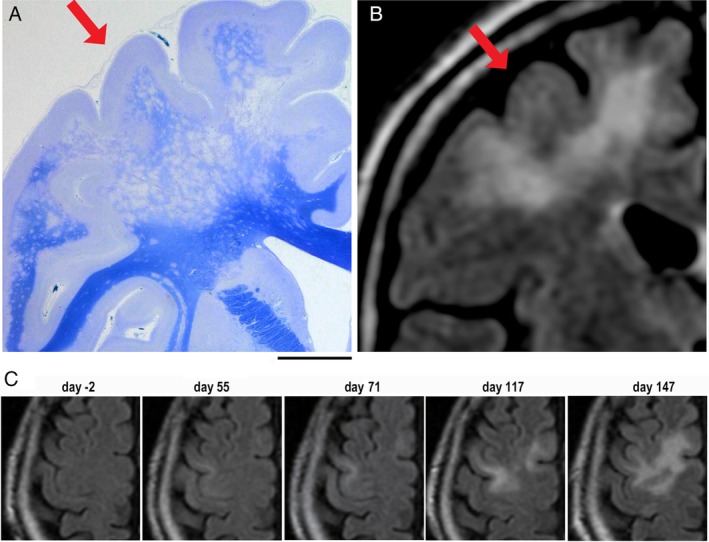
Progression of white matter lesions in the right frontoparietal lobe. (A) A semimacroscopic image of the right cerebral hemisphere displays a large demyelinated lesion occupying the frontoparietal lobe. Small demyelinating lesions fused to form a huge lesion in the deep white matter. KB staining. (B) A FLAIR image on day 147. Hyperintense signals are distinct in the corresponding lesion (red arrows). (C) Axial FLAIR images of the precentral cortex in the right frontal lobe (days ‐2, 55, 71, 117, and 147). Hyperintense linear signals first developed along the corticomedullary junction on day 55 and later expanded deeply to the white matter. Scale bar: 1 cm.
FLAIR images of the right frontoparietal lobe, especially those of the precentral gyrus, were reviewed (Fig. 2C). Although the precentral gyrus was not affected at the time of the first visit (on day ‐2), hyperintense linear signals developed nearly 2 months later (on day 55) at juxtacortical/subcortical regions. The hyperintense signals extended deeply to the white matter, where the signals from adjacent gyri were fused to form larger ones (on days 117, 147). This corresponded well with the neuropathology of the post‐mortem brain (Fig. 2A). These observations indicate that PML demyelinating lesions first appear, in part, in the cerebral cortex and/or juxtacortical/subcortical white matter. These demyelinating lesions likely later extend deeply to the white matter and then expand and fuse to form larger demyelinating lesions.
“Extension” likely regulates JCV spread and disease progression
To further investigate developing patterns of PML lesions, cortical and juxtacortical/subcortical white matter areas were more carefully examined all over the brain. Small demyelinating lesions were scattered mostly in the juxtacortical/subcortical white matter and in cortical regions everywhere in the brain, including the right and left frontal, parietal, temporal, and occipital lobes (Fig. 3A–D). However, most small demyelinating lesions were locally present around the corticomedullary junction, and extension of demyelinating lesions to the deeper white matter was seen in relatively restricted areas.
Figure 3.
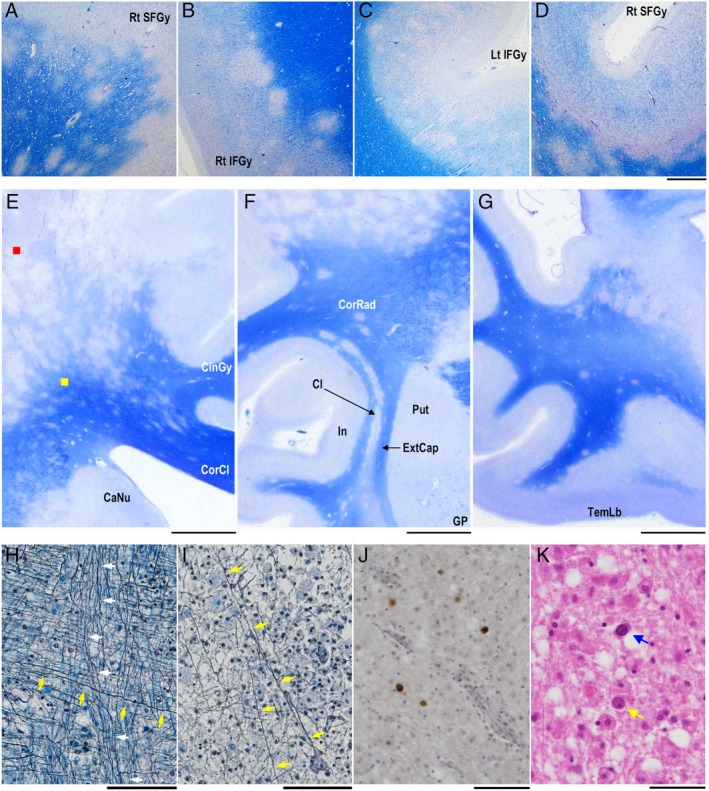
Small demyelinating lesions in the cortical gray matter and juxtacortical/subcortical white matter. (A–D) Small demyelinating lesions in the cortical gray matter and juxtacortical/subcortical white mater are observed all over the brain. KB staining. (E, F, G) Coronal sections of the right cerebrum. Demyelinating lesions extend to the corpus callosum in panel E and are aligned with the claustrum in panel F, or are rather close to the cortex in the temporal lobe in panel G. KB staining. (H) Axons with and without myelin sheaths in the deep white matter. Axons extending toward the corpus callosum (yellow arrows) and those toward the internal capsule (white arrows) cross each other in the deep white matter (yellow square in panel E). Fibers extending toward the corpus callosum are demyelinated, but those toward the internal capsule are not. Bodian + Luxol fast blue (LFB) staining. (I) Axon destruction in advanced demyelinated lesion (red square in panel E). Although a few fibers are observed (yellow arrows), most axons are fragmented and numerous myelin‐laden macrophages appear. Bodian + LFB staining. (J) Immunostaining for JCV capsid proteins reveal the presence of infected glial cells in the borders of expanding demyelinating lesions. (K) JCV‐infected glial cells with full inclusions (blue arrow) and cells with dot‐shaped intranuclear structures (yellow arrow). HE staining. CaNu, caudate nucleus; Cl, claustrum; CinGy, cingulate gyrus; CorCl, corpus callosum; CorRad, corona radiata; ExtCap, external capsule; GP, globus pallidus; In, insula; Lt IFGy, left inferior frontal gyrus; Rt IFGy, right inferior frontal gyrus; Put, putamen; Rt SFGy, right superior frontal gyrus; TemLb, temporal lobe. Scale bars: 1 mm (A‐D), 1 cm (E), 5 mm (F, G), 100 μm (H‐J), 50 μm (K).
In the frontoparietal lobe, multiple demyelinating lesions extended deeply to the white matter and some lesions skipped toward the corpus callosum (Fig. 3E). Small demyelinating lesions were also aligned with the claustrum, but these lesions were discontiguous from the huge demyelinated lesions in the frontoparietal lobes (Fig. 3F). In the temporal lobe, small demyelinating lesions were also scattered in the juxtacortical/subcortical area, but they hardly extended to the deep white matter (Fig. 3G).
Demyelinated axonal fibers were evaluated using Bodian plus Luxol fast blue (LFB) staining. In the borders of expanding lesions (Fig. 3E, yellow square), axonal fibers extending toward the corpus callosum were demyelinated (Fig. 3H, yellow arrows), while the myelin sheaths of those extending toward the internal capsule were intact (Fig. 3H, white arrows). In contrast, in advanced lesions (Fig. 3E, red square), most demyelinated axons were fragmented with infiltration of numerous myelin‐laden macrophages. Few demyelinated fibers remained in these areas (Fig. 3I, yellow arrows). JCV‐positive oligodendroglias were usually seen in the borders of expanding lesions (Fig. 3J). The nuclei of oligodendroglia‐like cells were relatively small, and some cells contained dot‐shaped inclusions described as early signs of JCV infection.19
The brainstem includes white matter tracts for “extension” of demyelination
In the right putamen and thalamus, the earliest MRI signals appeared on day ‐2, but the lesion did not grow larger for almost 6 months (inactive lesion). In contrast, hyperintense FLAIR signals later developed in the left thalamus on day 55, and this lesion grew larger until the patient's death (active lesion) (Fig. 1B). Thus, coronal FLAIR images were compared between day 55 and day 117. The signals in brainstem and bilateral thalami seemed discontiguous on day 55, but contiguous disease extension involving the left thalamus, brainstem, and bilateral cerebellar hemispheres was suggested on day 117 (Fig. 4A).
Figure 4.
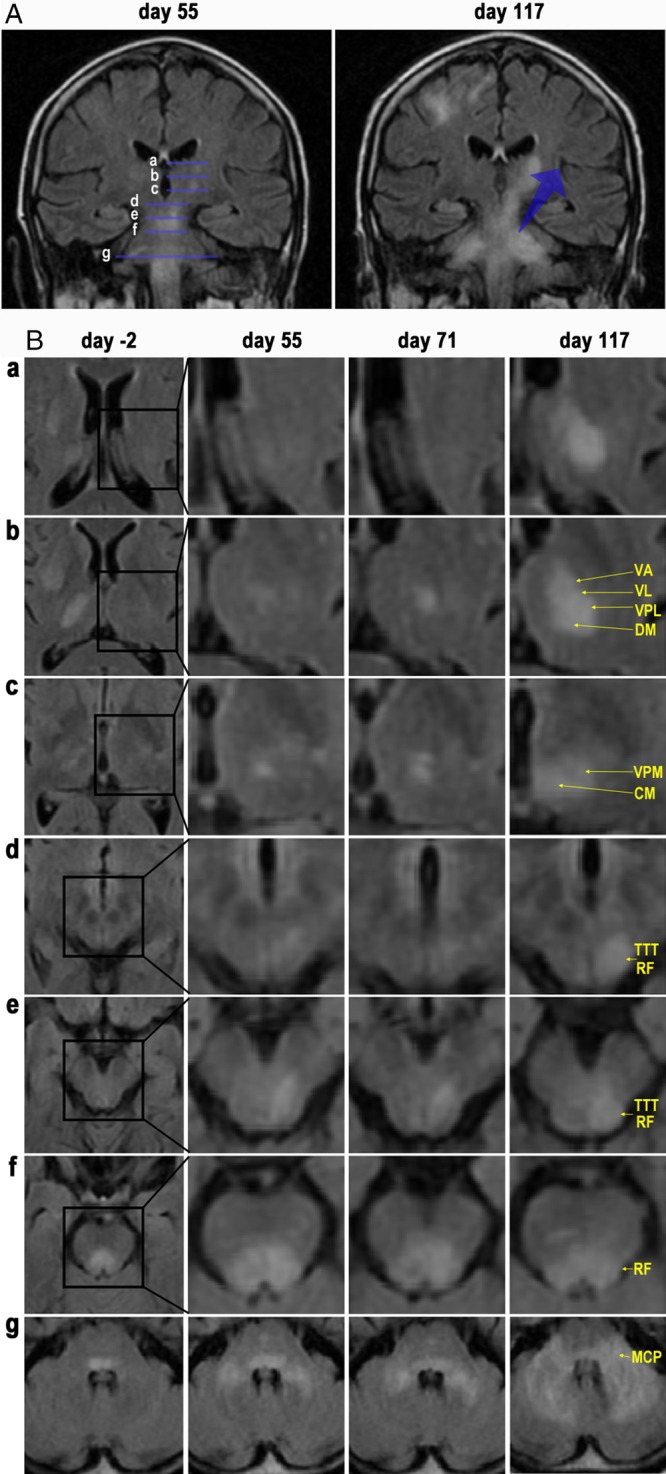
FLAIR images of the central and infratentorial lesions. (A) Coronal FLAIR images obtained on day 55 and day 117. PML lesions extend from the brainstem to the left thalamus. The lesions also extend from the pons to the bilateral cerebellar hemispheres. Lines a–g correspond to the sections a–g, in panel B, respectively. (B) Axial FLAIR images on days ‐2, 55, 71, and 117 indicate that the hyperintense lesions extend from the pontine tegmentum to the left thalamus. The lesions also expand from the MCPs to the pontine basilar regions and the cerebellar hemispheres. CM, centromedian nucleus; DM, dorsomedial nucleus; MCP, middle cerebellar peduncle; RF, reticular formation; TTT, trigeminothalamic tract; VA, ventral anterior nucleus; VL, ventral lateral nucleus; VPL, ventral posterolateral nucleus; VPM, ventral posteromedial nucleus.
MR images from day ‐2 to day 117 also revealed continuous and contiguous extension of hyperintense FLAIR signals. These signals were initiated in the pontine tegmentum, extended via the midbrain including the reticular formation (RF) and trigeminothalamic tract (TTT), and seemed to reach the centromedian nucleus (CM) and ventral posteromedial nucleus (VPM) of the left thalamus, where they further expanded to adjacent thalamic nuclei (Fig. 4B).
Neuropathology revealed marked demyelination in the brainstem, including the midbrain, pons, and medulla oblongata. KB staining demonstrated uneven distribution of demyelinating lesions, indicating involvement of specific nerve fibers or tracts (Fig. 5). In the left dorsal region of the midbrain, the RF and TTT were severely degraded, but interestingly the bilateral oculomotor nuclei (OMN), medial longitudinal fasciculus (MLF), and superior cerebellar peduncle decussation (SCPD) were clearly intact (Fig. 5A and B). In the pontine tegmentum, diffuse demyelinating areas were observed in regions such as the RF and TTT, but the MLF was clearly preserved within the severely demyelinated region. In the pontine basilar region, pontocerebellar fibers (PCF) were markedly degraded, and the lesions were contiguous to the MCPs extending toward the cerebellar white matter. Pontine longitudinal fibers (LF) were also severely damaged (Fig. 5C and D). The demyelinating lesions in the pontine basilar regions were not contiguous with those in the pontine tegmentum, and the medial lemniscus (ML) was well preserved between the two demyelinating lesions. This indicates that pontine basilar regions and tegmentum are involved in distinct tract‐dependent disease extension.
Figure 5.
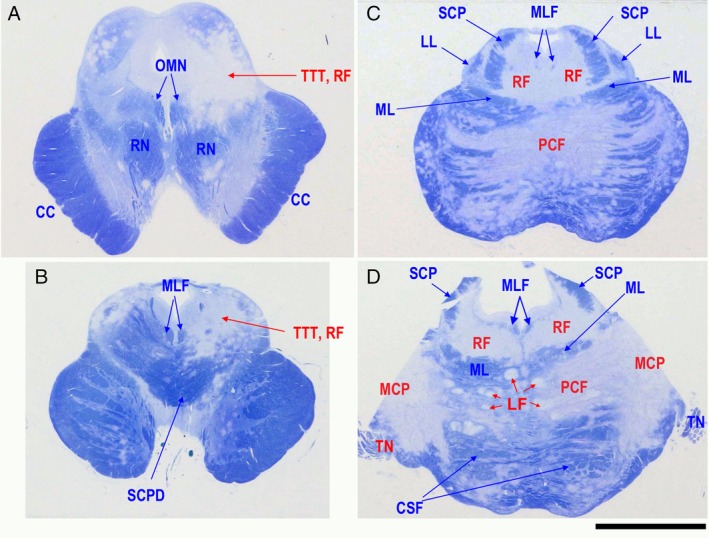
Semimacroscopic images of sections from the midbrain (A, B) and pons (C, D) sections stained with Klüver‐Barrera. In the midbrain and pontine tegmentum, the white matter, including nerve fibers such as trigeminothalamic tract (TTT) and reticular formation (RF) is markedly degenerated. In contrast, medial longitudinal fasciculus (MLF) and oculomotor nucleus (OMN) are well preserved. In the pontine base region, demyelination expands from the middle cerebellar peduncle (MCP) to both directions of pontocerebellar fibers (PCF) and the cerebellar white matter. CC, crus cerebri; LF, longitudinal fibers; LL, lateral lemniscus; ML, medial lemniscus; RN, red nucleus; SCP, superior cerebellar peduncle; SCPD, superior cerebellar peduncle decussation; TN, trigeminal nerve. Scale bar: 1 cm.
PML lesions involve basal ganglia and cerebellar dentate nuclei
The basal ganglia were carefully evaluated. In the bilateral thalami, lateral regions, including the ventral posterolateral nucleus (VPL) were severely affected, although less severe degeneration was also observed in the bilateral dorsomedial nucleus (DM). Tissue damage was more distinct in the active left thalamic lesion, which was contiguous with the left red nucleus (RNu). Mildly demyelinating lesions were also observed in the right RNu and bilateral substantia nigra (SN) (Fig. 6A). In the pontine tegmentum, the bilateral loci coerulei were relatively well preserved (Fig. 6B), but in the pontine basilar region, the pontine nuclei were moderately affected. Advanced PML lesions were observed in the cerebellar white matter. As a result, the left dentate nucleus was no longer recognizable (Fig. 6C), although the right dentate nucleus was barely observable (data not shown). Histopathologically, neurons in these lesions were severely degenerated or lost (Fig. 6D, E), and thus, PML is not only a white matter disease, but also involves gray matter.
Figure 6.
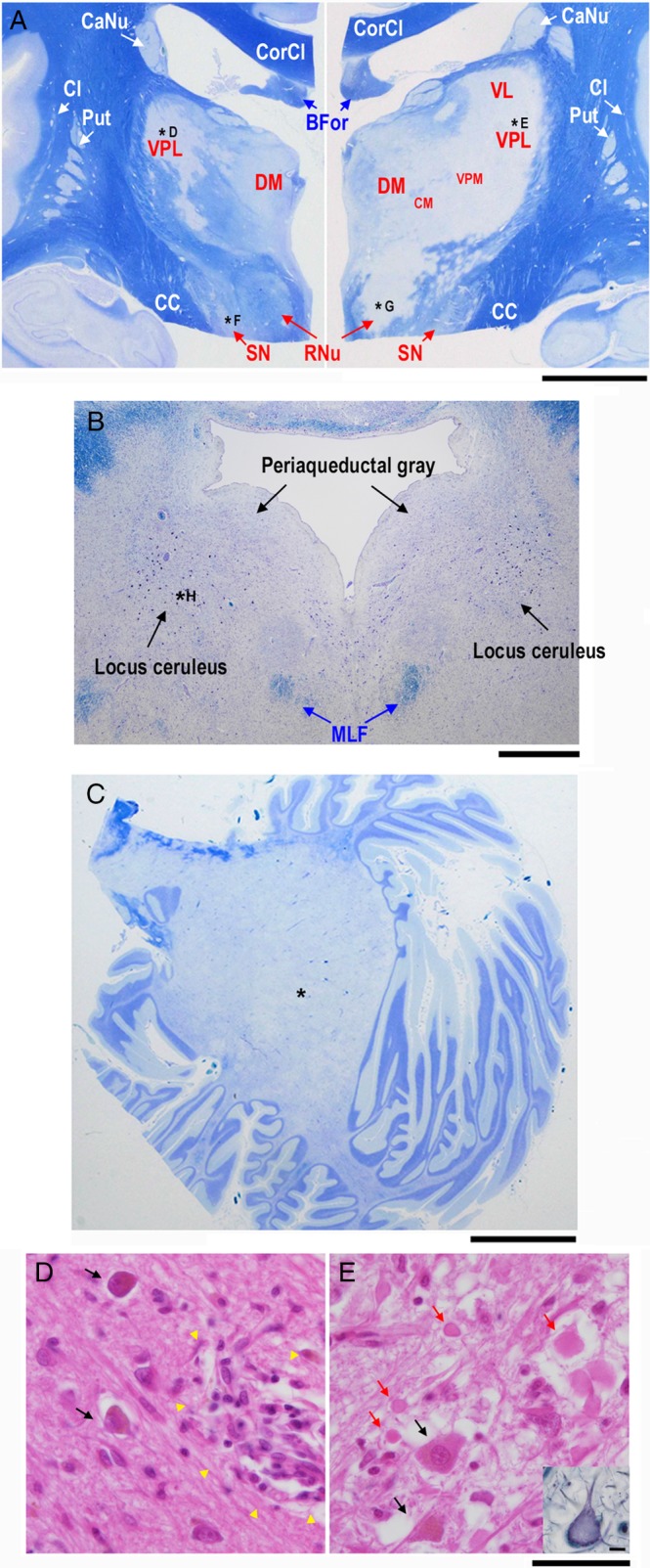
Semimacroscopic images of the basal ganglia and thalamus (A), pontine tegmentum (B), and cerebellar hemisphere (C) sections stained with Klüver‐Barrera as well as microscopic images of right (D) and left (E) thalami sections stained with hematoxylin‐eosin (D, E) and Bodian (inset in E). Demyelinating lesions are apparent in both sides of the thalami, red nuclei (RNu), substantia nigra (SN), pontine tegmenta, and cerebellar hemispheres. Neurons in both sides of the thalami are severely degenerated or lost. Ghost neurons are especially prominent in the ventral posterolateral nucleus (VPL) of the left thalamus (inset in E). CaNu, caudate nucleus; CC, crus cerebri; Cl, claustrum; CM, centromedian nucleus; CorCl, corpus callosum; DM, dorsomedial nucleus; Put, putamen; VL, ventral lateral nucleus; VPM, ventral posteromedial nucleus. Scale bars: 1 cm (A‐C), 10 μm (D, E).
Early and small demyelinating lesions in the deep white matter
Even though our 1.5T MRIs did not suggest the presence of PML lesions in the temporal and occipital lobes in the clinical setting (Fig. 7A), the post‐mortem study revealed early and small demyelinating lesions scattered all over the brain. KB staining in the occipital lobe revealed markedly small demyelinating lesions on a background of diffuse myelin pallor (Fig. 7B). The pathological features resembled “starry” or “Milky Way” appearance, which is known as early NTZ‐PML lesions on 7T MRI.10 Early and small demyelinating lesions developed along with perivascular and parenchymal edema, and blood vessels were not necessarily intralesionally observed (Fig. 7C–E). These findings were in good agreement with 7T MRI findings in patients with early NTZ‐PML,10 and were distinct from MS pathology characterized by the central intralesional vein.20 Interestingly, perivascular edema in the thalamic lesion resembled a lacunar infarction (Fig. 7F). The early and small demyelinating lesions were approximately 400 μm in diameter (Fig. 7G–I), and oligodendroglia‐like cells with small but swollen nuclei were accompanied around the early and small demyelinating lesions. Intranuclear features with dot‐shaped structures suggested the presence of JCV‐infected cells in the early stage (Fig. 7J); however, typical JCV‐infected cells with amphophilic viral inclusions in markedly enlarged nuclei had not yet developed.
Figure 7.
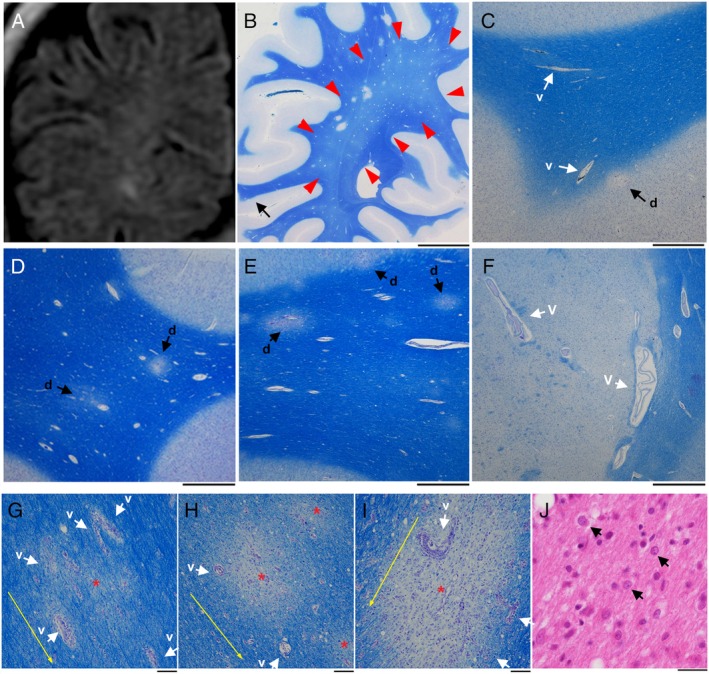
Early and small demyelinating lesions with perivascular edema. (A) An MRI FLAIR image (1.5 T). A coronal view of the right occipital lobe on day 147 suggests unclear signal intensity in the deep white matter. (B‐I) Semimacroscopic images of autopsied brain sections stained with Klüver‐Barrera. The lesions are scattered, and red arrows indicate myelin pallor (B). At a higher magnification of a part of panel B (black arrow in panel B), the lesions are present at the corticomedullary border (indicated by “d” and black arrow), and mildly enlarged blood vessels are also seen in this region (indicated by “v” and white arrow) (C). The lesions are also seen in the frontal lobe (indicated by “d” and black arrows) along with mildly enlarged blood vessels (D, E). Blood vessels in the left thalamus are enlarged and perivascular edema is seen. This resembles a lacular infarct (indicated by “v” and white arrows) (F). The lesions are present along with vascular proliferation in the surrounding area (indicated by “v” and white arrows), and red asterisks and yellow arrows indicate the centers of demyelinating lesions and the direction of nerve fiber extension, respectively (G‐I). (J) A microscopic image of a brain section stained with hematoxylin‐eosin. Oligodendroglia‐like cells with mildly enlarged nuclei are present (arrows) around early and small demyelinating lesions. Some of these cells bear dot‐shaped intrauclear structures. Scale bars: 1 cm (B), 1 mm (C‐F), 100 μm (G‐I), 25 μm (J).
DISCUSSION
JCV infects mostly oligodendrocytes, and propagates progenies in the enlarging nuclei. Previous studies have demonstrated that JCV‐infected glial cells activate the cell cycle from a S‐to‐G2‐like state, in which punctate intranuclear domains, called promyelocytic nuclear bodies (PML‐NBs) provide scaffolds for viral proliferation.19 Thus, intranuclear punctate structures (dot‐shaped inclusions), reflecting JCV proliferation at PML‐NBs, are early signs of JCV infection. The reproduced viruses later spread to fill the markedly enlarged nuclei (full inclusions) and finally lyse the host cells.19, 21, 22, 23, 24 Progressive demyelinating lesions develop with continuous viral infection. However, most studies thus far have focused on the process in which JCV propagates within the nucleus.25 It is thus not yet well known how JCV is later spread within the brain and progresses as demyelinating disease. Here, we propose that dynamic PML evolution consists of three steps: (1) initiation, (2) extension/expansion of demyelinating lesions, and (3) fusion of the demyelinating lesions (Figs 8, 9).
Figure 8.
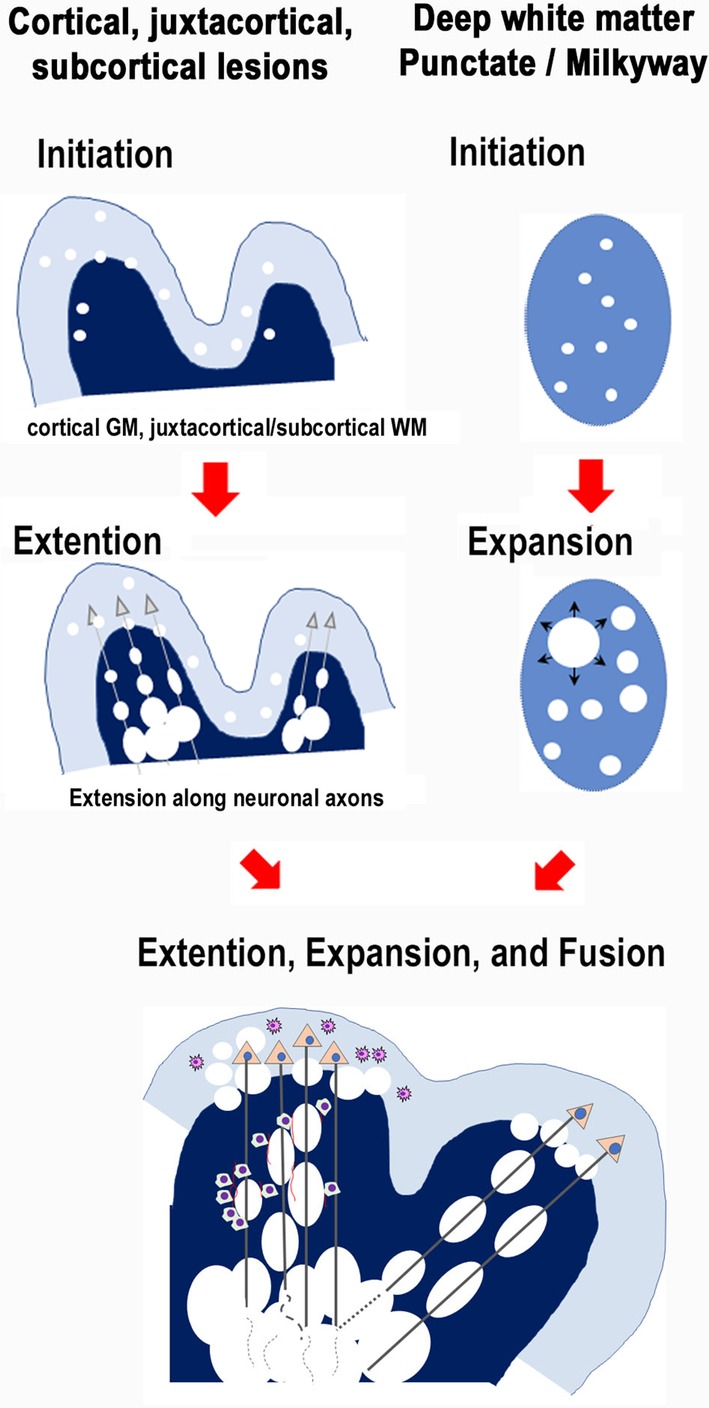
Evolutional patterns of PML demyelinating lesions with schematic illustrations including “initiation”, “extension”, and “fusion”. Initiation of early and small demyelinating lesions likely occurs in vascular‐rich areas. Thus, the cortical gray matter, juxtacortical/subcortical white matter, and even the basal ganglia are likely initiation sites for early demyelination. Demyelinating lesions extend discontiguously at distant locations along nerve fibers or expand locally. The lesions fuse each other (fusion) to form larger lesions.
Figure 9.
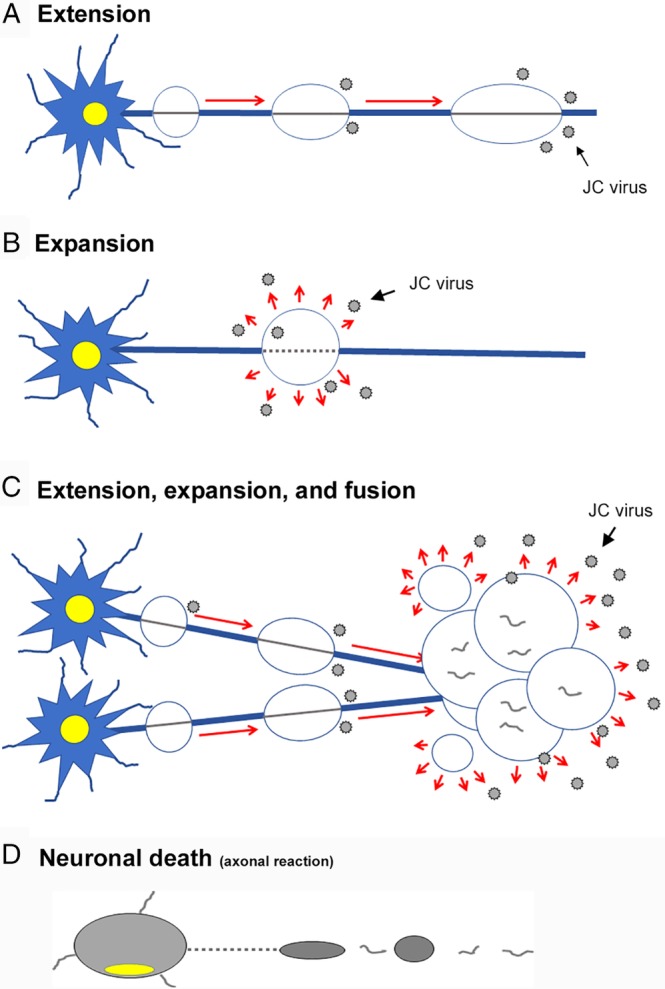
Schematic diagrams showing pathologic processes of PML. How do the PML lesions spread in a tract‐dependent manner? JCV progenies likely spread along nerve axons. (A) In the extension stage, JCV forms proliferation foci along axons in a skipping manner. JCV‐infected cells are usually seen in the borders of extending lesions. (B) In the expansion stage, JCV proliferates locally, leading to expansion of the demyelinating areas. JCV‐infected cells are seen in the areas around expanding lesions. (C) In the fusion stage, the demyelinating lesions fuse with one another to form larger lesions. The brain tissue is severely damaged with axonal destruction. (D) In neuronal death stage, neurons die due to axonal destruction.
First step: “Initiation” as a small demyelinating lesion
We hypothesize that the maternal virus likely reaches the brain via blood vessels, and thus the areas rich in vasculature would be the first targets of virus proliferation. In this case, linear hyperintense FLAIR signals developed in the precentral corticomedullary borders, where pathology revealed the appearance of small demyelinating lesions. Indeed, the small demyelinating lesions around the corticomedullary border were seen all over the brain. These lesions are highly likely the initial PML lesions, in agreement with a previous description of “early pathological changes” in asymptomatic non‐AIDS cases.26
In the present case, hyperintense FLAIR signals also first appeared in the basal ganglia, including the putamen and thalamus, as well as in the pontine tegmentum. In addition, punctate small and early demyelinating lesions were scattered in the deep white matter all over the brain. MRI findings, which were consistent with those described above have been previously reported as variations of PML lesions.9, 14, 15, 27, 28 Therefore, the “initiation” of PML lesions can occur in multiple locations, if vasculature exists. The next steps, “extension” and “expansion” thus seem very important to define the progression of demyelinating lesions.
Second step: “Extension” and “expansion” of demyelinating lesions
We propose the existence of three patterns in the progression of multifocal demyelinating lesions: “extension”, “expansion”, and “fusion” (Fig. 8). In the present case, alignment of demyelinating lesions along nerve fibers was revealed using Bodian + LFB staining, indicating that discontiguous virus proliferation foci increased in number along neuronal axons (in a skipping manner). Therefore, “extension” of demyelinating lesions is tract‐dependent. In good agreement, neuropathology and MRI findings in this study indicated three major PML lesions defined with “extension” patterns; (A) typical cerebral lesion extending from the corticomedullary border to the deep white matter and partly to the corpus callosum in the frontoparietal lobe; (B) central lesion extending from the pontine tegmentum to the left thalamus, including the RF and TTT; and (C) infratentorial lesion extending from the bilateral MCPs to both pontine basilar fields and cerebellar hemispheres. The extension of PML lesions has been also reported in the superior longitudinal fasciculus, and thalamic lesions may also extend to the neocortical gray matter via the corticospinal/pyramidal tract,15, 29 although they were not observed in the present case.
Each of the demyelinating lesions can also expand in size due to local proliferation of JCV. During “expansion”, the replicated progeny viruses lyse the host cell and infect the neighboring cells. The demyelinating lesions may also fuse with one another to form larger lesions. During “extension”, neuronal axons are preserved and spread the replicated progeny viruses to distant locations. Tissue damage is relatively mild, and JCV‐infected oligodendroglia are usually present in the borders of the extending lesions. In contrast, during “expansion”, viruses propagate locally, and tissue damage would be more severe (Fig. 9).
Third step: “Fusion” of demyelinating lesions, resulting in axonal destruction
Huge demyelinating lesions were formed with “fusion” of small demyelinating lesions, where the brain tissue is severely damaged, and neuronal axons are finally fragmented. JCV‐infected cells are not any more present in the severely degraded lesions.
Although the condition is referred to as “leukoencephalopathy”, the gray matter is also known to be affected in PML.11, 15 It is noteworthy that neurons in the bilateral thalami were severely degenerated or lost in the present case (Fig. 6D, E). Indeed, in advanced PML lesions, axonal fibers are usually fragmented, and axon destruction in PML has also been reported by other investigators.30 However, the disease extension along the white matter tracts that directly relate to the thalamic neurons was not apparent. Neuronal pathology has not been extensively discussed in the context of PML.
JCV‐infected cells distribute in the borders of extending or expanding lesions
JCV‐infected cells were always present in the borders of extending or expanding demyelinating lesions. This is not unexpected from the viewpoint of virology, since the JCV life cycle is mostly dependent on host cell machinery due to the virus's limited genomic capacity.31 Thus, glial proliferation of the oligodendroglia‐like morphology is usually observed in the borders of extending or expanding lesions. Activated glial cells had small but mildly swollen nuclei, some of which contained granular or punctate intranuclear structures suggestive of early signs of JCV infection.19, 23, 32
PML is the most common clinical manifestation of JCV infectious diseases. Other CNS manifestations include JCV encephalopathy, (JCVE), JCV granule cell neuronopathy (JCV‐GCN), and JCV meningitis (JCVM),33, 34, 35, 36 and they may also overlap with PML.37 Although JCV infection of neurons is observed in JCVE and JCV‐GCN, the present case was characterized by selective infection of glial cells (both oligodendroglia and astroglia), and therefore led to the final diagnosis, “progressive multifocal leukoencephalopathy”. Oligodendroglia is most permissive for JCV infection, and abundant progeny virions may spread in myelin sheaths (oligodendroglial processes) along axons, which likely relate to tract‐dependent extension of demyelinating lesions.
In conclusion, development of PML demyelinating lesions was elucidated in post‐mortem brain, in comparison with pre‐mortem MR images. We propose that the three major lesions were developed with the following three steps: (1) initiation, (2) extension/expansion, and (3) fusion of the demyelinating lesions. Maternal viruses likely reach the initial infection sites via the bloodstream and form small demyelinating lesions. The demyelinating lesions extend along white matter tracts or locally expand themselves. The growing demyelinating lesions then fuse together, resulting in huge lesions. These pathological observations corresponded well to the MRI findings, and would greatly contribute to early diagnosis of PML.
ACKNOWLEDGMENTS
The authors thank Ayako Nakamura, Yoshinari Yamamoto, Hitomi Yokota, and Mayumi Yokotsuka for excellent technical assistance. We also thank Drs. Toshitaka Nagao, Nobuo Sanjo and Kazuo Nakamichi for support, helpful suggestions or comments.
Funding
This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to Y. S.‐H.) (18K07397).
REFERENCES
- 1. Whiteman ML, Post MJ, Berger JR, Tate LG, Bell MD, Limonte LP. Progressive multifocal leukoencephalopathy in 47 HIV‐seropositive patients: Neuroimaging with clinical and pathologic correlation. Radiology 1993; 187: 233–240. [DOI] [PubMed] [Google Scholar]
- 2. Mark AS, Atlas SW. Progressive multifocal leukoencephalopathy in patients with AIDS: Appearance on MR images. Radiology 1989; 173: 517–520. [DOI] [PubMed] [Google Scholar]
- 3. Chalkley JJ, Berger JR. Progressive multifocal leukoencephalopathy in multiple sclerosis. Curr Neurol Neurosci Rep 2013; 13: 408. [DOI] [PubMed] [Google Scholar]
- 4. Nishiyama S, Misu T, Shishido‐Hara Y et al Fingolimod‐associated PML with mild IRIS in MS: A clinicopathologic study. Neurol Neuroimmunol Neuroinflamm 2018; 5: e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berger JR, Cree BA, Greenberg B et al Progressive multifocal leukoencephalopathy after fingolimod treatment. Neurology 2018; 90: e1815–e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong‐Si T, Richman S, Wattjes MP et al Outcome and survival of asymptomatic PML in natalizumab‐treated MS patients. Ann Clin Transl Neurol 2014; 1: 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Major EO, Yousry TA, Clifford DB. Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: A decade of lessons learned. Lancet Neurol 2018; 17: 467–480. [DOI] [PubMed] [Google Scholar]
- 8. Wijburg MT, Kleerekooper I, Lissenberg‐Witte BI et al Association of progressive multifocal leukoencephalopathy lesion volume with JC virus polymerase chain reaction results in cerebrospinal fluid of Natalizumab‐treated patients with multiple sclerosis. JAMA Neurol 2018; 75: 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hodel J, Darchis C, Outteryck O et al Punctate pattern: A promising imaging marker for the diagnosis of natalizumab‐associated PML. Neurology 2016; 86: 1516–1523. [DOI] [PubMed] [Google Scholar]
- 10. Sinnecker T, Othman J, Kuhl M et al 7T MRI in natalizumab‐associated PML and ongoing MS disease activity: A case study. Neurol Neuroimmunol Neuroinflamm. 2015; 2: e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wijburg MT, Witte BI, Vennegoor A et al MRI criteria differentiating asymptomatic PML from new MS lesions during natalizumab pharmacovigilance. J Neurol Neurosurg Psychiatry 2016; 87: 1138–1145. [DOI] [PubMed] [Google Scholar]
- 12. Yousry TA, Pelletier D, Cadavid D et al Magnetic resonance imaging pattern in natalizumab‐associated progressive multifocal leukoencephalopathy. Ann Neurol 2012; 72: 779–787. [DOI] [PubMed] [Google Scholar]
- 13. Wattjes MP, Wijburg MT, Vennegoor A et al MRI characteristics of early PML‐IRIS after natalizumab treatment in patients with MS. J Neurol Neurosurg Psychiatry 2016; 87: 879–884. [DOI] [PubMed] [Google Scholar]
- 14. Wattjes MP, Verhoeff L, Zentjens W et al Punctate lesion pattern suggestive of perivascular inflammation in acute natalizumab‐associated progressive multifocal leukoencephalopathy: Productive JC virus infection or preclinical PML‐IRIS manifestation? J Neurol Neurosurg Psychiatry 2013; 84: 1176–1177. [DOI] [PubMed] [Google Scholar]
- 15. Wattjes MP, Richert ND, Killestein J et al The chameleon of neuroinflammation: Magnetic resonance imaging characteristics of natalizumab‐associated progressive multifocal leukoencephalopathy. Mult Scler 2013; 19: 1826–1840. [DOI] [PubMed] [Google Scholar]
- 16. Phan‐Ba R, Lommers E, Tshibanda L et al MRI preclinical detection and asymptomatic course of a progressive multifocal leucoencephalopathy (PML) under natalizumab therapy. J Neurol Neurosurg Psychiatry 2012; 83: 224–226. [DOI] [PubMed] [Google Scholar]
- 17. Metz I, Radue EW, Oterino A et al Pathology of immune reconstitution inflammatory syndrome in multiple sclerosis with natalizumab‐associated progressive multifocal leukoencephalopathy. Acta Neuropathol 2012; 123: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleinschmidt‐DeMasters BK, Miravalle A, Schowinsky J, Corboy J, Vollmer T. Update on PML and PML‐IRIS occurring in multiple sclerosis patients treated with natalizumab. J Neuropathol Exp Neurol 2012; 71: 604–617. [DOI] [PubMed] [Google Scholar]
- 19. Shishido‐Hara Y, Yazawa T, Nagane M et al JC virus inclusions in progressive multifocal leukoencephalopathy: Scaffolding promyelocytic leukemia nuclear bodies grow with cell cycle transition through an S‐to‐G2‐like state in enlarging oligodendrocyte nuclei. J Neuropathol Exp Neurol 2014; 73: 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maggi P, Absinta M, Grammatico M et al Central vein sign differentiates multiple sclerosis from central nervous system inflammatory vasculopathies. Ann Neurol 2018; 83: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shishido‐Hara Y. Progressive multifocal leukoencephalopathy and promyelocytic leukemia nuclear bodies: A review of clinical, neuropathological, and virological aspects of JC virus‐induced demyelinating disease. Acta Neuropathol 2010; 120: 403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shishido‐Hara Y, Higuchi K, Ohara S, Duyckaerts C, Hauw JJ, Uchihara T. Promyelocytic leukemia nuclear bodies provide a scaffold for human polyomavirus JC replication and are disrupted after development of viral inclusions in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 2008; 67: 299–308. [DOI] [PubMed] [Google Scholar]
- 23. Shishido‐Hara Y, Ichinose S, Uchihara T. JC virus intranuclear inclusions associated with PML‐NBs: analysis by electron microscopy and structured illumination microscopy. Am J Pathol 2012; 180: 1095–1106. [DOI] [PubMed] [Google Scholar]
- 24. Shishido‐Hara Y. Progressive multifocal leukoencephalopathy: Dot‐shaped inclusions and virus‐host interactions. Neuropathology 2015; 35: 487–496. [DOI] [PubMed] [Google Scholar]
- 25. Ferenczy MW, Marshall LJ, Nelson CD et al Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus‐induced demyelinating disease of the human brain. Clin Microbiol Rev 2012; 25: 471–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Astrom KE, Stoner GL. Early pathological changes in progressive multifocal leukoencephalopathy: A report of two asymptomatic cases occurring prior to the AIDS epidemic. Acta Neuropathol 1994; 88: 93–105. [DOI] [PubMed] [Google Scholar]
- 27. Ledoux S, Libman I, Robert F, Just N. Progressive multifocal leukoencephalopathy with gray matter involvement. Can J Neurol Sci 1989; 16: 200–202. [DOI] [PubMed] [Google Scholar]
- 28. Mathew RM, Murnane M. MRI in PML: Bilateral medullary lesions. Neurology 2004; 63: 2380. [DOI] [PubMed] [Google Scholar]
- 29. Sobha N, Sinha S, Taly AB et al Progressive multifocal leucoencephalopathy with discrete involvement of pyramidal tract. J Neurol Neurosurg Psychiatry 2005; 76: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wharton KA Jr, Quigley C, Themeles M et al JC polyomavirus abundance and distribution in progressive multifocal leukoencephalopathy (PML) brain tissue implicates myelin sheath in intracerebral dissemination of infection. PLoS One 2016; 11: e0155897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shishido‐Hara Y, Hara Y, Larson T, Yasui K, Nagashima K, Stoner GL. Analysis of capsid formation of human polyomavirus JC (Tokyo‐1 strain) by a eukaryotic expression system: Splicing of late RNAs, translation and nuclear transport of major capsid protein VP1, and capsid assembly. J Virol 2000; 74: 1840–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shishido‐Hara Y, Ichinose S, Higuchi K, Hara Y, Yasui K. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclear domain 10 for assembly into virions. J Virol 2004; 78: 9890–9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bag AK, Cure JK, Chapman PR, Roberson GH, Shah R. JC virus infection of the brain. Am J Neuroradiol 2010; 31: 1564–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wuthrich C, Cheng YM, Joseph JT et al Frequent infection of cerebellar granule cell neurons by polyomavirus JC in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 2009; 68: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wuthrich C, Dang X, Westmoreland S et al Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol 2009; 65: 742–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koralnik IJ, Wuthrich C, Dang X et al JC virus granule cell neuronopathy: A novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol 2005; 57: 576–580. [DOI] [PubMed] [Google Scholar]
- 37. Wijburg MT, van Oosten BW, Murk JL, Karimi O, Killestein J, Wattjes MP. Heterogeneous imaging characteristics of JC virus granule cell neuronopathy (GCN): A case series and review of the literature. J Neurol 2015; 262: 65–73. [DOI] [PubMed] [Google Scholar]