

Abstract
Non‐syndromic oculocutaneous albinism (nsOCA) is a group of genetically heterogeneous autosomal recessive disorders with complete lack or decrease pigmentation in skin, hair, and eyes. TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and LRMDA were reported to cause OCA1‐4 and OCA6‐7, respectively. By sequencing all the known nsOCA genes in 114 unrelated Chinese nsOCA patients combined with In silico analyses, splicing assay, and classification of variants according to the standards and guidelines of American College of Medical Genetics and Genomics, we detected seventy‐one different OCA‐causing variants separately in TYR, OCA2, SLC45A2, and SLC24A5, including thirty‐one novel variants (13 in TYR, 11 in OCA2, and 7 in SLC45A2). This study shows that OCA1 is the most common (75/114) and OCA2 ranks the second most common (16/114) in Chinese. 99 patients of our cohort were caused by variants of all the known nsOCA genes. Cutaneous phenotypes of OCA1, OCA2, and OCA4 patients were shown in this study. The second OCA6 case in China was identified here. These data expand the spectrum of OCA variants as well phenotype and facilitate clinical implement of Chinese OCA patients.
Keywords: genes, oculocutaneous albinism, phenotype, variants
Significance.
With comprehensive analysis of all the known nsOCA genes in 114 unrelated Chinese nsOCA patients, we identified thirty‐one novel OCA‐causing variant and reported the prevalence of different types of OCA in Chinese population: OCA1 (65.79%, 75/114) as the most common type, 16 OCA2 (14.03%, 16/114) as the second most common, 7 OCA4 (6.14%, 7/114), 1 OCA6 (0.88%, 1/114), and 15 OCA with unknown or unclassified variants. In this study, the second Chinese OCA6 case was identified and cutaneous phenotype of OCA1, OCA2, and OCA4 patients was present, which are helpful to facilitate clinic implement.
1. INTRODUCTION
Oculocutaneous albinism (OCA) is a group of autosomal recessive diseases with high heterogeneity, characterized with reduced or lost melanin in eyes, skin, and hair, often accompanied by photophobia, strabismus, poor visual acuity, and nystagmus, with an estimated worldwide prevalence of 1:17,000 (Gronskov, Ek, & Brondum‐Nielsen, 2007; Hutton & Spritz, 2008b; Witkop, 1979), 1:18,000 in Chinese Han population and 3.80% are carriers based on the survey in Shandong Province (Gong, Shao, Zheng, Chen, & Guo, 1994). OCA presents either isolated or in syndromic forms in clinic (Tomita & Suzuki, 2004). Six genes (TYR/OCA1, OCA2/OCA2, TYRP1/OCA3, SLC45A2/OCA4, SLC24A5/OCA6, and LRMDA/OCA7) were identified to be associated with non‐syndromic OCA (nsOCA; Boissy et al., 1996; Durham‐Pierre et al., 1994; Gronskov et al., 2013; Morice‐Picard et al., 2014; Newton et al., 2001; Tomita, Takeda, Okinaga, Tagami, & Shibahara, 1989; Wei et al., 2013), and a locus OCA5 is mapped to chromosome 4q24 in a consanguineous Pakistani family whose causative gene is not yet known (Kausar, Bhatti, Ali, Shaikh, & Ahmed, 2013). So far, over 125 genes were found involved in pigmentation regulation, at least 25 of which affecting the production, metabolism, distribution, or function of melanin, including the melanosomal proteins like RAB7, RAB38, TYRP2, and SILV, have been considered as potential OCA candidate genes (Montoliu et al., 2014; Sitaram & Marks, 2012), and novel OCA genes are going to be uncovered in the near future with the advancement of high‐throughput sequencing.
Among six known nsOCA genes, both TYR and TYRP1 encode key melanogenic enzymes, whose defect caused OCA1 and OCA3 (MIM 203290), respectively. According to phenotype, OCA1 can be classified as two subtypes of OCA1A (MIM 203100) or less severe OCA1B (MIM 606952; Ainger, Jagirdar, Lee, Soyer, & Sturm, 2017; Dolinska et al., 2017; Tomita et al., 1989). OCA3 is reported to be rare in Chinese. Proteins encoded by OCA2, SLC45A2, and SLC24A5 are ion transporters on melanosomal membranes to maintain melanosomes homeostasis, in which deleterious variants correspondingly lead to OCA2 (MIM 203200; Durham‐Pierre et al., 1994; Park et al., 2015), OCA4 (MIM 606574; Newton et al., 2001), and OCA6 (MIM 113750; Morice‐Picard et al., 2014; Wei et al., 2013). LRMDA is involved in melanocyte differentiation, of which the defect can cause OCA7 (MIM 615179; Gronskov et al., 2013). Less than 10 OCA6 cases have been reported worldwide (Bertolotti et al., 2016; Morice‐Picard et al., 2014; Veniani et al., 2016; Wei et al., 2013) and OCA7 was currently reported only in a Lithuanian family and individuals from Faroe Islands, suggesting both types are rare (Gronskov et al., 2013). OCA clinical traits differ among patients with variants in different genes or the different variants in the same genes, while OCA patients with different variants can also have some overlap or similar phenotype. The molecular classification is more accurate in nsOCA subtype (Gronskov et al., 2007).
The prevalence of nsOCA subtypes varies with different populations. OCA1 has been previously reported to be the most common in Asian (Suzuki & Tomita, 2008; Wei et al., 2010), Dane (Gronskov et al., 2009), non‐Hispanic Caucasians (Hutton & Spritz, 2008a), and a mixed population composed of Africans, Asians, and Europeans (Rooryck et al., 2008), and is very uncommon in African‐Americans (Gronskov et al., 2007), while OCA2 is the most frequent in nsOCA patients of African ethnic origin (King, Hearing, Creel, & Oetting, 2001). The prevalence of other OCA subtypes differs in different populations (Gronskov et al., 2009; Hutton & Spritz, 2008a; Suzuki & Tomita, 2008; Wei et al., 2010).
In this study, 114 nsOCA patients are recruited from 18 provinces of China and comprehensive molecular analysis was conducted to reveal spectral distribution of Chinese nsOCA in all known OCA causative genes.
2. MATERIALS AND METHODS
2.1. Study subjects
In this study, a total of 114 unrelated subjects diagnosed as nsOCA by dermatologic specialists and ophthalmological specialists were enrolled from the 18 provinces of China. Most patients were born after 2010. White skin, white to light blond hair, pink or blue to gray irises, and mild to severe nystagmus were observed in all 114 OCA patients. Unrelated Patient 4002701, 4008301, 4008401, and 4008601 have consanguineous parents. Another cohort ascertained in this study as normal control comprises of 100 ethnically matched unrelated individuals. Detailed ocular and skin examinations for OCA diagnosis and routine physical examinations to exclude anomaly in other organ were carried out for clinical data of these participants in this study. This study adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review board (IRB) of Medicine School of Tongji University in Shanghai, China (registration number: 2013YXY12). Written informed consent was obtained, and approximately 5 ml blood sample was voluntarily provided from all participating members.
2.2. PCR, sequencing, and in silico analysis
DNA extraction kits from Tiangen Biotech Company were used to extract total genomic DNA. Touchdown PCR amplification procedures were used with an annealing temperature of 60–57°C for all primers. Primers for screening all known nsOCA genes TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and LRMDA and for constructing wild‐type plasmid as well as mutant plasmid are available on request (Table S1). PCR amplified all the exons and their flanking regions, purified, and analyzed using the ABI 3730 automated sequencer (Applied Biosystems). The allele frequency data of identified variants were checked in NHLBI ExAC database and GnomAD database. The following is the access number of OCA genes—TYR (ENSP00000263321), OCA2 (ENSP00000346659), SLC45A2 (ENSP00000296589), and SLC24A5 (ENSP00000341550). Three in silico methods SIFT, PolyPhen‐2, and MutationTaster were used to evaluate the pathogenicity of variants in protein level.
2.3. In vitro splicing assay
To evaluate the impact on mRNA splicing of the variants, in vitro splicing assay was performed based on the comparative assay about the splicing pattern of genomic fragment of wild‐type (WT) and mutant (MUT), respectively, constructed into minigene plasmid pCAS2 (a kind gift from Prof. A. Martins, University of Rouen in France; Soukarieh et al., 2016). For each variant, wild‐type exons were amplified from human genomic DNA together with about 150bp of flanking sequences and the fragments were inserted into the MluI and BamHI cutting sites of pCAS2. MUT minigene vectors were prepared by site‐directed mutagenesis with overlap PCR method and vectors pCAS2‐WT OCA2 or SLC45A2 as template, respectively (Table S1). Sequencing the inserts of constructs was to verify sequence accuracy. 1 µg WT or MUT plasmids were parallel and transiently transfected into cell lines at a density of about 3 × 105 per well, respectively. Total RNAs were isolated 24 hr after transfection with TRIzol reagent according to the manufacturer's instructions. Then, minigene transcripts were analyzed by reverse transcription PCR (RT‐PCR) with a pair of primers (Table S1). PCR products with different sizes were separated on a 2% agarose gel by electrophoresis. Three independent experiments were carried out. In vitro splicing assay was performed in HeLa cells and ARPE‐19. The detailed procedure of in vitro splicing assay was performed according to the description in Soukarieh et al. (2016).
3. RESULTS
3.1. Classification of variants
In our study, seventy‐seven different variant alleles were identified separately in TYR, OCA2, SLC45A2, and SLC24A5 genes in 107 nsOCA patients (Table 1) after comprehensive analysis of all known nsOCA genes (TYR, OCA2, TYRP1, SLC45A2/OCA4, SLC24A5, and LRMDA) in total 114 Chinese nsOCA patients, including forty‐three variant alleles reported previously to be associated with nsOCA (Dolinska et al., 2017; Fokkema et al., 2011; Lasseaux et al., 2018; Wei et al., 2010, 2013) and thirty‐four novel alleles. All the novel variant alleles were not found in any of our 100 Chinese normal controls. The novel 34 different variants in this study include eleven missense (TYR_c.636A>T, TYR_c.937C>A, TYR_c.1169A>G, TYR_c.1234C>A, TYR_c.1325C>A, OCA2_c.849C>A, OCA2_c.1342C>T, OCA2_c.1504G>A, OCA2_c.2030T>G, OCA2_c.2244G>A, and SLC45A2_c.133A>G), nine nonsense (TYR_c.21C>A, TYR_c.24C>A, TYR_c.324G>A, TYR_c.653G>A, TYR_c.944C>G, OCA2_c.247C>T, OCA2_c.2195C>G, SLC45A2_c.529G>T, and SLC45A2_c.844G>T), ten indels (insertions or deletions) (TYR_c.456delC, TYR_c.561_562insCATTATTATGTGTCAAATTATCCCC, TYR_c.572dupG, OCA2_c.1010dupT, OCA2_c.2165delT, OCA2_c.2204_2205insCGGT, OCA2_c.2373_2375delCGT, SLC45A2_c.152_153delTG, SLC45A2_c.869dupA, and SLC45A2_c.1273delC), and four in splicing site (OCA2_c.646+3A>G, OCA2_c.2140‐2A>G, OCA2_c.2245‐11T>G, and SLC45A2_c.1032+1G>T). In addition, two known variants identified in this study, OCA2_c.808‐3C>G and SLC24A5_c.1361dupT, are firstly reported to be homozygous (Figure 1). The frequency of novel variants all is 0 in Exome Aggregation Consortium (ExAC) except for OCA2_c.849C>A (p.Ser283Arg; its frequency 0.000008238) and SLC45A2_c.1273delC (its frequency 0.00003295). Among the novel variants, ten are nonsense variants which could result in truncated, dysfunctional proteins and could be classified as pathogenic variants according to the standards and guidelines of American College of Medical Genetics and Genomics (ACMG; Richards et al., 2015). Among ten novel indels, nine frameshift indels can be classified as pathogenic variants and a variant OCA2_c.2373_2375delCGT (p.Val792del) can only be classified as a variant with uncertain significance (VUS). Eight variants are in or flank splicing site, including three reported previously to be related to OCA (Marti et al., 2018; Rimoldi et al., 2014), and five novel variants (OCA2_c.646+3A>G, OCA2_c.2140‐2A>G, OCA2_c.2245‐11T>G, OCA2_c.808‐3C>G, and SLC45A2_c.1032+1G>T), four (OCA2_c.646+3A>G, OCA2_c.2140‐2A>G, OCA2_c.808‐3C>G, and SLC45A2_c.1032+1G>T) of which in vitro splicing assay compared with WT in HeLa and ARPE‐19 cell lines, demonstrated that brought about change in splicing (Figure 2) and no change was observed between WT and MUT for analysis of variant OCA2_c.2245‐11T>G (Data not shown). Therefore, seven splicing can be classified as pathogenic variants and OCA2_c.2245‐11T>G can only be classified as a VUS at the current stage. Of eleven novel missense variants, ten were predicated to be pathogenic with three in silico methods while OCA2_c.849C>A (p.Ser283Arg) (its frequency 0.000008238) can be classified as a VUS and predicting it as the benign in protein level with three analyses. Therefore, among seventy‐seven different variant alleles, seventy‐four may be nsOCA‐causing and three are VUS.
Table 1.
Variants identified in a Chinese cohort of OCA patients
| Variants info. | Pathogenicity prediction in protein level | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene name | Patients no. | Variant | EX. | Status | Type | Polyphen‐2 | SIFT | MutationTaster | ExAC/ GnomAD | Reported or not | OCA type |
| TYR | 4001801 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HET. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | OCA1 |
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4002001 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HOM. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| 4002401 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HET. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4002601 | c.229C>G(p.Arg77Gly) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4002801 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1037G>A(p.Gly346Glu) | EX3 | HET. | Missense | PRD | D | DC | 0.000008299/0.00001633 | YES | |||
| 4002901 | c.230G>A(p.Arg77Gln) | EX1 | HET. | Missense | PRD | D | DC | 0.00009925/0.00007949 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4003201 | c.230G>A(p.Arg77Gln) | EX1 | HET. | Missense | PRD | D | DC | 0.00009925/0.00007949 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4003401 | c.820‐3C>G | INV1 | HET. | Splicing | — | — | — | 0.000008264/0 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4003501 | c.229C>G(p.Arg77Gly) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.715C>T(p.Arg239Trp) | EX1 | HET. | Missense | PRD | D | DC | 0.00004131/0.0000285 | YES | |||
| 4004001 | c.896G>A(p.Arg299His) | EX2 | HOM. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| 4004101 | c.230G>A(p.Arg77Gln) | EX1 | HET. | Missense | PRD | D | DC | 0.00009925/0.00007949 | YES | ||
| c.1204C>T(p.Arg402X) | EX4 | HET. | Nonsense | — | — | — | 0.00004984/0.0000326 | YES | |||
| 4004701† | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.944C>G(p.Ser315X) | EX2 | HET. | Nonsense | — | — | — | 0/0 | NO | |||
| 4004901 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4005001 | c.229C>G(p.Arg77Gly) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4005101† | c.636A>T(p.Arg212Ser) | EX1 | HET. | Missense | PRD | D | DC | 0/0.000004065 | NO | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4005501 | c.896G>A(p.Arg299His) | EX2 | HOM. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| 4005601 | c.758G>A(p.Gly253Glu) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4005701 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4005801 | c.929dupC(p.Arg311LysfsX7) | EX2 | HOM. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| 4005901 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.425A>T(p.Lys142Met) | EX1 | HET. | Missense | PRD | D | DC | 0.00000824/0.00001219 | YES | |||
| 4006101† | c.324G>A(p.Trp108X) | EX1 | HET. | Indel. | — | — | — | 0/0 | NO | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4006201† | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.456delC(p.Ile153X) | EX1 | HET. | Indel. | — | — | — | 0/0 | NO | |||
| 4006301 | c.929dupC(p.Arg311LysfsX7) | EX2 | HOM. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| 4006501 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4006601† | c.1168C>G(p.His390Asp) | EX3 | HET. | Missense | PRD | D | DC | 0.000008251/0.000004068 | YES | ||
| c.1325C>A(p.Ser442Tyr) | EX4 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4006701† | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.1169A>G(p.His390Arg) | EX3 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4007101 | c.832C>T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | ||
| c.1168C>G(p.His390Asp) | EX3 | HET. | Missense | PRD | D | DC | 0.000008251/0.000004068 | YES | |||
| 4007401 | c.896G>A(p.Arg299His) | EX2 | HOM. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| 4007501 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.1204C>T(p.Arg402X) | EX4 | HET. | Nonsense | — | — | — | 0.00004984/ 0.0000326 | YES | |||
| 4007601† | c.653G>A(p.Trp218X) | EX1 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4007801 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4007901 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4008201 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4008401 | c.929dupC(p.Arg311LysfsX7) | EX2 | HOM. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| 4008601 | c.229C>G(p.Arg77Gly) | EX1 | HOM | Missense | PRD | D | DC | 0/0 | YES | ||
| 4009001 | c.896G>A(p.Arg299His) | EX2 | HOM. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| 4009101 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HET. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/ 0.00003622 | YES | |||
| 4009201 | c.71G>A(p.Cys24Tyr) | EX1 | HET. | Missense | PRD | D | DC | 0.000008238/0.000004061 | YES | ||
| c.820‐3C>G | INV1 | HET. | Splicing | — | — | — | 0.000008264/0 | YES | |||
| 4009301 | c.832C > T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4009401 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1204C>T(p.Arg402X) | EX4 | HET. | Nonsense | — | — | — | 0.00004984/0.0000326 | YES | |||
| 4009501† | c.24C>A(p.Cys8X) | EX1 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| c.895C>A(p.Arg299Ser) | EX2 | HET. | Missense | PRD | D | DC | 0.000008249/0.00002439 | YES | |||
| 4009701 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1425G>A(p.Trp475X) | EX5 | HET. | Nonsense | — | — | — | 0.00001648/0.000008131 | YES | |||
| 4009801 | c.929dupC(p.Arg311LysfsX7) | EX2 | HOM. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| 4009901 | c.895C>T(p.Arg299Cys) | EX2 | HET. | Missense | PRD | D | DC | 0.00002475/0.00001626 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4010001 | c.655G>A(p.Glu219Lys) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.832C>T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | |||
| 4010101 | c.820‐3C>G | INV1 | HET. | Splicing | — | — | — | 0.000008264/0 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4010601 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4010801 | c.655G>A(p.Glu219Lys) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.832C>T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | |||
| 4011001† | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HET. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| c.1234C>A(p.Pro412Thr) | EX4 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4011201 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4011301 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1037‐2A>T | INV2 | HET. | Splicing | — | — | — | 0/0 | YES | |||
| 4011601 | c.655G>A(p.Glu219Lys) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4011801 | c.71G>A(p.Cys24Tyr) | EX1 | HET. | Missense | PRD | D | DC | 0.000008238/0.000004061 | YES | ||
| c.164G>A(p.Cys55Tyr) | EX1 | HET. | Missense | PRD | D | DC | 0/0.00003231 | YES | |||
| 4011901 | c.1A>G(p.Met1?) | EX1 | HET. | Missense | — | — | — | 0.00003299/0.00006494 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4012001 | c.1199G>T (p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | ||
| c.1204C>T (p.Arg402X) | EX4 | HET. | Nonsense | — | — | — | 0.00004984/0.0000326 | YES | |||
| 4012101 | /0(p.Gly253Glu) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4012201 | c.832C>T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4012401 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HOM. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| 4001901 | c.703T>C(p.Tyr235His) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4003301 | c.895C>T(p.Arg299Cys) | EX2 | HET. | Missense | PRD | D | DC | 0.00002475/0.00001626 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/ 0.00003622 | YES | |||
| 4003601† | c.572dupG(p.Ser192IlefsX2) | EX1 | HET. | Indel. | — | — | — | 0/0 | NO | ||
| c.820‐3C>G | INV1 | HET. | Splicing | — | — | — | 0.000008264/0 | YES | |||
| 4003701 | c.71G>A(p.Cys24Tyr) | EX1 | HET. | Missense | PRD | D | DC | 0.000008238/0.000004061 | YES | ||
| c.1265G>A(p.Arg422Gln) | EX4 | HET. | Missense | PRD | D | DC | 0.00004968/0.00005709 | YES | |||
| 4003801 | c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| c.1037‐7A>T | INV2 | HET. | Splicing | — | — | — | 0.0008838/0.0008789 | YES | |||
| c.1037‐10_11delTT | INV2 | HET. | Splicing | — | — | — | 0/0 | YES | |||
| 4004201† | c.561_562insCATTATTATGTGTCAAATTATCCCC (p.Gly190CysfsX12) | EX1 | HET. | Indel. | — | — | — | 0/0 | NO | ||
| c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | |||
| 4004301 | c.230_232dupGGG(p.Arg77_Glu78insGly) | EX1 | HET. | Indel. | — | — | — | 0.000008268/0.00002529 | YES | ||
| c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | |||
| 4005201 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.819G>C(p.Gln273His) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | |||
| 4005301 | c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/ 0.00003622 | YES | |||
| 4006001 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| c.1265G>A(p.Arg422Gln) | EX4 | HET. | Missense | PRD | D | DC | 0.00004968/0.00005709 | YES | |||
| 4008101† | c.446A>G(p.Tyr149Cys) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.937C>A(p.Pro313Thr) | EX2 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4008501 | c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4008801 | c.896G>A(p.Arg299His) | EX2 | HOM. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | ||
| 4010201 | c.229C>T(p.Arg77Trp) | EX1 | HET. | Missense | PRD | D | DC | 0.00003307/0.00003251 | YES | ||
| c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | |||
| 4010301 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| c.895C>A(p.Arg299Ser) | EX2 | HET. | Missense | PRD | D | DC | 0.000008249/0.00002439 | YES | |||
| 4011401 | c.832C>T(p.Arg278X) | EX2 | HET. | Nonsense | — | — | — | 0.00019/0.000177 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4012301 | c.230G>A(p.Arg77Gln) | EX1 | HET. | Missense | PRD | D | DC | 0.00009925/0.00007949 | YES | ||
| c.1199G>T(p.Trp400Leu) | EX4 | HET. | Missense | PRD | D | DC | 0.00002493/0.00003622 | YES | |||
| 4008901 | c.896G>A(p.Arg299His) | EX2 | HET. | Missense | PRD | D | DC | 0.00007424/0.0000614 | YES | OCA1? | |
| ? | ? | ? | ? | — | — | — | — | — | |||
| 4009601 | c.346C>T(p.Arg116X) | EX1 | HET. | Nonsense | — | — | — | 0.00002473/0.00002887 | YES | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| 4010701 | c.929dupC(p.Arg311LysfsX7) | EX2 | HET. | Indel. | — | — | — | 0.00004948/0.00004066 | YES | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| 4011101† | c.21C>A(p.Tyr7X) | EX1 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| OCA2 | 4000401† | c.1426A>G(p.Asn476Asp) | EX14 | HET. | Missense | PRD | D | DC | 0/0 | YES | OCA2 |
| c.2030T>G(p.Val677Gly) | EX19 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4000601 | c.1262G>C(p.Arg421Pro) | EX13 | HET. | Missense | PRD | D | DC | 0/0 | YES | ||
| c.632C>T(p.Pro211Leu) | EX6 | HET. | Missense | PRD | D | DC | 0.0001494/0.0001228 | YES | |||
| 4001201† | c.1010dupT(p.Leu338ProfsX11) | EX9 | HET. | Indel. | — | — | — | 0/0 | NO | ||
| c.1504G>A(p.Gly502Ser) | EX15 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4002701 | c.2228C>T(p.Pro743Leu) | EX21 | HOM | Missense | PRD | D | DC | 0.00009078/0.0001263 | YES | ||
| 4003001† | c.1342C>T(p.Leu448Phe) | EX13 | HET. | Missense | PRD | D | DC | 0/0 | NO | ||
| c.2204_2205insCGGT(p.Ser736GlyfsX6) | EX21 | HET. | Indel. | — | — | — | 0/0 | NO | |||
| 4003101† | c.406C>T(p.Arg136X) | EX4 | HET. | Nonsense | — | — | — | 0.00003295/0.000004061 | YES | ||
| c.1342C>T(p.Leu448Phe) | EX13 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4003901† | c.406C>T(p.Arg136X) | EX4 | HET. | Nonsense | — | — | — | 0.00003295/0.000004061 | YES | ||
| c.646+3A>G | INV6 | HET. | Splicing | — | — | — | 0/0 | NO | |||
| 4004601† | c.2195C>G(p.Ser732X) | EX21 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| c.1441G>A(p.Ala481Thr) | EX14 | HET. | Missense | POD | T | DC | 0.007751/0.008502 | YES | |||
| 4005401† | c.247C>T(p.Gln83X) | EX3 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| c.2344G>A(p.Gly782Arg) | EX23 | HET. | Missense | PRD | D | DC | 0/0.000007215 | YES | |||
| 4007301† | c.2165delT(p.Ile722LysfsX17) | EX21 | HET. | Indel. | — | — | — | 0/0 | NO | ||
| c.2244G>A(p.Met748Ile) | EX21 | HET. | Missense | PRD | D | DC | 0/0 | NO | |||
| 4007701 | c.1255C>T(p.Arg419Trp) | EX13 | HET. | Missense | PRD | D | DC | 0.0002452/0.000264 | YES | ||
| c.1349C>T(p.Thr450Met) | EX13 | HET. | Missense | PRD | D | DC | 0.00001678/0.00003231 | YES | |||
| 4008301* | c.808‐3C>G | INV7 | HOM. | Splicing | — | — | — | 0/0.000004061 | NO | ||
| 4010501 | c.1182+1G>A | INV11 | HOM. | Missense | — | — | — | 0.00009144/0.00005775 | YES | ||
| 4010901 | c.1441G>A(p.Ala481Thr) | EX14 | HET. | Missense | POD | T | DC | 0.007751/0.008502 | YES | ||
| c.1503+5G>A | INV14 | HET. | Splicing | — | — | — | 0.000008237/0.00001082 | YES | |||
| 4011501† | c.2140‐2A>G | INV20 | HET. | Splicing | — | — | — | 0/0 | NO | ||
| c.632C>T(p.Pro211Leu) | EX6 | HET. | Missense | PRD | D | DC | 0.0001494/0.0001228 | YES | |||
| 4002501 | c.1182+1G>A | INV11 | HET. | Splicing | — | — | — | 0.00009144/0.00005775 | YES | ||
| c.1714C>T(p.Arg572Cys) | EX16 | HET. | Missense | PRD | D | DC | 0.00006945/0.00005862 | YES | |||
| 4006901† | c.2245‐11T>G | INV21 | HET. | Splicing | — | — | — | 0/0 | NO | OCA2? | |
| c.2373_2375delCGT(p.Val792del) | EX23 | HET. | Indel. | — | — | — | 0/0.000008121 | NO | |||
| 4004501† | c.849C>A(p.Ser283Arg) | EX8 | HET. | Missense | Benign | T | P | 0.000008238/0.00002525 | NO | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| 4008001 | c.406C>T(p.Arg136X) | EX4 | HET. | Nonsense | — | — | — | 0.00003295/0.000004061 | YES | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| 4008701 | c.808‐3C>G | INV7 | HET. | Splicing | — | — | — | 0/0.000004061 | NO | ||
| ? | ? | ? | ? | — | — | — | — | — | |||
| SLC45A2 | 4000501† | c.1032+1G>T | INV4 | HET. | Splicing | — | — | — | 0/0 | NO | OCA4 |
| c.1045G>A(p.Gly349Arg) | EX5 | HET. | Missense | PRD | D | DC | 0.0001318/0.00007311 | YES | |||
| 4001001† | c.133A > G(p.Arg45Gly) | EX1 | HOM. | Missense | PRD | D | DC | 0/0 | NO | ||
| 4001601† | c.529G>T(p.Glu177X) | EX2 | HET. | Nonsense | — | — | — | 0/0 | NO | ||
| c.844G>T(p.Glu282X) | EX3 | HET. | Nonsense | — | — | — | 0/0 | NO | |||
| 4002201† | c.152_153delTG(p.Val51GlyfsX82) | EX1 | HET. | Indel. | — | — | — | 0/0 | YES | ||
| c.1045G>A(p.Gly349Arg) | EX5 | HET. | Missense | PRD | D | DC | 0.0001318/0.00007311 | YES | |||
| 4004801† | c.133A>G(p.Arg45Gly) | EX1 | HET. | Missense | PRD | D | DC | 0/0 | NO | ||
| c.478G>C(p.Asp160His) | EX2 | HET. | Missense | PRD | D | DC | 0/0.000004061 | YES | |||
| 4007201† | c.478G>C(p.Asp160His) | EX2 | HET. | Missense | PRD | D | DC | 0/0.000004061 | YES | ||
| c.1273delC(p.Leu425TrpfsX9) | EX6 | HET. | Indel. | — | — | — | 0.00003295/0.00001218 | NO | |||
| 4011701† | c.478G>C(p.Asp160His) | EX2 | HET. | Missense | PRD | D | DC | 0/0.000004061 | YES | ||
| c.869dupA(p.Asn290LysfsX6) | EX3 | HET. | Indel. | — | — | — | 0/0.000008145 | NO | |||
| SLC24A5 | 4007001* | c.1361dupT(p.Leu454PhefsX33) | EX9 | HOM. | Indel. | — | — | — | 0/0 | NO | OCA6 |
The proband marked with † sign carries novel variant or variants; the proband with * sign carries known variants but its homozygosity reported firstly in this study. The variants reported to be associated with OCA at first time are in the bold. The items without data available are marked with backslash. Variants marked with hyphen are not necessary to be predicted or improper to be predicted their pathogenicity in protein level via SIFT, Polyphen‐2, and MutationTaster. Grayish lattices were splicing mutations and non‐pathogenic results in protein level with all or two of three in silico approaches. The allele frequency of ExAC or GnomAD data here refers to all individuals.
B, benign; D, Damaging; DC, Disease causing; P, polymorphism; POD, possibly damaging; PRD, Probably damaging; T, tolerated.
Figure 1.
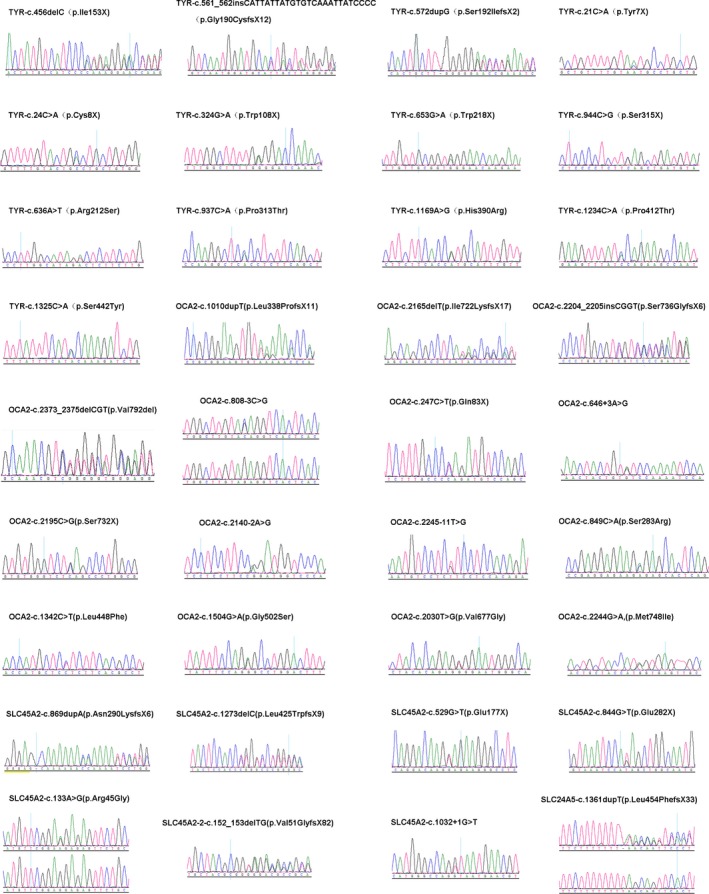
Sequence chromatograms of novel variants in TYR, OCA2, SLC45A2, and SLC24A5
Figure 2.
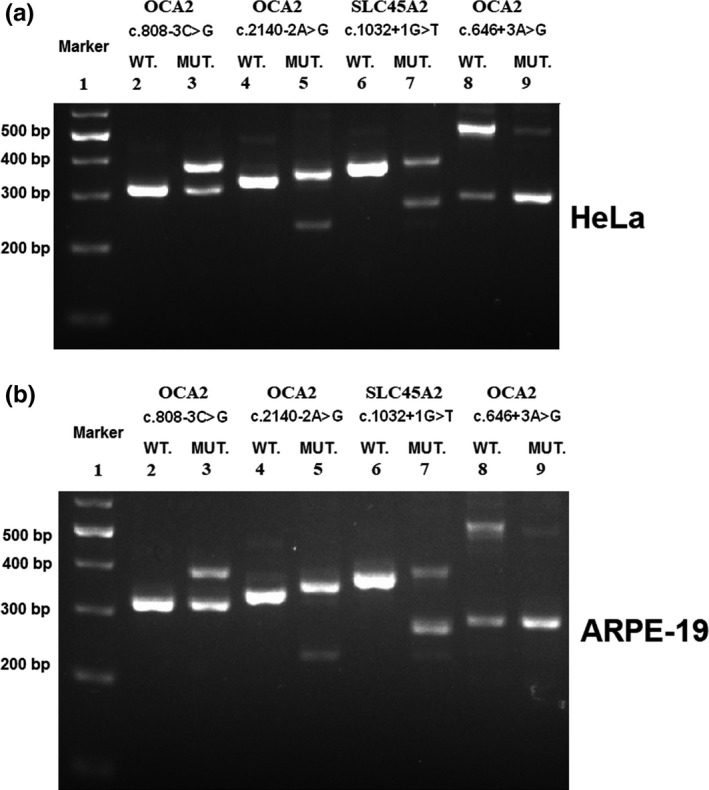
Splicing assay shows variant‐induced change in OCA2 or SLC45A2 splicing. Gel electrophoresis of RT‐PCR products for all tested constructs. Lane 1: 100bp marker, splicing assay is based on comparative assay about the splicing pattern of genomic fragment of wild‐type (WT) and mutant (MUT), respectively. Lane 2 and lane 3 as a group are to evaluate the change in splicing which the variant OCA2_c.808‐3C>G brought about. Lane 4 and lane 5 are for OCA2_c.2140‐2A>G; lane 6 and lane 7 are for SLC45A2_c.1032+1G>T; lane 8 and lane 9 are for OCA2_c.646+3A>G. The differences in the size and composition of band(s) between WT and MUT demonstrate the variant‐induced aberrant in mRNA level. (a) In vitro splicing assay in HeLa cell line. (b) In vitro splicing assay in ARPE‐19 cell line
3.2. Spectral distribution of variants of all known OCA genes in Chinese nsOCA patients
In our cohort of 114 Chinese nsOCA patients, 7 were identified without variants in all known nsOCA genes, 6 were identified to carry one pathogenic allele and unknown variant on second chromosome, 2 patients were identified to carry one or two compound VUS, and other 99 were identified to carry two or more pathogenic alleles including 17 patients with homozygous variants and 82 patients with compound heterozygous variants. Among 17 patients with homozygous variants, four unrelated patients 4002701 carried OCA2_c.2228C>T (p.Pro743Leu), 4008301 carried OCA2_c.808‐3C>G, 4008401 carried c.929dupC (p.Arg311LysfsX7), and 4008601 carried c.229C>G (TYR_p.Arg77Gly) have consanguineous parents. Molecular diagnosis shows that in our cohort, there are 75 OCA1 patients (65.79%, 75/114), 16 OCA2 (14.03%, 16/114), 7 OCA4 (6.14%, 7/114), 1 OCA6 (0.88%, 1/114), and 15 OCA with unknown or unclassified variants (13.16%, 15/114; Figure 3a).
Figure 3.
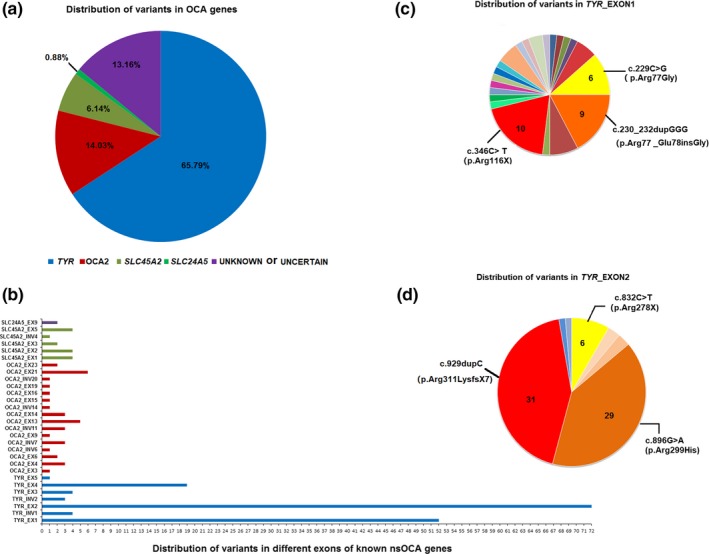
Spectral distribution of variants of all known OCA genes in Chinese nsOCA patients. (a) The prevalence of OCA types in our cohort. (b) Distribution of variants in exons of known OCA genes identified in this study. (c) Distribution of variants in exon 1 of TYR. (d) Distribution of variants in exon 2 of TYR
Of 79 TRY‐related OCA patients, 75 were found to have two mutational alleles and 4 (Patient 4008901, Patient 4009601, Patient 4010701, and Patient 4011101) were found to have one mutational allele in TYR and second allele unknown (Table 1). We identified thirty‐nine different mutational alleles of TYR in our cohort (Table 1), thirteen of which have not been previously reported: c.21C>A (p.Tyr7X), c.24C>A (p.Cys8X), c.324G>A (p.Trp108X), c.456delC (p.Ile153X), c.561_562insCATTATTATGTGTCAAATTATCCCC (p.Gly190CysfsX12), c.572dupG (p.Ser192IlefsX2), c.636A>T (p.Arg212Ser), c.653G>A (p.Trp218X), c.937C>A (p.Pro313Thr), c.944C>G (p.Ser315X), c.1169A>G (p.His390Arg), c.1234C>A (p.Pro412Thr), and c.1325C>A (p.Ser442Tyr; Figure 1, Table 1). In OCA1, 27 of 39 different mutational alleles are clustered on exon 1 and exon 2 of TYR, accounting for 81.1% (131 of 155) of the total TYR mutational alleles (Figure 3b). Among these mutational alleles of TYR, c.929dupC and p.Arg299His account for 20.00% (31 of 155) and 18.71% (29 of 155), respectively (Figure 3c,d), the variant in exon 4 p.Trp400Leu ranks third with 7.10% (11 of 155; Figure 3b), and other variants with higher sequence are p.Arg116X (6.45%, 10 of 155), p.Arg77_Glu78insGly (5.81%, 9 of 155), p.Arg77Gly (3.87%, 6 of 155), and p.Arg278X (3.87%, 6 of 155; Figure 3c,d). The above seven alleles account for 65.81% (102 of 155) of the mutational TYR alleles in our cohort of Chinese OCA patients.
In our cohort, sixteen nsOCA patients were diagnosed as OCA2, and four are uncertain including Patient 4006901 identified to carry two compound VUS (OCA2_c.2245‐11T>G and OCA2_c.2373_2375delCGT), Patient 4004501 with one VUS (OCA2_c.849C>A) plus Patient 4008001 and Patient 4008001 only identified to carry one pathogenic variant in OCA2 (Table 1). Twenty‐four different OCA2‐causing variants identified in 16 patients, of which 11 mutational alleles were novel: c.247C>T (p.Gln83X), c.646+3A>G, c.1010dupT (p.Leu338ProfsX11), c.1342C>T (p.Leu448Phe), c.1504G>A (p.Gly502Ser), c.2030T>G (p.Val677Gly), c.2140‐2A>G, c.2195C>G (p.Ser732X), c.2165delT (p.Ile722LysfsX17), c.2204_2205insCGGT (p.Ser736GlyfsX6), and c.2244G>A (p.Met748Ile), plus 13 were recurrent (Table 1). Among 13 known variants, homozygous variants c.808‐3C>G are firstly reported.
Seven nsOCA patients were diagnosed as OCA4, and total fourteen variant alleles are identified in SLC45A2. All OCA4 patients in this study carried at least one novel variant. Together, nine different OCA4 causative variants were found in this study including two recurrent variants—c.478G>C (p.Asp160His) and c.1045G>A, and seven novel variants—c.133A>G (p.Arg45Gly), c.152_153delTG (p.Val51GlyfsX82), c.529G>T (p.Glu177X), c.844G>T (p.Glu282X), c.869dupA (p.Asn290LysfsX6), c.1032+1G>T, and c.1273delC (p.Leu425TrpfsX9).
In addition, homozygous variants c.1361dupT (p.Leu454PhefsX33) in SLC24A5 were detected in Patient 4007001—the second molecularly diagnosed as OCA6 case in Chinese population. Homozygous variants SLC24A5_c.1361dupT are firstly reported here (Figure 1). No OCA3 patient and OCA7 patient were identified in our cohort.
3.3. Cutaneous phenotype of OCA1, OCA2, and OCA4 patients
Patient 4005001, diagnosed as OCA1 with compound variants c.229C>G (p.Arg77Gly) and c.929dupC (p.Arg311LysfsX7) in TYR, presents milky white hair and skin (Figure 4), and does not tan, and his irises are light blue as well as full transillumination, photophobia, has poor visual acuity (Figure 4). Patient 4003001, diagnosed as OCA2 with two novel heterozygous variants c.1342C>T (p.Leu448Phe) and c.2204_2205insCGGT (p.Ser736GlyfsX6) in OCA2, had blond hair at birth, and hair darken into brown at four‐year age (Figure 4), plus light blue irises at birth were recorded in the medical history and brown irises were observed currently. Patient 4003101, diagnosed as OCA2 with a reported variant c.406C>T (p.Arg136X) and a novel variant c.1342C>T (p.Leu448Phe) in OCA2, had blond hair (Figure 4) and brown irises at the time when she was recruited. Patient 4003901, diagnosed as OCA2 with a reported variant c.406C>T (p.Arg136X) and a novel variant c.646+3A>G in OCA2, had golden hair. Patient 4004801, diagnosed as OCA4 with a reported variant c.478G>C (p.Asp160His) and a novel variant c.133A>G (p.Arg45Gly) in SLC45A2, had light blond hair, white skin (Figure 4), and irises translucency.
Figure 4.
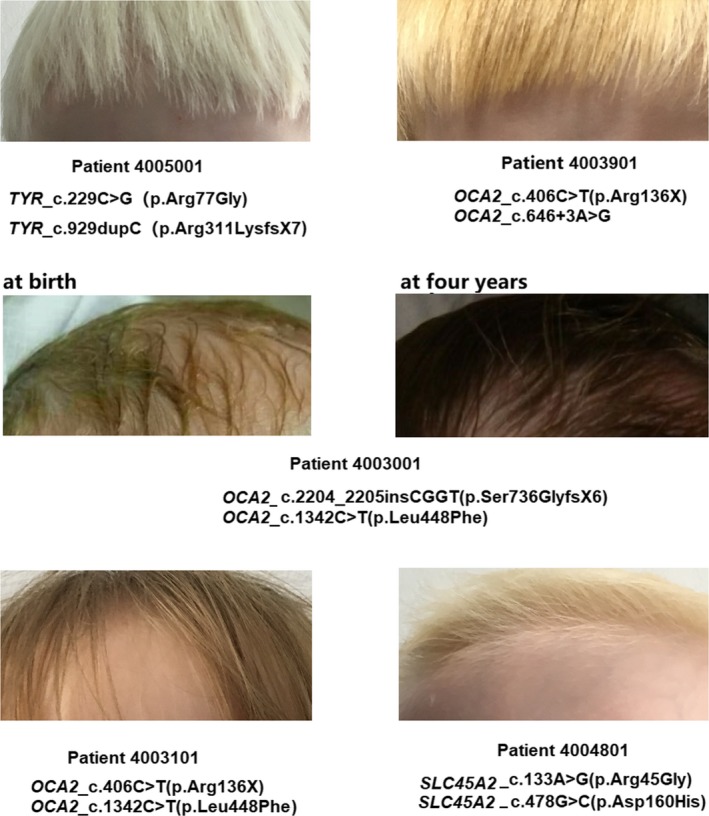
Phenotypes in skin and hairs of patients with different types of nsOCA
4. DISCUSSION
The oxidation and polymerization of tyrosine synthesized in epidermal melanocytes originate a macromolecular biopolymer of melanin. Melanin is found in several tissues such as skin, hair and iris, choroid, and retina of the eye. Melanin can be transferred to the surrounding cells to protect them from the effect of UV radiation at sun exposure. Two types of melanin are produced in melanosomes, including brown or black photoprotective eumelanins, and yellow or red phototoxic pheomelanins, whose ratio of each type depends on the enzymatic activity of TYR (tyrosinase) and the availability of cysteine which is one of the rate‐limiting factors in glutathione metabolism. Melanosomal ion transport proteins and pH are crucial for the genesis and function of melanosome. Low TYR activity presents in melanosomes from individuals with fair skin color displaying more acidic. In addition, low TYR activity and/or low concentrations of cysteine lead to phototoxic pheomelanins and high TYR activity and/or high concentrations of cysteine lead to photoprotective eumelanins. Any defect in melanocytes, dysfunction of melanocytes, and impairment in producing and transferring of melanin can cause albinism, and its specific involvement of skin, hair, and eyes is called as nsOCA. To date, TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and LRMDA are six known nsOCA‐causing genes, corresponding to OCA1, OCA2, OCA3, OCA4, OCA6, and OCA7 (Boissy et al., 1996; Durham‐Pierre et al., 1994; Gronskov et al., 2013; Newton et al., 2001; Tomita et al., 1989; Wei et al., 2013).
In this study, comprehensive analysis of all currently known nsOCA genes (TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and LRMDA) in 114 nsOCA patients recruited from 18 provinces in China shows the prevalence of OCA1, OCA2, OCA4, and OCA6 is 65.79%, 14.03%, 6.14%, and 0.88%, respectively, and the left nsOCA with uncertain causative defect of molecule (13.16%). In our cohort, OCA1 is the most common type of nsOCA and OCA2 ranks as the second most common type of nsOCA, which are in accordance with what reported in Japanese (Suzuki & Tomita, 2008), non‐Hispanic Caucasians (Hutton & Spritz, 2008a), Danes (Gronskov et al., 2009), in the population of a European setting at the albino day hospital (Marti et al., 2018), and in the group of the patients mainly from France who were originated from different countries worldwide (Lasseaux et al., 2018). In this study, we failed to find the second variants in 7 patients and after sequencing all the exons and their flanking regions, which could be ascribed to the following possibilities of an undetected large indel in another allele or variants in deep‐intronic regions or in regulatory elements located far away from those six known nsOCA genes. Plus, additional 8 patients without found pathogenic variants in all known nsOCA genes might suggest the possibility of uncovering novel OCA genes. In addition, if OCA2_c.2245‐11T>G, OCA2_c.2373_2375delCGT, and OCA2_c.849C>A were confirmed to be pathogenic in further function study, two patients with those variants would be OCA2.
The spectrum of variants in each nsOCA genes has been reported to vary with populations. In OCA1, 27 of 39 different mutational alleles are clustered on exon 1 and exon 2 of TYR, accounting for 81.1% (131 of 155) of the total TYR mutational alleles (Figure 3b), suggesting exon 1 and exon 2 is mutational hotspots in Chinese nsOCA, which is consistent with the report of Wei et al.'s study (Wei et al., 2010). In addition, both c.929idupC and p.Arg299His, which is the most frequent alleles in this study, are on exon 2. In this study, twenty‐four different OCA2‐causing variants identified in 16 patients are sparsely distributed in OCA2, and no apparent mutational hotspots can be observed in our cohort, which has also been observed in Wei et al's study (Wei et al., 2010). Although we supported the recommendation about prioritizing the sequencing of hotspots in exons 1 and 2 of TYR in diagnosis of OCA pointed out by Wei et al. (2010), the priority of SLC45A2 over OCA2 in sequencing may be uncertain. The frequency of a SLC45A2 mutational allele c.478G>C (p.Asp160His) was 3/14 in this study while Wei et al. (2010) described that it accounts for 55.6% (15/27) of the mutational alleles of SLC45A2 in their study, which may suggest that it is possible that kind of difference exists between the ancestors of patients in this study and those in Wei et al.’ study although all the patients in both studies are Chinese.
The defect in OCA2, SLC45A2, and SLC24A5 functioning as ion transporters on melanosomal membranes to maintain melanosomes homeostasis (Durham‐Pierre et al., 1994; Morice‐Picard et al., 2014; Newton et al., 2001; Park et al., 2015; Wei et al., 2013) can cause OCA2, OCA4, and OCA6, respectively. The hair color of Patient 4003001 who was molecularly diagnosed as OCA2 darkened with age from blond hair at birth to brown at four years old (Figure 4, Table 1) and the color of irises darken too. Patient 4003001 and Patient 4003101 of whom both carry one same novel variant allele of OCA2_c.1342C>T (p.Leu448Phe) have different hair color, which might be due to difference in impact between c.2204_2205insCGGT (p.Ser736GlyfsX6) and c.406C>T (p.Arg136X) (Figure 4, Table 1). Similarly, the difference in hair color also exists between Patient 4003101 and Patient 4003901 of whom both carry one same variant allele OCA2_c.406C>T (p.Arg136X) but the second allele is different between them (Figure 4, Table 1). The hair of Patient 4005001 and Patient 4004801 look to have less pigment than that of other 3 OCA2 patients. To be sure, genotype–phenotype correlation analysis based on more patients with different types of OCA is helpful to answer whether there is correlation between genotype and phenotype in OCA1, OCA2, and OCA4. The defect in SLC24A5 can cause OCA6, and less than 10 OCA6 cases have been reported worldwide (Bertolotti et al., 2016; Morice‐Picard et al., 2014; Veniani et al., 2016; Wei et al., 2013). Skin phenotype of OCA6 cases is heterogeneous, and their hair color ranges from dark brown to white. Ophthalmologic anomalies of OCA6 cases have been reported to include severe hypopigmentation in retina and foveal hypoplasia and extensive iris transillumination (Bertolotti et al., 2016; Montoliu et al., 2014; Morice‐Picard et al., 2014; Veniani et al., 2016; Wei et al., 2013). In this study, Patient 4007001 has been firstly diagnosed as OCA2 with light brown hair and white skin, and without severe ocular problems except for nystagmus and photophobia when being recruited at toddler stage, actually caused by homozygous variants in SLC24A5 and was molecularly diagnosed as OCA6 in our study. This OCA6 case expands the currently limited spectrum of OCA6 since less than ten OCA6 cases had been reported.
Herein, we described the prevalence of nsOCA types in Chinese population and thirty‐one different novel OCA causative variants, which expands the spectrum of nsOCA variants. In addition, the second OCA6 case in Chinese population was detected in our cohort. These findings may facilitate molecular diagnosis and genetic counseling for OCA patients.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Supporting information
ACKNOWLEDGEMENTS
We appreciated all the volunteers for participating in this study. Thanks to Prof. A. Martins and group members from University of Rouen in France for sharing pCAS2 vector with us. This work was supported by the National Key Basic Research Program of China (973 Program 2015CB964601); National Natural Science Foundation of China (81371062); and Thousand Youth Talents Program of China (to J.C.).
Zhong Z, Gu L, Zheng X, et al. Comprehensive analysis of spectral distribution of a large cohort of Chinese patients with non‐syndromic oculocutaneous albinism facilitates genetic diagnosis. Pigment Cell Melanoma Res. 2019;32:672–686. 10.1111/pcmr.12790
Contributor Information
Jun Zhang, Email: junzhang@tongji.edu.cn, Email: chenjianjun@tongji.edu.cn.
Jianjun Chen, Email: chenjianjun@tongji.edu.cn.
REFERENCES
- Ainger, S. A. , Jagirdar, K. , Lee, K. J. , Soyer, H. P. , & Sturm, R. A. (2017). Skin Pigmentation Genetics for the Clinic. Dermatology, 233(1), 1–15. 10.1159/000468538 [DOI] [PubMed] [Google Scholar]
- Bertolotti, A. , Lasseaux, E. , Plaisant, C. , Trimouille, A. , Morice‐Picard, F. , Rooryck, C. , … Arveiler, B. (2016). Identification of a homozygous mutation of SLC24A5 (OCA6) in two patients with oculocutaneous albinism from French Guiana. Pigment Cell and Melanoma Research, 29(1), 104–106. 10.1111/pcmr.12425 [DOI] [PubMed] [Google Scholar]
- Boissy, R. E. , Zhao, H. , Oetting, W. S. , Austin, L. M. , Wildenberg, S. C. , Boissy, Y. L. , … Nordlund, J. J. (1996). Mutation in and lack of expression of tyrosinase‐related protein‐1 (TRP‐1) in melanocytes from an individual with brown oculocutaneous albinism: A new subtype of albinism classified as "OCA3". American Journal of Human Genetics, 58(6), 1145–1156. [PMC free article] [PubMed] [Google Scholar]
- Dolinska, M. B. , Kus, N. J. , Farney, S. K. , Wingfield, P. T. , Brooks, B. P. , & Sergeev, Y. V. (2017). Oculocutaneous albinism type 1: Link between mutations, tyrosinase conformational stability, and enzymatic activity. Pigment Cell and Melanoma Research, 30(1), 41–52. 10.1111/pcmr.12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham‐Pierre, D. , Gardner, J. M. , Nakatsu, Y. , King, R. A. , Francke, U. , Ching, A. , … Brilliant, M. H. (1994). African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nature Genetics, 7(2), 176–179. 10.1038/ng0694-176 [DOI] [PubMed] [Google Scholar]
- Fokkema, I. F. , Taschner, P. E. , Schaafsma, G. C. , Celli, J. , Laros, J. F. , & den Dunnen, J. T. (2011). LOVD v.2.0: The next generation in gene variant databases. Human Mutation, 32(5), 557–563. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Gong, Y. , Shao, C. , Zheng, H. , Chen, B. , & Guo, Y. (1994). Study on genetic epidemiology of albinism. Yi Chuan Xue Bao, 21(3), 169–172. [PubMed] [Google Scholar]
- Grønskov, K. , Dooley, C. M. , Østergaard, E. , Kelsh, R. N. , Hansen, L. , Levesque, M. P. , … Rosenberg, T. (2013). Mutations in c10orf11, a melanocyte‐differentiation gene, cause autosomal‐recessive albinism. American Journal of Human Genetics, 92(3), 415–421. 10.1016/j.ajhg.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronskov, K. , Ek, J. , & Brondum‐Nielsen, K. (2007). Oculocutaneous albinism. Orphanet Journal of Rare Diseases, 2, 43 10.1186/1750-1172-2-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønskov, K. , Ek, J. , Sand, A. , Scheller, R. , Bygum, A. , Brixen, K. , … Rosenberg, T. (2009). Birth prevalence and mutation spectrum in Danish patients with autosomal recessive albinism. Investigative Ophthalmology and Visual Science, 50(3), 1058–1064. 10.1167/iovs.08-2639 [DOI] [PubMed] [Google Scholar]
- Hutton, S. M. , & Spritz, R. A. (2008a). Comprehensive analysis of oculocutaneous albinism among non‐Hispanic caucasians shows that OCA1 is the most prevalent OCA type. The Journal of Investigative Dermatology, 128(10), 2442–2450. 10.1038/jid.2008.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton, S. M. , & Spritz, R. A. (2008b). A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Investigative Ophthalmology and Visual Science, 49(3), 868–872. 10.1167/iovs.07-0791 [DOI] [PubMed] [Google Scholar]
- Kausar, T. , Bhatti, M. A. , Ali, M. , Shaikh, R. S. , & Ahmed, Z. M. (2013). OCA5, a novel locus for non‐syndromic oculocutaneous albinism, maps to chromosome 4q24. Clinical Genetics, 84(1), 91–93. 10.1111/cge.12019 [DOI] [PubMed] [Google Scholar]
- King, R. A. , Hearing, V. J. , Creel, D. J. , & Oetting, W. S. (2001). Albinism In Beaudet A. L., Scriver C. R., Valle D., & Sly W. S. (Eds.), The metabolic & molecular basis of inherited disease (8th ed., pp. 5587–5627). New York, NY: McGraw‐Hill. [Google Scholar]
- Lasseaux, E. , Plaisant, C. , Michaud, V. , Pennamen, P. , Trimouille, A. , Gaston, L. , … Arveiler, B. (2018). Molecular characterization of a series of 990 index patients with albinism. Pigment Cell and Melanoma Research, 31(4), 466–474. 10.1111/pcmr.12688 [DOI] [PubMed] [Google Scholar]
- Marti, A. , Lasseaux, E. , Ezzedine, K. , Léauté‐Labrèze, C. , Boralevi, F. , Paya, C. , … Morice‐Picard, F. (2018). Lessons of a day hospital: Comprehensive assessment of patients with albinism in a European setting. Pigment Cell and Melanoma Research, 31(2), 318–329. 10.1111/pcmr.12651 [DOI] [PubMed] [Google Scholar]
- Montoliu, L. , Grønskov, K. , Wei, A.‐H. , Martínez‐García, M. , Fernández, A. , Arveiler, B. , … Li, W. (2014). Increasing the complexity: New genes and new types of albinism. Pigment Cell and Melanoma Research, 27(1), 11–18. 10.1111/pcmr.12167 [DOI] [PubMed] [Google Scholar]
- Morice‐Picard, F. , Lasseaux, E. , François, S. , Simon, D. , Rooryck, C. , Bieth, E. , … Arveiler, B. (2014). SLC24A5 mutations are associated with non‐syndromic oculocutaneous albinism. The Journal of Investigative Dermatology, 134(2), 568–571. 10.1038/jid.2013.360 [DOI] [PubMed] [Google Scholar]
- Newton, J. M. , Cohen‐Barak, O. , Hagiwara, N. , Gardner, J. M. , Davisson, M. T. , King, R. A. , & Brilliant, M. H. (2001). Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. American Journal of Human Genetics, 69(5), 981–988. 10.1086/324340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Morya, V. K. , Nguyen, D. H. , Singh, B. K. , Lee, H. B. , & Kim, E. K. (2015). Unrevealing the role of P‐protein on melanosome biology and structure, using siRNA‐mediated down regulation of OCA2. Molecular and Cellular Biochemistry, 403(1–2), 61–71. 10.1007/s11010-015-2337-y [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimoldi, V. , Straniero, L. , Asselta, R. , Mauri, L. , Manfredini, E. , Penco, S. , … Primignani, P. (2014). Functional characterization of two novel splicing mutations in the OCA2 gene associated with oculocutaneous albinism type II. Gene, 537(1), 79–84. 10.1016/j.gene.2013.11.102 [DOI] [PubMed] [Google Scholar]
- Rooryck, C. , Morice‐Picard, F. , Elcioglu, N. H. , Lacombe, D. , Taieb, A. , & Arveiler, B. (2008). Molecular diagnosis of oculocutaneous albinism: New mutations in the OCA1‐4 genes and practical aspects. Pigment Cell & Melanoma Research, 21(5), 583–587. 10.1111/j.1755-148X.2008.00496.x [DOI] [PubMed] [Google Scholar]
- Sitaram, A. , & Marks, M. S. (2012). Mechanisms of protein delivery to melanosomes in pigment cells. Physiology (Bethesda), 27(2), 85–99. 10.1152/physiol.00043.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukarieh, O. , Gaildrat, P. , Hamieh, M. , Drouet, A. , Baert‐Desurmont, S. , Frébourg, T. , … Martins, A. (2016). Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genetics, 12(1), e1005756 10.1371/journal.pgen.1005756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , & Tomita, Y. (2008). Recent advances in genetic analyses of oculocutaneous albinism types 2 and 4. Journal of Dermatological Science, 51(1), 1–9. 10.1016/j.jdermsci.2007.12.008 [DOI] [PubMed] [Google Scholar]
- Tomita, Y. , & Suzuki, T. (2004). Genetics of pigmentary disorders. American Journal of Medical Genetics, 131C(1), 75–81. 10.1002/ajmg.c.30036 [DOI] [PubMed] [Google Scholar]
- Tomita, Y. , Takeda, A. , Okinaga, S. , Tagami, H. , & Shibahara, S. (1989). Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochemical and Biophysical Research Communications, 164(3), 990–996. 10.1016/0006-291X(89)91767-1 [DOI] [PubMed] [Google Scholar]
- Veniani, E. , Mauri, L. , Manfredini, E. , Gesu, G. P. , Patrosso, M. C. , Zelante, L. , … Primignani, P. (2016). Detection of the first OCA6 Italian patient in a large cohort of albino subjects. Journal of Dermatological Science, 81(3), 208–209. 10.1016/j.jdermsci.2015.11.012 [DOI] [PubMed] [Google Scholar]
- Wei, A. , Wang, Y. U. , Long, Y. , Wang, Y. I. , Guo, X. , Zhou, Z. , … Li, W. (2010). A comprehensive analysis reveals mutational spectra and common alleles in Chinese patients with oculocutaneous albinism. The Journal of Investigative Dermatology, 130(3), 716–724. 10.1038/jid.2009.339 [DOI] [PubMed] [Google Scholar]
- Wei, A.‐H. , Zang, D.‐J. , Zhang, Z. , Liu, X.‐Z. , He, X. , Yang, L. , … Li, W. (2013). Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. The Journal of Investigative Dermatology, 133(7), 1834–1840. 10.1038/jid.2013.49 [DOI] [PubMed] [Google Scholar]
- Witkop, C. J. (1979). Albinism: Hematologic‐storage disease, susceptibility to skin cancer, and optic neuronal defects shared in all types of oculocutaneous and ocular albinism. Alabama Journal of Medical Sciences, 16(4), 227–330. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials