

Abstract
Methotrexate (MTX) is recognized as the anchor drug in the algorithm treating chronic arthritis (RA, psoriatic arthritis), as well as a steroid sparing agent in other inflammatory conditions (polymyalgia rheumatica, vasculitis, scleroderma). Its main mechanism of action has been related to the increase in extracellular adenosine, which leads to the effects of A2A receptor in M1 macrophages that dampens TNFα and IL12 production and increases IL1Ra and TNFRp75. By acting on A2B receptor on M2 macrophages it enhances IL10 synthesis and inhibits NF‐kB signaling. MTX has also been shown to exert JAK inhibition of JAK2 and JAK1 when tested in Drosophila melanogaster as a model of kinase activity and in human cell lines (nodular sclerosis Hodgkin's lymphoma and acute myeloid leukemia cell lines). These effects may explain why MTX leads to clinical effects similar to anti‐TNFα biologics in monotherapy, but is less effective when compared to anti‐IL6R in monotherapy, which acting upstream exerts major effects downstream on the JAK1‐STAT3 pathway. The MTX effects on JAK1/JAK2 inhibition also allows to understand why the combination of MTX with Leflunomide, or JAK1/JAK3 inhibitor leads to better clinical outcomes than monotherapy, while the combination with JAK1/JAK2 or JAK1 specific inhibitors does not seem to exert additive clinical benefit.
Keywords: adenosine, JAK inhibition, Methotrexate
Review on Methotrexate inhibitory effects on JAK 1‐2 in several cell lines at concentrations used in the clinic.
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- b‐DMARD
biological synthetic disease modified antirheumatic drugs
- cs‐DMARD
conventional synthetic disease modified antirheumatic drugs
- DHFR
dihydrofolate reductase
- ET
essential thrombocythemia
- GPI
glucose‐6‐phosphate isomerase
- IL6R
IL6 receptor
- MTX
methotrexate
- PsA
psoriatic arthritis
- PV
policythemia vera
- TCZ
tocilizumab
- ts‐DMARDs
targeted synthetic DMARDs
1. INTRODUCTION
Methotrexate (MTX) has become the anchor drug in RA and psoriatic arthritis (PsA) since 1962, when Black et al. showed that it was clinically effective in 21 patients with PsA1 and Hoffmeister in 1972 published an abstract in which he showed that 11 of 29 patients with RA had major clinical improvement at 10–15 mg/weekly2 and that the decrease of MTX dose led to 80% of disease relapse.
MTX is now the best first‐line treatment in all the recommendations by the Scientific Societies and has a retention rate at 2 years of more than 60% both in RA and in PsA, associated with an important clinical benefit.3 In clinical practice, once patients are consider to be incomplete responders (MTX‐IR), MTX is used in combination with conventional synthetic disease modified antirheumatic drugs (cs‐DMARDs),4 biologicals (b‐DMARDs),5 or targeted synthetic DMARDs (ts‐DMARDs).6, 7
Once biologics are compared directly with MTX in clinical trials, only tocilizumab (TCZ) and etanercept (to a lower degree, only for the ACR 20) appear to be superior to MTX, whereas ts‐DMARDs as JAK inhibitors showed a higher efficacy than MTX monotherapy.8, 9
How can we explain the similar efficacy of MTX to anti‐TNF α in monotherapy?
1.1. Intracellular and extracellular mechanisms of action of MTX
Being a chemotherapy agent, MTX acts as a folate antagonist that inhibits dihydrofolate reductase (DHFR) and ultimately purine synthesis, blocking highly replicative cells in the S phase. These effects can be bypassed and the clinical effects blocked by folinic acid supplementation acting on the folate pathway downstream of DHFR and relieving the inhibition of purine synthesis. However, at the lower doses used in RA, folinic acid reduces the side effects associated with MTX treatment without reverting its anti‐inflammatory and immune‐suppressive actions but maintaining its clinical efficacy, proving that other mechanisms of action are responsible for its DMARD activity. In addition, MTX blocks the 5‐aminoimidazole‐4‐carboxamide ribonucleotide transformylase (ATIC) that converts 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) to formyl‐AICAR (FAICAR). AICAR leads to enhanced release of adenine nucleotides into the extracellular space and converted to adenosine, which is thought to be the major driver of the pharmacologic action of MTX in RA.10, 11 Adenosine inhibits oxygen radical by neutrophils, suppresses monocyte‐derived cytokine/chemokine (among which TNF‐α and IL12) production acting on A2A, and through A2B AR increases IL10 production and promotes M2 macrophage activation.12
These pharmacobiologic effects may explain why the combination therapy of MTX + anti‐TNFα is more effective than MTX alone or anti‐TNF alone.5 Moreover, rituximab (RTX) and abatacept (CTLA4‐Ig) led to a better ACR 50 response or Boolean remission rate compared to MTX alone, yet it did not reach statistical significance,13, 14 whereas in RA, only TCZ monotherapy showed to be clearly superior to MTX monotherapy.15
2. MTX AND CYTOKINE SYNTHESIS
The histopathological analysis of synovial tissue inflammation in RA has shown that RA synovitis can be roughly divided into at least three major groups, myeloid, lymphoid, and fibroid pathotypes, respectively. If after MTX therapy the synovial inflammation still persists, the first subset responds better to TNF‐I and the second to IL6‐R inhibition.16 Why does not IL6 inhibition receives any additive effect by MTX?
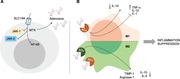
In Jurkat cells stimulated with TNF, MTX suppressed NF‐kB activation by inhibiting IkBα degradation and suppressing IkBα phosphorylation, effects all determined by adenosine.17 MTX leads to the downregulation of TNFα and IL1β and to an increase of IL1Ra and TNFRp75.18 It also inhibits NF‐kB activity in T cells, via BH4 depletion, and reduces activation of JNK, p53, and NF‐kB activity in fibroblast‐like synoviocytes through the release of adenosine and adenosine receptor activation, normalizing in vivo elevated NF‐kB activity.19 In addition to these effects, in collagen‐induced arthritis, Neurath et al. demonstrated that intraperitoneal MTX injection, prior to the development of arthritis, significantly suppressed IL15‐induced synthesis in splenic mononuclear cells of TNF, indirectly. However, no significant effects were seen on IL6.20 In the glucose‐6‐phosphate isomerase‐induced (GPI) model of arthritis, MTX inhibits GPI‐induced arthritis, with a vanishing effect, showing its ability to significantly reduce IL6 and IL6 receptor (IL6R) expression. Nevertheless this effect was mirrored by the progressive decrease of SLC19A1 expression, which is an important folate carrier for MTX intracellular uptake (Fig. 1A). When SCL19A1 expression decreases, MTX does not exert its effects anymore. Therefore, IL6 is the major driver of MTX unresponsiveness in the GPI model of arthritis.21 These effects may explain the similar efficacy of MTX compared to TNF inhibitors used in monotherapy but when added to TNF‐inhibitors (TNF‐I) MTX because of the possible increase of IL1Ra and TNFRp75, can allow a further control of inflammation. The MOA of TNF‐I is out of the scope of this review, yet it is important to consider that the biopharmacology may have practical translation into the clinics. If after MTX therapy the synovial inflammation still presents a lymphoid phenotype where a stronger IL6 inhibition is likely needed, MTX will exert little more effect.
Figure 1.
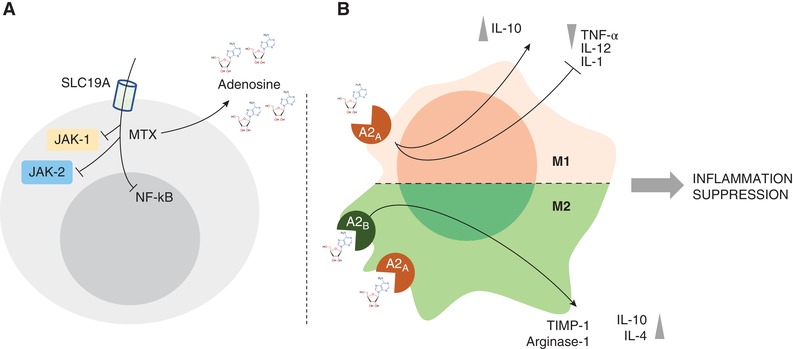
(A, B) MTX mechanisms of action and anti‐inflammatory properties of adenosine on macrophages. (A) Intracellular uptake through SLC19A receptor, extracellular increase in Adenosine and repression of NF‐kB activity. These effects and the inhibition of JAK‐1/JAK‐2 repress the autoimmune inflammation. (B) Schematic view of the anti‐inflammatory activities of adenosine on M1 and M2 macrophages
MTX exerts its suppression on cytokines promoting the release of adenosine producing its modulatory effect on inflammatory signals through A2A and A2B receptors on macrophages leading to the suppression of proinflammatory cytokines release (i.e., TNF‐α) and promotion of anti‐inflammatory molecules (i.e., IL‐10) (Fig. 1B).22 Moreover, MTX was demonstrated to repress the inflammatory signal in RA due to its inhibitory effect on another proinflammatory mediator, as High‐mobility group box 1 (HMGB1). In particular, MTX interferes with HMGB1 and RAGE ligand, at molecular level, acting via JAK/STAT pathway with a repression of TNF‐α overproduction.23, 24
3. LEFLUNOMIDE, AURANOFIN, AND MTX: JAK‐STAT INHIBITION
The first cs‐DMARD showing a potential intracellular effect on transcription factors of the JAK/STAT pathway was Leflunomide (LFN). In 1998, Siemasko showed that in spleen cells from B10 E mice,25 LFN diminished the tyrosine phosphorylation of JAK3 and STAT6 at concentrations of 100–200µM. Moreover, another cs‐DMARD as Auranofin, markedly inhibited IL‐6‐induced phosphorylation of JAK1 at concentrations of 1.3µM and STAT3 in human Hep2 cell lines.26 Auranofin inhibited constitutive and IL6‐induced activation of JAK2 and phosphorylation of STAT3 in the range of 6 mg/day in humans. The most interesting results on csDMARDs were presented in 2015, when Thomas reported that MTX inhibited JAK/STAT pathway activity in Drosophila melanogaster used as a low complexity model system to screen the action of small molecules on JAK‐STAT pathways.27 When tested in human cells lines (HDLM2—Hodgkin's lymphoma derived, HEL—Acute myeloid leukemia expressing JAK2 V617F variant, a gain of function mutation associated with most human myeloproliferative neoplasms), MTX significantly reduced JAK1 phosphorylation and STAT1 and STAT5 phosphorylation at concentrations of 0.4–0.8µM in RA. When tested in the human Hodgkin's lymphoma‐derived cell lines, MTX inhibited phosphorylation of STAT 1 and STAT5 but not of STAT3. Similarly, taken orally, 1µM ruxolitinib, a JAK1/JAK2 selective inhibitor produced a deeper suppression of STAT5 phosphorylation. Interestingly, transgenic mice in which the JAK2 locus was replaced by the human JAK2 V617F allele, developing essential thrombocythemia (ET)‐ and policythemia vera (PV)‐like neoplasms showed a significant reduction of STAT3‐STAT5 phosphorylation in splenocytes of homozygous mice treated with MTX,28 while in silico modeling suggested that this inhibition may be the result of a direct binding of MTX to the JAK2 kinase domain ATP binding pocket. Accordingly, in two patients with myeloproliferative disorders as PV and ET, MTX at 10 mg weekly allowed the reduction of constitutional symptoms and the normalization of hematological values within 2 months and their maintenance.29 Finally, molecular studies performed on RA patients and SLE patients demonstrated that low‐dose MTX reduced in vivo JAK/STAT pathway activity,30 supporting the hypothesis that MTX may act via inhibition of the JAK/STAT signaling.
4. MTX, TNF‐I, AND TOCILIZUMAB IN CLINICAL TRIALS
In all clinical trials, including TNFα inhibitors (i.e., infliximab, adalimumab, golimumab, certolizumab, and etanercept), the rate of ACR50 or DAS‐remission achievement was not statistically different when comparing patients treated with monoclonals alone and MTX alone. On the other hand, the combination of b‐DMARD and MTX, was associated with a significant reduction in joint damage compared to MTX alone and in MTX naive patients only.31 Considering mechanisms of action other than TNF inhibition, only TCZ proved to be clearly superior to MTX.9
Therefore, we hypothesize that MTX has similar efficacy to anti‐TNF monotherapy through Adenosine and JAKs inhibition. However, selective JAK inhibition does not directly suppress TNF synthesis by immune cells.32 Therefore, the additive role of MTX, when used in combination with TNF inhibitors, may be supported in vivo in RA by the MTX suppressive effects on IL1β, IL3, IL5, IFNγ, and as expected from the JAK1 inhibition, also on IL6.33
In clinical trials, TCZ was demonstrated to be superior to MTX, and to be more successful in RA patients who are incomplete responders to TNF inhibition.9 In fact, IL6 signaling through IL6‐IL6R‐gp130 binding, leads to JAK1/JAK2/STAT3 activation.32 Therefore, TCZ acting upstream of JAKs and blocking the interaction of IL6 with its receptors, can more strongly suppress the inflammatory signal and once MTX is added, acting downstream, limited additional effects are registered. These insights may allow to understand why TCZ appears to be the only b‐DMARD superior to MTX alone.34
5. JAK INHIBITORS AND MTX
The inhibition of JAK‐STAT phosphorylation has been tested in Drosophila as a cell model to screen small molecules, as well as in several immune cells, like splenocytes, T lymphocytes, and whole blood or directly on enzyme assays.35, 36, 37, 38 When JAK inhibitors are tested, strong inhibition of JAK1 also inhibits JAK1/JAK3‐dependent IL‐15 signaling. When assessed in kinase assays using 1 mM ATP, none of the compounds are only JAK3 selective and all inhibit both JAK1/JAK3‐dependent IL‐15 signaling and JAK1/TYK2‐dependent interferon α signaling.39 This explains why at high concentrations compounds inhibiting JAK1‐3 may also inhibit JAK2, thus, displaying a pan‐JAK inhibition. JAK1‐2‐3‐TYK2 inhibition and the effects on the inflammatory pathways have been comprehensively discussed in several reviews.39, 40 JAK3 appears to be mainly involved in hematopoietic cells and has a crucial role in lymphocyte biology while JAK1,2 and TYK2 are expressed ubiquitously.37 For this reason, JAK3 inhibition appeared to be a first rational target in rheumatic inflammatory diseases. The randomized clinical trial comparing tofacitinib and MTX showed a higher effectiveness of tofacitinib at 5 and 10 mg compared to MTX at 10–20 mg/week,6 suggesting that specific inhibition of JAK1/JAK3, by tofacitinib, appears to be superior to MTX, which acts by inhibiting JAK1/JAK2.
When used in combination, MTX plus tofacitinib showed similar clinical effect to MTX plus adalimumab,41 suggesting that the specific inhibition led by tofacitinib combined with JAK1/JAK2 inhibition by MTX may have additive effects. The same results did not occur with baricitinib, which acts on JAK1/JAK2. In fact, clinical trial showed that the addition of MTX to baricitinib does not result in significantly better clinical benefits, already obtained by baricitinib alone, which can rescue MTX incomplete responders owing to its specific inhibition, though the combination led to a better control of erosions.42 Finally, given the different JAK targets being inhibited, it can be hypothesized that the combination of baricitinib plus LFN might be of clinical interest considering that LFN inhibits JAK3 and this may explain also the additive effect of LFN when used in combination with MTX.43 The same should occur with Upadacitinib and Filgotinib.44, 45 When examining the side effects associated to JAK inhibitors, the major one is represented by the occurrence of Herpes Zoster infection.46 Despite discordant results have been described in the literature,47 yet most recent data suggest that Zoster infection may be a side effect of MTX therapy.48 All these data suggest that MTX may indeed display JAK1/JAK2 inhibition in vivo.
6. PERSPECTIVES
MTX is a fundamental part of the therapeutic strategy in several inflammatory and autoimmune diseases. Its efficacy is recognized as a fundamental first step approach in several inflammatory diseases. Its effects on the purinergic pathways through adenosine modulation and on NF‐kB signal together with emerging insights into its action on JAK1/JAK2 pathways (Fig. 2) suggest that it will certainly remain the anchor drug of excellence in the clinics.
Figure 2.
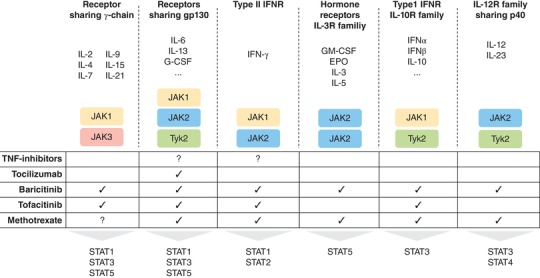
Schematic view of the effects on JAKs played by conventional synthetic (MTX), targeted synthetic (tofacitinib/baricitinib) and biological disease modifying anti‐rheumatic drugs (TNF‐inhibitor/tocilizumab). MTX: Methotrexate
DISCLOSURES
EG and GF conceived and structured the review; EG, SA, BT, MPZ and GF performed research and wrote the manuscript; EG, SA, BT, MPZ and GF read and approved the final version of the manuscript. The authors declare no conflicts of interest.
Gremese E, Alivernini S, Tolusso B, Zeidler MP, Ferraccioli G. JAK inhibition by methotrexate (and csDMARDs) may explain clinical efficacy as monotherapy and combination therapy. J Leukoc Biol. 2019;106:1063–1068. 10.1002/JLB.5RU0519-145R
Contributor Information
Elisa Gremese, Email: elisa.gremese@unicatt.it.
Gianfranco Ferraccioli, Email: gianfranco.ferraccioli@unicatt.it.
REFERENCES
- 1. Black RL, O'Brien WM, Van Scott EJ, et al. Methotrexate therapy in psoriatic arthritis. JAMA. 1964;189:743‐747. [PubMed] [Google Scholar]
- 2. Hoffmeister RT. Methotrexate in rheumatoid arthritis. Arthritis Rheumatol. 1972;15:114. [Google Scholar]
- 3. O'Dell JR, Haire CE, Erikson N, et al. Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications. N Engl J Med. 1996;334:1287‐1291. [DOI] [PubMed] [Google Scholar]
- 4. Ferraccioli GF, Gremese E, Tomietto P, et al. Analysis of improvements, full responses, remission and toxicity in rheumatoid patients treated with step‐up combination therapy (methotrexate, cyclosporin A, sulphasalazine) or monotherapy for three years. Rheumatology. 2002;41:892‐898. [DOI] [PubMed] [Google Scholar]
- 5. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: a multicenter, randomized, double‐blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26‐37. [DOI] [PubMed] [Google Scholar]
- 6. Van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508‐519. [DOI] [PubMed] [Google Scholar]
- 7. Taylor PC, Keystone EC, van der Heijde D, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:852‐862. [DOI] [PubMed] [Google Scholar]
- 8. Nam JL, Takase‐Minegishi K, Ramiro S, et al. Efficacy of biological disease‐modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2017;76:1113‐1136. [DOI] [PubMed] [Google Scholar]
- 9. Tarp S, Furst DE, Dossing A, et al. Defining the optimal biological monotherapy in rheumatoid arthritis: a systematic review and meta‐analysis of randomised trials. Semin Arthritis Rheum. 2017;46:699‐708. [DOI] [PubMed] [Google Scholar]
- 10. Haskó G, Bruce Cronstein B. Regulation of inflammation by Adenosine. Frontiers Immunol. 2013;4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedman B, Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 2019;86:301‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koscsó B, Csóka B, Kokal E, et al. Adenosine augments IL‐10‐induced STAT3 signaling in M2c macrophages. J Leukoc Biol. 2013;94:1309‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Edwards JCW, Szczepanski L, Szechi´nski J, et al. Efficacy of B‐cell–targeted therapy with Rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572‐2581. [DOI] [PubMed] [Google Scholar]
- 14. Emery P, Burmester G, Bykerk V, et al. Evaluating drug‐free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active‐controlled AVERT nstudy of 24 months, with a 12‐month, double‐blind treatment period. Ann Rheum Dis. 2015;74:19‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emery P, Pope JE, Kruger K, et al. Efficacy of monotherapy with biologics and JAK inhibitors for the treatment of rheumatoid arthritis: a systematic review. Adv Ther. 2018;35:1535‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dennis G, Jr , Holweg CT, Kummerfeld SK, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. 2014;16:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Majumdar S, Aggarwal BB. Methotrexate suppresses NF‐kB activation through inhibition of IkBa phosphorylation and degradation. J Immunol. 2001;167:2911‐2920. [DOI] [PubMed] [Google Scholar]
- 18. Seitz M, Zwicker M, Loetscher P. Effects of MTX on differentiation of monocytes and production of cytokine inhibitors by monocytes. Arthr Rheum. 1998;41:2032‐2038. [DOI] [PubMed] [Google Scholar]
- 19. Spurlock CF, 3rd , Gass HM, 4th , Bryant CJ, et al. Methotrexate‐mediated inhibition of nuclear factor κB activation by distinct pathways in T cells and fibroblast‐like synoviocytes. Rheumatology. 2015;54:178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neurath MF, Hildner K, Becker C, et al. Methotrexate specifically modulates cytokine production by T cells and macrophages in murine collagen‐induced arthritis (CIA): a mechanism for methotrexate‐mediated immunosuppression. Clin Exp Immunol. 1999;115:42‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hashizume M, Yoshida H, Tanaka K, et al. Interleukin‐6 regulates anti‐arthritic effect of methotrexate via reduction of SLC19A1 expression in a mouse arthritis model. Arthritis Res Ther. 2012;14:R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasko G, Pacher P. Regulation of macrophage function by adenosine. Aterioscler Thromb Vasc Biol. 2012;32:865‐869. 10.1161/ATVBAHA.111.226852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Xu P, Xu K, et al. Methotrexate affects HMBG1 expression in rheumatoid arthritis, and the downregulation of HMGB1 prevents rheumatoid arthritis progression. Mol Cell Biochem. 2016;420:161‐170. [DOI] [PubMed] [Google Scholar]
- 24. Kuroiwa J, Takakusagi Y, Kusayanagi T, Kuramochi K, et al. Identification and characterization of the direct interaction between methotrexate (MTX) and high‐mobility group box 1 (HMGB1) protein. POLS One. 2013;8:e63073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siemasko K, Chong AS, Jäck HM, et al. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol. 1998;160:1581‐1588. [PubMed] [Google Scholar]
- 26. Kim NH, Lee MY, Park SJ, et al. Auranofin blocks interleukin‐6 signalling by inhibiting phosphorylation of JAK1 and STAT3‐. Immunology. 2007;122:607‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas S, Fisher KH, Snowden JA, et al. Methotrexate is a JAK/STAT pathway inhibitor. PLos One. 2015;10:e0130078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chinnaiya K, Lawson MA, Thomas S, et al. Low dose methotrexate in myeloproliferative neoplasm model. Haematologica. 2017 Sep;102(9):e336‐e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palandri F, Labate C, Sabattini E, et al. Low‐dose methotrexate as treatment of myeloproliferative neoplasms: proof of principle of clinical activity. Am J Hematol. 2016;91:e329‐30. [DOI] [PubMed] [Google Scholar]
- 30. Goropevšek A, Holcar M, Pahor A, et al. STAT signaling as a marker of SLE disease severity and implications for clinical therapy. Autoimmun Rev. 2018;18:144‐154. [DOI] [PubMed] [Google Scholar]
- 31. Tarp S, Jurgensen TS, Furst TS, et al. Added value of combining methotrexate with a biological agent compared to biological monotherapy in rheumatoid arthritis patients: a systematic review and meta‐analysis of randomised trials. Semin Arthr. Rheum. 2019;48:958‐966. [DOI] [PubMed] [Google Scholar]
- 32. O'Shea JJ, Gadina M. Selective Janus kinase inhibitors come of age. Nat Rev Rheumatol. 2019;15:74‐75. [DOI] [PubMed] [Google Scholar]
- 33. Kremer JM, Lawrence DA, Hamilton R, McInnes IB. Long‐term study of the impact of methotrexate on serum cytokines and lymphocyte subsets in patients with active rheumatoid arthritis: correlation with pharmacokinetic measures. RMD Open. 2016;2:e000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bijlsma JWJ, Welsing PMJ, Woodworth TG, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U‐Act‐Early): a multicentre, randomised, double‐blind, double‐dummy, strategy trial. Lancet. 2016;388:343‐355. [DOI] [PubMed] [Google Scholar]
- 35. Spindler SR, Li R, Dhahbi JM. Novel protein kinase signaling systems regulating lifespan identified by small molecule library screening using Drosophila . PLoS One. 2012;7(2):e29782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dowty ME, Jesson MI, Ghosh S, et al. Preclinical to clinical translation of tofacitinib, a janus kinase inhibitor, in rheumatoid arthritis. J Pharmacol Exp Ther. 2014;348:165‐173. [DOI] [PubMed] [Google Scholar]
- 37. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus Kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57:5023‐5038. [DOI] [PubMed] [Google Scholar]
- 39. Gadina M, Le MT, Schwartz DM, et al. Janus kinases to Jakinibs: from basic insights to clinical practice. Rheumatology. 2019;58:i4‐i16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery. 2017;16:843‐862. [DOI] [PubMed] [Google Scholar]
- 41. Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double‐blind, head‐to‐head, randomised controlled trial. Lancet. 2017;390:457‐468. [DOI] [PubMed] [Google Scholar]
- 42. Fleischmann R, Schiff M, van der Heijde D, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease‐modifying antirheumatic drug treatment. Arthritis Rheumatol. 2017;69:506‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hazlewood GS, Barnabe C, Tomlinson G, et al. Methotrexate monotherapy and methotrexate combination therapy with traditional and biologic diseasemodifying antirheumatic drugs for rheumatoid arthritis: a network metaanalysis (Review). Cochrane Database Syst Rev. 2016; ;353:i1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐ controlled phase 3 trial. Lancet. 2018;391:2503‐2512. [DOI] [PubMed] [Google Scholar]
- 45. Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS‐6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose‐ finding study (DARWIN 1). Ann Rheum Dis. 2017;76:998‐100. [DOI] [PubMed] [Google Scholar]
- 46. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13:234‐243. [DOI] [PubMed] [Google Scholar]
- 47. Pappas DA, Hooper MM, Kremer JM, et al. Herpes zoster reactivation in patients with rheumatoid arthritis: analysis of disease characteristics and disease‐modifying anti‐rheumatic drugs. Arthr Care Res. 2015;67:1671‐1678. [DOI] [PubMed] [Google Scholar]
- 48. Nakajima A, Urano W, Inoue E, et al. Incidence of herpes zoster in Japanese patients with rheumatoid arthritis from 2005 to 2010. Modern Rheumatol. 2015;25:558‐561. [DOI] [PubMed] [Google Scholar]