

Abstract
Aims
The gut microbiota is believed to play important roles in the health of pregnant mammals, including their nutrient metabolism, immune programming and metabolic regulation. However, until recently, the shifts in gut microbiota composition and faecal and blood metabolic activity during different stages of pregnancy had not been investigated.
Methods and Results
We investigated the shifts in backfat thickness, plasma and faecal metabolites and gut microbiota on days 30, 60, 90 and 110 of pregnancy and on day 21 after parturition (weaning) in sows. The backfat thickness of sows did not significantly differ among the different stages of pregnancy. The plasma concentrations of lipid metabolites, including triacylglycerol (TG), total cholesterol, high‐density lipoprotein‐cholesterol, low‐density lipoprotein‐cholesterol and calcium were reduced (P < 0·05) during pregnancy. In addition, the concentration of these metabolites, except TG, reached their maximum at the time of weaning. We also found that Tenericutes, Fibrobacteres and Cyanobacteria varied significantly according to the stages of pregnancy in sows (P < 0·05). Most of the genera, such as Clostridiales, Desulfovibrio, Mogibacteriaceae and Prevotella, increased (P < 0·05) with the progression of pregnancy and decreased (P < 0·05) at weaning. The alpha diversity values (i.e., Shannon diversity and observed species) of sow gut microbiota increased (P < 0·05) from pregnancy to weaning. Pregnancy stages also significantly influenced (P < 0·05) the community structure (beta diversity) of gut microbiota. The progression of pregnancy was associated with changes in lipid metabolism and several carbohydrate‐degradation bacteria (i.e., Prevotella, Succinivibrio, Bacteroides and Parabacteroides).
Conclusions
Although causal links between the measured parameters remain hypothetical, these findings suggest that the increased diversity and concentration of beneficial gut microbes are associated with the metabolism of pregnant sows.
Significance and Impact of the Study
Manipulation of the sow gut microbiota composition may potentially influence metabolism and health during pregnancy.
Keywords: gut microbiota, metabolite, microbial diversity, sows, stages of pregnancy
Introduction
Gastrointestinal bacterial communities are known to play critical roles in the functioning and health of their hosts, including their nutrient absorption, metabolism, immune programming and protection from pathogens (Dethlefsen et al. 2007). These bacterial communities are influenced by various host and environmental factors, such as host genetics (Goodrich et al. 2014), obesity (Ridaura et al. 2013), dietary intake (Wu et al. 2011), environmental parameters (Grzeskowiak et al. 2012), use of prebiotics, probiotics and antibiotics and pregnancy (Dethlefsen and Relman 2011; Debelius et al. 2016). The relationships between pregnancy and gut microbiota are of particular interest since pregnancy is characterized by dramatic changes in hormones, immune functions and metabolism to support the growth of the mother and foetoplacental unit (Newbern and Freemark 2011). The analysis of biological fluids, especially the blood, has been used to identify typical compounds to evaluate the underlying metabolic changes that occur during pregnancy (Shen et al. 2016). In addition, the maternal gut microbiota also undergoes dramatic changes throughout the gestation period (Kuperman and Koren 2016). Among the reported changes, changes in the amino acid and lipid content, energy metabolism, as well as gut microbiota composition and metabolic activity have been linked to pregnancy (Dai et al. 2015; DiGiulio et al. 2015; Mandal et al. 2016). The metabolic activity of the microbiota allows for the synthesis of various compounds, including short‐chain fatty acids (SCFA), indoles, ammonia, bioamines, gaseous compounds and vitamins (Blachier et al. 2017). These compounds, after intestinal absorption, can be modified by the host and may be actively involved in the host cells as co‐metabolites.
Recently, most studies have focused on changes that occur in the microbiota during pregnancy and have revealed an overall increase in Proteobacteria and Actinobacteria and a reduced diversity in the human gut (Koren et al. 2012). However, DiGiulio et al. (2015) showed that the taxonomic composition and diversity of the microbiota community remained remarkably stable in the vagina, distal gut and saliva during pregnancy. The exact role of intestinal microbiota and the balance of these microbes to the complex process of sustaining the conceptus in utero and in maintaining an adequate pregnant period is not yet known (Nelson et al. 2016). Several reports have evaluated the relationship between pregnancy and gut microbiota in humans, but few studies have explored the shifts in gut microbiota and other metabolites in sows throughout pregnancy until weaning.
Although previous studies have detected the ileal and colonic microbiota composition in Huanjiang mini‐pigs during pregnancy (Ji et al. 2016; Kong et al. 2016), such studies did not report the corresponding changes in plasma metabolites and the faecal microbiota in the pregnant and postpartum conditions. Therefore, in the present study, we investigated such factors in Yorkshire × Dutch Landrace sows. Pregnant sows were fed a restricted diet, in accordance with the practical production in order to avoid the unhealthy effects of obesity. The plasma and faeces metabolite composition and gut microbial composition were determined on days 30, 60, 90 and 110 of pregnancy and on day 21 after parturition (i.e., at weaning). We mainly addressed three questions: (i) How do the plasma and faeces metabolites change throughout the pregnancy and weaning stages? (ii) Do the alpha and beta diversity values of gut microbiota change during pregnancy and weaning? (iii) What are the relationships between gut microbiota and metabolic products? Our results have important significance for understanding the relationship between gut microbiota and pregnancy.
Materials and methods
Experimental design and ethical standards
This study was approved by the animal welfare committee of the Institute of Subtropical Agriculture, Chinese Academy of Sciences. The experiment was conducted between June and October 2016. In total, 40 Yorkshire × Dutch Landrace crossbred sows with second‐ or third‐parity (New Wellful Co. Ltd, Hunan, China) were used. After insemination, the sows were housed individually in crates (2·4 × 0·7 m) from day 1 to day 107 of pregnancy and then were housed in farrowing crates (2·2 × 1·8 m) until weaning. The sows were fed a commercial gestational diet (3100 kcal kg−1, DE) from mating until day 100 of pregnancy and received a commercial lactation diet (3300 kcal kg−1, DE) from day 101 of pregnancy to weaning at 21 days in the postfarrowing period. Diet formulations used in this study are presented in Table 1.
Table 1.
Formulation and chemical composition of diets
| Items | Gestational diet | Lactation diet |
|---|---|---|
| Ingredient, % | ||
| Corn | 58·67 | 60·46 |
| Wheat bran | 24·50 | 5·00 |
| Ground wheat | – | 2·00 |
| Soybean oil | 1·00 | 3·00 |
| Soybean meal | 11·50 | 19·50 |
| Albumen powder | – | 3·00 |
| Imported fish | – | 2·50 |
| Lysine | 0·12 | 0·30 |
| Threonine | 0·03 | 0·05 |
| Antioxidants | 0·02 | 0·03 |
| Antimildew agent | 0·06 | 0·06 |
| Detoxification cagent | 0·10 | 0·10 |
| Premix* | 4·00 | 4·00 |
| Nutritional value † | ||
| DE (MJ kg−1) | 13·25 | 15·00 |
| CP | 14·00 | 18·00 |
| Lys | 0·75 | 1·10 |
| Met | 0·20 | 0·35 |
| Thr | 0·55 | 0·79 |
Supplied the following amounts per kilogram of diet: vitamin A, 10 000 IU; vitamin D3, 2000 IU; vitamin E, 100 IU; vitamin K3, 2 mg; thiamine, 2 mg; riboflavin, 4 mg; nicotinic acid, 15 mg; d‐pantothenic acid, 10 mg; pyridoxine, 3 mg; d‐biotin, 0·2 mg; folic acid, 3 mg; vitamin B12, 0·02 mg; choline, 500 mg; Na (NaCl), 1·38 g; Fe (FeSO4·H2O), 80 mg; Cu (CuSO4·5H2O), 20 mg; Mn (MnSO4·H2O), 40 mg; Zn (ZnSO4·H2O), 100 mg; Co (CoCl), 0·1 mg; I (KI), 0·6 mg; and Se (Na2SeO3), 0·25 mg.
Calculated values.
The sows were fed 1·6 kg of the gestational diet after insemination, i.e., days 0 to 3 and then, fed 2·5 kg of the same diet according to the body condition of the pregnant sows from days 4 to 100. Sows received 3·5 kg of the lactation diet from days 101 to 113 and then received 2·5 kg on the day of parturition. Sows were fed twice per day (9:00 and 16:00) from insemination until 3 days after parturition. Thereafter, sows were gradually fed more until the feed was available ad libitum. From day 4 of lactation until weaning, sows were fed four times per day (9:00, 14:00, 18:00 and 22:00). Water was available ad libitum throughout the entire experiment.
Sample collection
Blood (n = 8) and fresh faecal samples (n = 8) were collected on days 30, 60, 90 and 110 of the gestation period and on day 21 after parturition (weaning). At the same time, the backfat thickness (n = 20) was measured. A single blood sample was collected into heparinized tubes from each sow 2 h after feeding by jugular venipuncture using a 10 ml syringe. Plasma was collected after centrifugation for 10 min at 3000 g and 4°C and stored at −20°C. The fresh faecal samples were immediately collected after defecation and transported in a refrigerated container (−20°C).
Measurements of carcass traits, plasma biochemical parameters and faecal metabolites
The backfat thickness (P2 site) was measured using a Renco lean‐meter (Renco Corporation, Golden Valley, MN). The concentrations of plasma metabolites, including total protein (TP), albumin (ALB), globulin (GLB), triacylglycerol (TG), total cholesterol (TC), high‐density lipoprotein‐cholesterol (HDL‐C), low‐density lipoprotein‐cholesterol (LDL‐C), calcium (Ca) and phosphorus (P) were analysed using a CX‐4 Automatic Biochemical Analyzer (Beckman Inc., Fullerton, CA) and commercial kits (Leadman Biochemistry Technology Company, Beijing, China) according to the manufacturers’ instructions. In addition, we also measured the activity (U l−1) of plasma enzymes, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and α‐amylase (α‐AMY). The faecal SCFAs, including straight‐chain fatty acids (acetate, propionate, butyrate, and pentanoate) and branched chain fatty acids (BCFA; isobutyrate and isopentanoate) were analysed using gas chromatography as described previously (Ji et al. 2018). The bioamines, including 1,7‐heptyl diamine, cadaverine, phenylethylamine, putrescine, tryptamine, tyramine, spermidine and spermine were measured using high‐performance liquid chromatography as described previously (Ji et al. 2018).
DNA extraction and 16S rRNA gene sequencing
DNA extraction and 16S rRNA gene sequencing were conducted at the Environmental Genomic Platform of Chengdu Institute of Biology. The total microbial genomic DNA from faeces was extracted using a QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The DNA concentration of each sample was measured with a NanoDrop® ND‐1000 instrument (NanoDrop Technologies Inc., Waltham, MA). The protocols of PCR amplification, gel extraction and sequencing library construction used in this study were described by Li et al. (2016). The extracted DNA was diluted to 10 ng µl−1 for the PCR amplifications. The universal primers: 515F (5′‐GTGYCAGCMGCCGCGGTA‐3′) and 909R (5′‐CCCCGYCAATTCMTTTRAGT‐3′), with a 12 nt unique barcode at 5′‐end of the 515F primer (Tamaki et al. 2011), were used to amplify the V4 region of microbial 16S rRNA gene. After PCR amplification, amplicons were extracted from 1·2 agarose gels and purified using the SanPrep DNA Gel Extraction Kit (Sangon Biotech, Shanghai, China) and quantified with a NanoDrop ND‐1000 instrument (NanoDrop Technologies Inc., Waltham, MA, USA). Purified amplicons of equimolar concentrations were then pooled together and paired‐end sequenced using an Illumina MiSeq sequencer (MiSeq Reagent Kit V.2,500 cycles) according to standard protocols (Caporaso et al. 2012).
Bioinformatics analysis
Bioinformatics analyses followed previous study (Li et al. 2016). Briefly, the raw sequences were analysed using QIIME Pipeline‐ver. 1.7.0 (http://qiime.org/tutorials/tutorial.html). All sequences were trimmed and assigned to each sample based on their unique barcodes (barcode mismatches = 0). The overlapping paired‐end reads were merged using the flash‐1.2.8 software (Caporaso et al. 2012). The merged sequences with high quality (read length >300 bp, without an ambiguous base ‘N’ and an average base quality score >30) were used for further analysis. The aligned 16S rRNA gene sequences were used for a chimera check using the Uchime algorithm (Edgar et al. 2011). Operational taxonomic units (OTUs) were clustered at a 97% identity threshold using CD‐HIT (Li and Godzik 2006). Singleton OTUs were filtered out. Each sample was rarefied to the same number of reads (8467). The most abundant sequences within each OTU were designated as ‘representative sequences’ and aligned against the core set of Greengenes 13_8 reference database (DeSantis et al. 2006) using the PyNAST tool. The representative sequences were taxonomically classified using the Ribosomal Database Project classifier in the QIIME platform (Wang et al. 2007). The alpha diversity indices, including observed species and Shannon diversity, were calculated. The rarefaction curves of all samples were generated from the observed OTUs at the OTU level. To assess the beta diversity, the weighted UniFrac distance metrics, which use phylogenetic information to calculate community similarity (Lozupone and Knight 2005), were produced through the QIIME pipeline. Principal coordinates analysis (PCoA) plots of the dissimilarity metrics were also visualized using origin 8.5.
Statistical analysis
An analysis of similarity (anosim) was used to reveal whether the beta diversity was significantly different across the pregnancy and parturition stages based on the weighted UniFrac matrices using the ‘vegan’ package in the R program (Li et al. 2017). The effects of the different stages on backfat thickness, plasma biochemical parameters, the apparent relative abundances of communities at the phylum and genus level and the alpha diversity indices were analysed using a one‐way analysis of variance with LSD post hoc test in SAS (SAS Institute, Inc., Cary, NC). P‐values <0·05 were considered to indicate statistical significance and P‐values: 0·05 ≤ P < 0·10 were considered to indicate a trend. Linear discriminant analysis effect size (LEfSe) was used to identify bacterial biomarkers at the different stages, based on P‐values < 0·05 and LDA scores >2·0. These analyses were performed online in the Galaxy workflow framework (http://huttenhower.sph.harvard.edu/galaxy/).
Nucleotide sequence accession numbers
The original 16S rRNA gene data are available at the European Nucleotide Archive: accession no. PRJEB26887 (http://www.ebi.ac.uk/ena/data/view/PRJEB26887).
Results
Backfat thickness and plasma biochemical parameters
The mean backfat thickness of 20 sows were 21·32, 21·24, 20·72, 19·62, and 18·32 mm on days 30, 60, 90 and 110 of pregnancy and on day 21 after parturition (weaning). The backfat thickness of sows on days 30 and 60 of pregnancy were greater (P < 0·05) than at weaning.
The evaluations of plasma metabolites revealed that the pregnancy stages affected the metabolism of protein, fat, Ca and P (Table 2). Plasma ALB and P concentrations progressively increased throughout pregnancy, and were reduced at the time of weaning (P < 0·05). Conversely, the GLB, HDL‐C, LDL‐C, TC, TG and Ca concentrations were reduced (P < 0·05) during pregnancy. In addition, the concentrations of these plasma metabolites, except TG, reached their maximum levels at the time of weaning (P < 0·05). With regard to enzyme activity in the plasma, the ALT activity was the highest on day 110 of gestation, but the lowest at the time of weaning (P < 0·05). The AST activity was higher on days 90 and 110 of gestation and lower on the other days (P < 0·05). The α‐AMY activity was higher (P < 0·05) on days 30, 60 and 110 of pregnancy. The LDH activity did not differ significantly (P> 0·05) with the progression of pregancy.
Table 2.
Plasma metabolite concentrations and enzyme activities of sows at different stages
| Items | Days of gestation in sows | Weaning | SEM | P‐values | |||
|---|---|---|---|---|---|---|---|
| 30 | 60 | 90 | 110 | ||||
| TP (g l−1) | 71·03 ± 0·66ab | 75·13 ± 2·25a | 59·87 ± 2·10b | 60·45 ± 2·37b | 76·05 ± 5·58a | 0·967 | 0·001 |
| ALB (g l−1) | 29·82 ± 0·64b | 30·17 ± 1·58b | 30·55 ± 1·51b | 35·83 ± 0·86a | 33·68 ± 0·57ab | 0·584 | 0·002 |
| GLB (g l−1) | 41·22 ± 0·93ab | 44·97 ± 1·98a | 29·32 ± 2·61bc | 24·62 ± 1·86c | 42·37 ± 5·60ab | 0·966 | 0·002 |
| TG (mmol l−1) | 0·25 ± 0·05ab | 0·32 ± 0·04a | 0·21 ± 0·03ab | 0·12 ± 0·02b | 0·14 ± 0·01b | 0·106 | 0·004 |
| TC (mmol l−1) | 1·42 ± 0·06abc | 1·47 ± 0·13ab | 1·12 ± 0·04bc | 1·03 ± 0·12c | 1·65 ± 0·10a | 0·172 | 0·001 |
| HDL‐C (mmol l−1) | 0·51 ± 0·02bc | 0·51 ± 0·04b | 0·41 ± 0·02bc | 0·35 ± 0·03c | 0·70 ± 0·07a | 0·108 | <0·001 |
| LDL‐C (mmol l−1) | 0·67 ± 0·04ab | 0·74 ± 0·07ab | 0·50 ± 0·04b | 0·55 ± 0·07ab | 0·77 ± 0·04a | 0·129 | 0·006 |
| Ca (mmol l−1) | 2·41 ± 0·03a | 2·48 ± 0·05a | 2·37 ± 0·05a | 2·18 ± 0·03b | 2·52 ± 0·03a | 0·108 | <0·001 |
| P (mmol l−1) | 1·61 ± 0·03c | 1·73 ± 0·06bc | 1·96 ± 0·06ab | 2·17 ± 0·08a | 1·78 ± 0·09bc | 0·143 | <0·001 |
| ALT (U l−1) | 31·28 ± 1·89ab | 30·50 ± 2·49ab | 25·40 ± 2·14b | 36·38 ± 1·52a | 24·50 ± 1·97b | 0·788 | 0·002 |
| AST (U l−1) | 19·08 ± 1·44b | 18·78 ± 0·84b | 31·02 ± 9·21a | 37·45 ± 4·17a | 23·30 ± 2·36ab | 0·001 | <0·001 |
| LDH (U l−1) | 193·17 ± 16·74 | 198·15 ± 10·88 | 256·73 ± 54·98 | 245·98 ± 20·64 | 212·70 ± 33·56 | 3·108 | 0·524 |
| α‐AMY (U l−1) | 3508·28 ± 336·53a | 3240·07 ± 605·66a | 1880·48 ± 398·75b | 3467·73 ± 190·30a | 1907·57 ± 476·36b | 11·404 | 0·014 |
The days 30, 60, 90 and 110 are after artificial fertilization and weaning (day 21) is after parturition. The results are obtained from five independent experiments involving 40 sows. Significant difference is indicated by different letters.
The concentrations of almost all the SCFAs and bioamines, except 1,7‐heptyl diamine, differed significantly among the different stages of pregnancy (Table 3). Notably, we found that acetate, propionate, butyrate, the total SCFAs and total SCFAs decreased from days 30 to 110 of pregnancy, but suddenly increased at weaning (P < 0·05).
Table 3.
The faecal concentrations of short‐chain fatty acids (SCFAs) and bioamines in sows at different stages
| Items | Days of gestation in sows | Weaning | SEM | P‐values | |||
|---|---|---|---|---|---|---|---|
| 30 | 60 | 90 | 110 | ||||
| SCFAs (mg g−1) | |||||||
| Acetate | 5·84 ± 0·50b | 5·39 ± 0·20b | 4·04 ± 0·27c | 2·67 ± 0·44d | 7·19 ± 0·48a | 0·332 | <0·001 |
| Propionate | 2·50 ± 0·13b | 2·41 ± 0·07b | 2·11 ± 0·13c | 1·33 ± 0·06d | 2·86 ± 0·11a | 0·104 | <0·001 |
| Isobutyrate | 0·27 ± 0·03ab | 0·35 ± 0·04a | 0·22 ± 0·04b | 0·22 ± 0·02b | 0·33 ± 0·01a | 0·017 | 0·01 |
| Butyrate | 1·71 ± 0·09a | 1·68 ± 0·15a | 1·05 ± 0·12b | 0·70 ± 0·06b | 1·61 ± 0·16a | 0·091 | <0·001 |
| Isovalerate | 0·58 ± 0·04a | 0·55 ± 0·05a | 0·34 ± 0·04b | 0·58 ± 0·05a | 0·67 ± 0·04a | 0·028 | <0·001 |
| Valerate | 0·36 ± 0·06a | 0·41 ± 0·04a | 0·14 ± 0·03b | 0·24 ± 0·02b | 0·36 ± 0·01a | 0·025 | <0·001 |
| Total BCFA | 0·84 ± 0·06b | 0·90 ± 0·07b | 0·55 ± 0·05c | 0·80 ± 0·07d | 1·01 ± 0·06a | 0·038 | <0·001 |
| Total straight‐chain fatty acids | 10·40 ± 0·69ab | 9·90 ± 0·28ab | 7·33 ± 0·43c | 5·45 ± 0·49b | 12·04 ± 0·70a | 0·489 | <0·001 |
| Total SCFA | 11·24 ± 0·71b | 10·80 ± 0·34b | 7·88 ± 0·48c | 6·25 ± 0·44c | 13·05 ± 0·71a | 0·473 | <0·001 |
| Bioamines (μg g−1) | |||||||
| 1,7‐heptyl diamine | 0·73 ± 0·06a | 0·81 ± 0·06a | 0·84 ± 0·05a | 0·67 ± 0·06a | 0·87 ± 0·09a | 0·031 | 0·228 |
| Cadaverine | 3·40 ± 0·46a | 1·23 ± 0·21c | 1·54 ± 0·12bc | 2·78 ± 0·23ab | 3·66 ± 0·87a | 0·265 | 0·002 |
| Phenylethylamine | 7·21 ± 0·61a | 3·70 ± 0·58b | 6·80 ± 0·86a | 6·08 ± 0·53a | 5·63 ± 0·75ab | 0·361 | 0·011 |
| Putrescine | 3·17 ± 0·23b | 2·83 ± 0·25b | 2·05 ± 0·29b | 4·45 ± 0·37a | 4·84 ± 0·74a | 0·259 | <0·001 |
| Spermidine | 7·53 ± 1·03b | 5·05 ± 0·86b | 5·01 ± 0·25b | 14·46 ± 1·46a | 6·20 ± 0·56b | 0·76 | <0·001 |
| Spermine | 1·25 ± 0·27a | 0·59 ± 0·08b | 0·73 ± 0·04b | 1·46 ± 0·22a | 0·54 ± 0·06b | 0·097 | 0·001 |
| Tryptamine | 2·47 ± 0·50a | 0·22 ± 0·05c | 1·16 ± 0·26b | 0·54 ± 0·19bc | 0·70 ± 0·14bc | 0·185 | <0·001 |
| Tyramine | 1·13 ± 0·23b | 0·47 ± 0·06b | 0·84 ± 0·25b | 1·08 ± 0·19b | 2·92 ± 0·72a | 0·219 | 0·001 |
| Total bioamine | 26·89 ± 1·72ab | 14·89 ± 1·68c | 18·99 ± 1·07c | 31·52 ± 1·61a | 25·36 ± 2·65bc | 1·33 | <0·001 |
The days 30, 60, 90 and 110 are after artificial fertilization and weaning (day 21) is after parturition. The results are obtained from five independent experiments involving 40 sows. Significant difference is indicated by different letters.
Alpha diversity and beta diversity of gut microbiota
To compare samples with different sequencing depths, each sample was rarefied to 8467 reads. Based on 97% sequence similarity, all the sequences were clustered into 11 587 bacterial OTUs. The OTU‐level rarefaction curve of observed OTUs across all samples reached stable values (Fig. S1), indicating that the sampling depth provided sufficient OTU coverage to accurately describe the faecal bacterial diversity in this study. To further dissect the changes in the gut bacterial communities throughout pregnancy until weaning, the alpha diversity indices, including observed species (Fig. 1a) and Shannon index (Fig. 1b) were investigated. These indices increased with the progress of gestation and exhibited the largest values at the time of weaning (P < 0·05).
Figure 1.
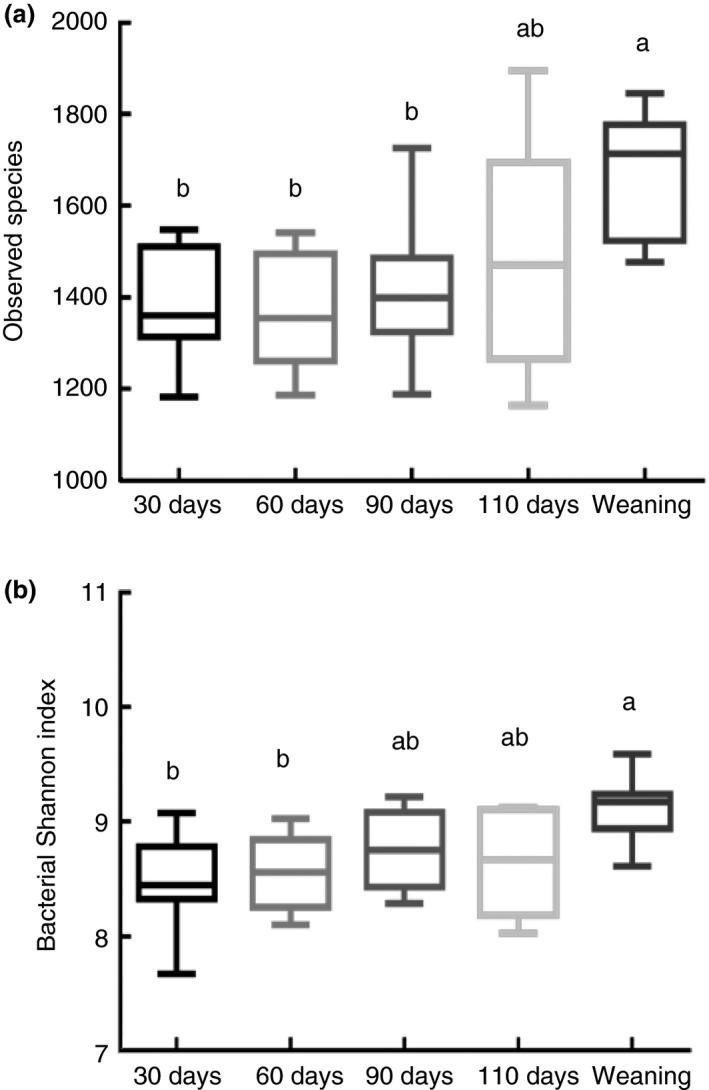
The changes of faecal microbial alpha diversity in different stages of sows. (a) The observed species of faecal bacteria in sows. (b) Shannon diversity index. Different letters above the bars denote a significantly different alpha diversity index among groups. The days 30, 60, 90 and 110 are after artificial fertilization and weaning (day 21) is after parturition. The results are obtained from five independent experiments involving 40 sows.
The PCoA based on the weighted UniFrac distance showed that the samples clustered together according to the stages of pregnancy and weaning, which indicated a shift in the gut bacterial community structure with the changes in the physiological state (Fig. 2). anosim analysis confirmed significant separation of gut bacterial communities among the different stages (R = 0·32, P < 0·001).
Figure 2.
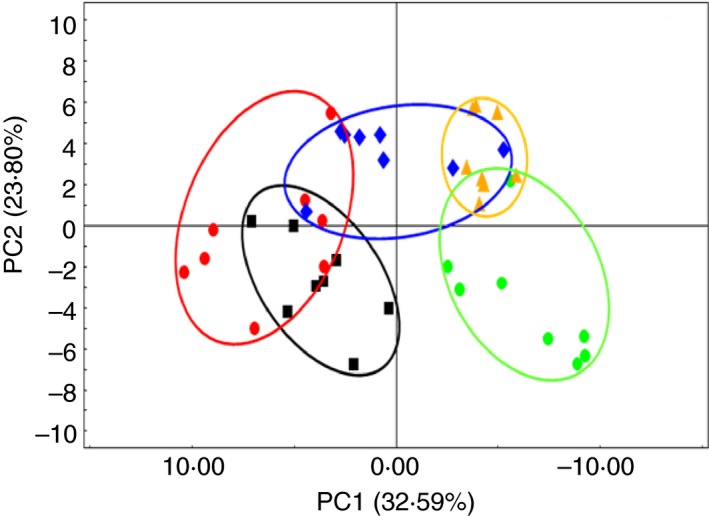
Scatterplot from PCoA of OTUs shown the differences in microbial community structures of sows among the five physiological stages. The days ( ) 30, (
) 30, ( ) 60, (
) 60, ( ) 90, and (
) 90, and ( ) 110 are after artificial fertilization and (
) 110 are after artificial fertilization and ( ) weaning (day 21) is after parturition. [Colour figure can be viewed at http://wileyonlinelibrary.com]
) weaning (day 21) is after parturition. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Taxonomic composition in the gut microbiota from pregnancy to weaning
In total, 23 phyla were identified within the faecal microbiota. The abundances of the 10 most abundant phyla (>0·5%) in each group are shown in Fig. 3a. Other phyla included: Acidobacteria, Chlamydiae, Chloroflexi, Deferribacteres, Elusimicrobia, Fusobacteria, Gemmatimonadetes, Lentisphaerae, OP8, Synergistetes, Thermi, TM7 and WPS‐2. Among the 23 phyla, Bacteroidetes and Firmicutes were detected as the dominant phyla regardless of the pregnancy stages and their total proportions reached as high as >60%. However, their relative abundance differed among the stages of pregnancy and weaning. Bacteroidetes increased linearly (P = 0·053) with the progression of pregnancy; becoming dominant on days 90 and 110 (42·11 and 45·56% respectively) of pregnancy. However, the abundance of Bacteroidetes decreased (P < 0·05) at weaning (38·67%). The abundance of Tenericutes was the lowest on day 60 of pregnancy and the highest at weaning (P < 0·05). Fibrobacteres increased (P < 0·05) linearly from days 30 to 90 of pregnancy (0·90, 1·55 and 2·52% respectively), but decreased (P < 0·05) to the lowest abundance on day 110 of pregnancy and at weaning (0·44 and 0·39% respectively). Cyanobacteria were found at the lowest abundance (0·28%) on day 60, but comprised 1% of the microbiota on day 110 of pregnancy (P < 0·05).
Figure 3.
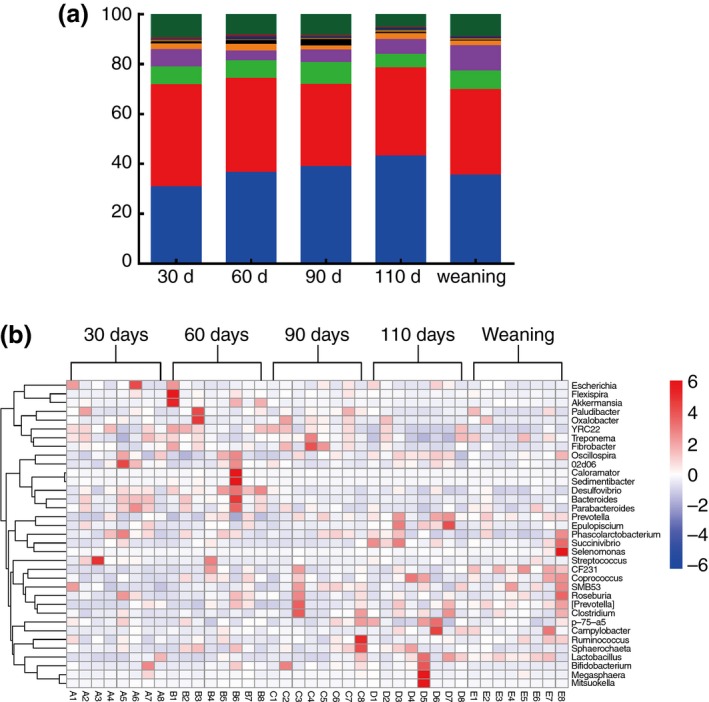
Taxonomic composition of faecal bacterial communities in different stages in sows. (a) Phylum‐level taxonomic composition of faecal bacterial communities in different stages in sows ( ) other bacteria (<0·5%); (
) other bacteria (<0·5%); ( ) unclassified bacteria;(
) unclassified bacteria;( ) Actinobacteria; (
) Actinobacteria; ( ) Verrucomicrobia; (
) Verrucomicrobia; ( ) Planctomycetes; (
) Planctomycetes; ( ) Cyanobacteria; (
) Cyanobacteria; ( ) Fibrobacteres; (
) Fibrobacteres; ( ) Proteobacteria; (
) Proteobacteria; ( ) Tenericutes; (
) Tenericutes; ( ) Spirochaetes; (
) Spirochaetes; ( ) Firmicutes; (
) Firmicutes; ( ) Bacteroidetes. (b) Heat map of genera in the relative abundances of sows faecal bacterial communities in five stages (30 d, 30 days of pregnancy; 60 d, 60 days of pregnancy; 90 d, 90 days of pregnancy; 110 d, 110 days of pregnancy; weaning, 21 days after parturition). [Colour figure can be viewed at http://wileyonlinelibrary.com]
) Bacteroidetes. (b) Heat map of genera in the relative abundances of sows faecal bacterial communities in five stages (30 d, 30 days of pregnancy; 60 d, 60 days of pregnancy; 90 d, 90 days of pregnancy; 110 d, 110 days of pregnancy; weaning, 21 days after parturition). [Colour figure can be viewed at http://wileyonlinelibrary.com]
To further investigate the taxonomic compositions of faecal samples, a total of 325 genera were identified. Among these genera, 35 had a mean relative abundance ≥0·5% of the total sequences in at least one sample (Fig. 3b). The cluster analysis based on a heat map demonstrated a higher similarity of the samples within groups than among groups. Prevotella (phylum: Bacteroidetes) was the most abundant genera in the faecal microbiota. The abundance of Prevotella linearly increased with the progression of pregnancy (i.e., represented 7·65, 7·51, 10·07 and 14·02% of the microbiota at 30, 60, 90 and 110 days of pregnancy respectively) and decreased at weaning (11·83%) (P < 0·05). The genus YRC22 was the most abundant on day 60 of pregnancy (5·66%), and decreased to the lowest levels at weaning (1·24%) (P < 0·05). The genus Bacteroides reached its maximum abundance (peaked) on day 60 of pregnancy (2·17%) (P < 0·05). The genus CF231 peaked at weaning (P < 0·05). Within the Firmicutes phylum, the genus Lactobacillus was more abundant on day 110 of pregnancy and at weaning (6·91% and 6·21% respectively) (P < 0·05). The genus Epulopiscium peaked on day 110 of pregnancy (P < 0·05). Fibrobacter (phylum: Fibrobacteres) peaked at 2·52% on day 90 of pregnancy (P < 0·05). In the Proteobacteria phylum, the genera Desulfovibrio and Succinivibrio peaked at 0·90 and 0·58% on days 60 and 110 of pregnancy respectively (P < 0·05). Akkermansia (phylum: Verrucomicrobia) peaked at 0·13% on day 60 of pregnancy (P < 0·05).
Microbial biomarkers of different pregnant stages
Linear discriminant analysis effect size analysis has been used to identify the biomarker species that distinguish the microbial communities at different stages. In our study, the dominant species from the faecal samples at 30 days of pregnancy were mainly from the classes Clostridiales and Moraxellaceae. The biomarker species at day 60 of pregnancy belonged to classes Coriobacteriaceae, Coriobacteriales, Bacteroides, YRC22, Bacteroidales, [Mogibacteriaceae], Pirellulaceae, Pirellulales, Aquabacterium, Desulfovibrio, Desulfovibrionales and Synergistales. The dominant species at day 90 of pregnancy were predominantly from the classes Fibrobacteraceae, Fibrobacterales, Lachnospiraceae, Veillonellaceae and Rhizobiales. The dominant species at day 110 of pregnancy were from the classes Bifidobacterium, Bifidobacteriales, S24‐7, p‐2534‐18B5, Mesonia, YS2, Lactobacillus, Peptococcus, Faecalibacterium, Anaerobibrio, [Eubacterium], Succinivibrio and Aeromonadales. The biomarker species at weaning were predominantly from the classes CF231, Lactobacillales, Lachnospira and Anaeroplasma (Fig. 4).
Figure 4.
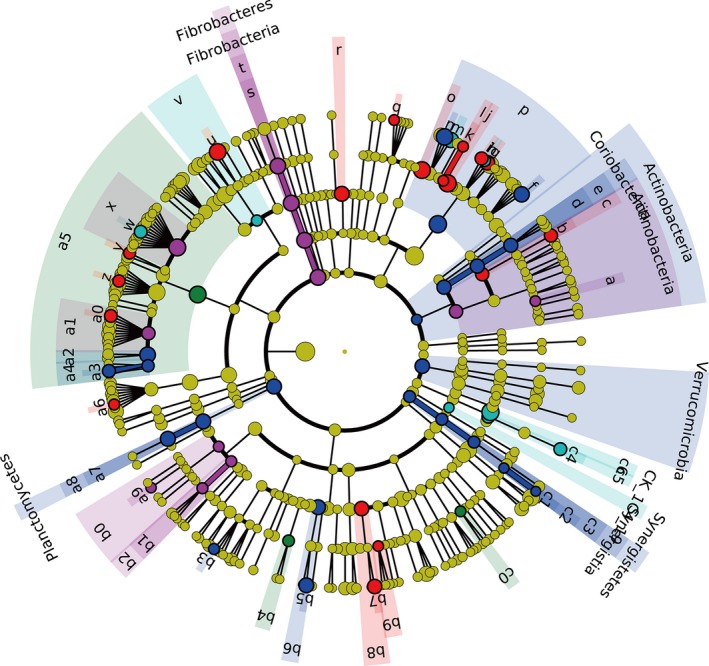
Cladogram plotted from LEfSe analysis indicated the biomarkers of the microbial community in different groups (P < 0·05; LDA score 2). ( ) 30 d, 30 days of pregnancy; (
) 30 d, 30 days of pregnancy; ( ) 60 d, 60 days of pregnancy; (
) 60 d, 60 days of pregnancy; ( ) 90 d, 90 days of pregnancy; (
) 90 d, 90 days of pregnancy; ( ) 110 d, 110 days of pregnancy; (
) 110 d, 110 days of pregnancy; ( ) weaning, 21 days after parturition (
) weaning, 21 days after parturition ( ) a: Bifidobacterium; (
) a: Bifidobacterium; ( ) b: Bifidobacteriales; (
) b: Bifidobacteriales; ( ) c: Coriobacteriaceae; (
) c: Coriobacteriaceae; ( ) d: Coriobacteriales; (
) d: Coriobacteriales; ( ) e: Bacteroides; (
) e: Bacteroides; ( ) f: S24_7; (
) f: S24_7; ( ) g: CF231; (
) g: CF231; ( ) h: YRC22; (
) h: YRC22; ( ) i: p_2534_18B5; (
) i: p_2534_18B5; ( ) j: Bacteroidales; (
) j: Bacteroidales; ( ) k: Mesonia; (
) k: Mesonia; ( ) l: YS2; (
) l: YS2; ( ) m: Fibrobacteraceae; (
) m: Fibrobacteraceae; ( ) n: Fibrobacterales; (
) n: Fibrobacterales; ( ) o: Lactobacillus; (
) o: Lactobacillus; ( ) p: Lactobacillales; (
) p: Lactobacillales; ( ) q: Lachnospira; (
) q: Lachnospira; ( ) r: Lachnospiraceae; (
) r: Lachnospiraceae; ( ) s: Peptococcus; (
) s: Peptococcus; ( ) t: Faecalibacterium; (
) t: Faecalibacterium; ( ) u: Anaerovibrio; (
) u: Anaerovibrio; ( ) v: Veillonellaceae; (
) v: Veillonellaceae; ( ) w: _Mogibacteriaceae_; (
) w: _Mogibacteriaceae_; ( ) x: Clostridiales; (
) x: Clostridiales; ( ) y: _Eubacterium_; (
) y: _Eubacterium_; ( ) z: Pirellulaceae; (
) z: Pirellulaceae; ( ) a0: Pirellulales; (
) a0: Pirellulales; ( ) a1: Rhizobiales; (
) a1: Rhizobiales; ( ) a2: Aquabacterium; (
) a2: Aquabacterium; ( ) a3: Desulfovibrio; (
) a3: Desulfovibrio; ( ) a4: Desulfovibrionales; (
) a4: Desulfovibrionales; ( ) a5: Succinivibrio; (
) a5: Succinivibrio; ( ) a6: Aeromonadales; (
) a6: Aeromonadales; ( ) a7: Moraxellaceae; (
) a7: Moraxellaceae; ( ) a8: Synergistales; (
) a8: Synergistales; ( ) a9: Anaeroplasma). [Colour figure can be viewed at http://wileyonlinelibrary.com]
) a9: Anaeroplasma). [Colour figure can be viewed at http://wileyonlinelibrary.com]
Relationship between the bacterial community and bacterial metabolites
Particular genera were found to be linked to faecal metabolite concentrations (Fig. 5). The genera Bacteroides and Parabacteroides were positively correlated with tryptamine, while the genera Akkermansia and Sedimentibacter were negatively correlated with phenylethylamine; YRC22 and Fibrobacter were also negatively, but Coprococcus was positively correlated with putrescine. Five genera were associated with cadaverine (positive correlations: [Prevotella], 02d06 and Roseburia; negative correlations: YRC22 and Fibrobacter); four genera were associated with tyramine (positive correlations: CF231 and Ruminococcus; negative correlations: YRC22 and Desulfovibrio); 14 genera were associated with spermidine (positive correlations: Prevotella, Lactobacillus, Oscillospira, p‐75‐a5, Clostridium, Succinivibrio, Megasphaera, Epulopiscium and Mitsuokella; negative correlations: Treponema, YRC22, Fibrobacter, Oxalobacter and Sedimentibacter); three genera were associated with spermine (positive correlations: Epulopiscium and Bifidobacterium; negative correlation: CF231); genus Parabacteroides was positively, but Sphaerochaeta was negatively correlated with acetate; three genera Clostridium, Succinivibrio and Epulopiscium were negatively correlated with propionate; two genera were associated with isobutyrate (positive correlation: Desulfovibrio; negative correlation: Bifidobacterium); four genera were associated with butyrate (positive correlations: Parabacteroides and Desulfovibrio; negative correlations: Succinivibrio and Bifidobacterium); the genera YRC22 and Fibrobacter were negatively, but Lactobacillus was positively correlated with isovalerate, while Desulfovibrio and Sedimentibacter were positively correlated with valerate. A total of 11 genera were associated with indole (positive correlations: Prevotella, Lactobacillus, Oscillospira, [Prevotella], Succinivibrio, Campylobacter and Selenomonas; negative correlations: YRC22, Bacteroides, Parabacteroides and Desulfovibrio).
Figure 5.
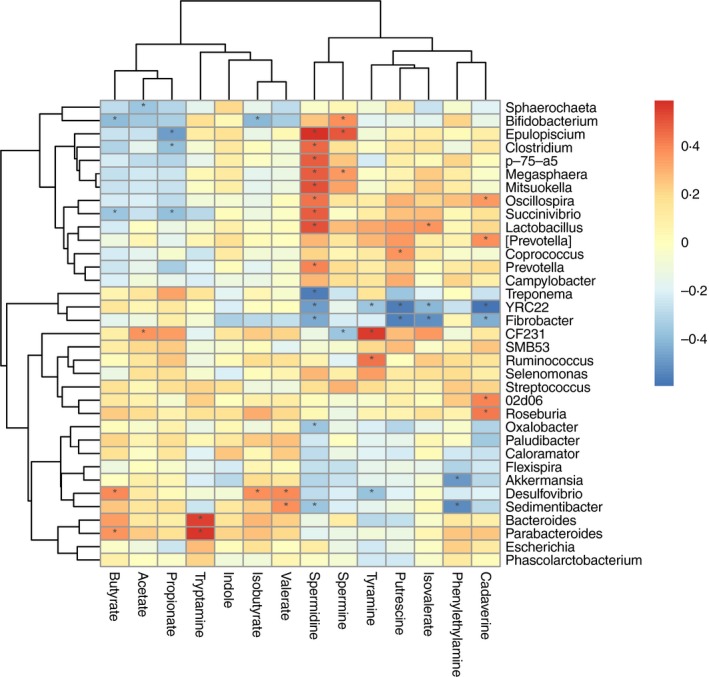
Correlations between the genera and the faecal metabolite concentrations. Only the genera for which abundance was significantly associated with the faecal concentrations of metabolites are presented; the red represents a significantly positive correlation, the blue represents a significantly negative correlation, and the white represents no significant correlation. * means the P value < 0.05. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Discussion
This study investigated the shifts in plasma and faecal metabolites and in the gut microbiota of Yorkshire × Dutch Landrace sows during pregnancy (days 30, 60, 90 and 110 after artificial fertilization) and at weaning (day 21 after parturition). The results revealed significant metabolic changes in the maternal plasma and faeces at different stages, mainly involving the changes in lipid and protein metabolism, SCFAs and bioamines, as well as shifts in the gut microbiota composition in sows from pregnancy to weaning. Our results were consistent with those of Liu et al. (2018), who also demonstrated shifts in the gut microbiota and SCFA profiles of sows during pregnancy. These results may help to improve animal health during pregnancy via the manipulation of gut microbial communities.
Assessments of the blood biochemical parameters of animals can provide information on the metabolic state of the animals and help to improve the health status of the animals (Friendship 1992). The present study showed that plasma concentrations of GLB and TP were reduced on days 90 and 110 of pregnancy and recovered at weaning. As expected, protein digestion and absorption in the maternal hepatic and adipose tissues were likely to have been activated to preserve energy needs for the foetus (Shen et al. 2016). Previously, TP has been shown to be associated with GLB, but not with ALB. In addition, the GLB and TP in the serum of sows were found to decrease when nearing parturition (Machadoneto et al. 1987; Goff and Horst 1997; Castillo et al. 2005). Thus, the contents of TP and GLB in the blood are expected to be dependent on the nutritional needs of the foetus.
Maternal lipid metabolism plays a pivotal role in the initiation and development of pregnancy (Pinto et al. 2015) and the lipid concentrations are very sensitive to the energy balance (Shen et al. 2016). Pregnancy might affect lipid metabolism and thus lead to changes in the levels of many lipid compounds, including LDL, VLDL, TC, and TG. The present study showed that the plasma concentrations of HDL‐C, LDL‐C, TC and TG all decreased with the progression of pregnancy and recovered at weaning (except TG). We speculated that sows need to provision more nutrients for foetal development during pregnancy, so that no excess fat is used to synthesize blood lipids. Our results are consistent with those of Metges et al. (2012) found that the plasma concentrations of LDL‐C and HDL‐C increased at −5, 24, 66 and 108 days post coitum regardless of differences in diet (i.e., low protein, adequate protein or high protein diets), and that TC decreased linearly in sows fed the adequate protein diet. The serum cholesterol concentration decreased linearly from 14 days to 2 weeks prepartum (Tumbleson et al. 1970). These changes might be related to some oestrogen, such as progesterone. In a study by Saleri et al. (2015), the plasma progesterone levels were low, but increased to 10‐fold and then reached a plateau during the early stages of gestation. The progesterone levels then decreased from day 86 of pregnancy until day 10 after parturition and thereafter remained at lower levels (Saleri et al. 2015). These results indicated that blood hormone concentrations are closely dependent on the pregnancy and parturition stages in sows.
The present study showed an increased alpha diversity in the gut microbiota throughout the progression of pregnancy until weaning. Diversity is known to improve the stability and performance of communities and is, therefore, very important in many ecosystems (Margalef 1963; McNaughton 1977; Tap et al. 2015; Li et al. 2018). In particular, gut microbial diversity has been proposed as a new biomarker of health and metabolic capacity (Clarke et al. 2014). We speculate that a diverse gut microbiota probably provides many metabolic capacities and functional redundancy in sows, which ensures the sufficient supply of nutrients for foetal growth and development. However, further research is needed to investigate this inference. Our results of the anosim and PCoA analyses and the clustering of samples also suggested that the gut microbiota differed among the stages of pregnancy. It is noteworthy that our findings demonstrated that the structure and composition of the gut microbiota can vary considerably over time. As previously reported (Lu et al. 2014; Pajarillo et al. 2015), the phyla Bacteroidetes and Firmicutes were the most abundant in the gut of sows, regardless of the stage of pregnancy. Jost et al. (2014) also reported that Firmicutes exhibited no detectable changes over the perinatal period (Jost et al. 2014). However, the findings of our study differed from those of Koren et al. (2012) who reported an overall increase in Proteobacteria and Actinobacteria. The present study demonstrated that the abundance of Tenericutes, Fibrobacteres and Cyanobacteria changed significantly among the different physiological stages of sows. In terms of the phylum level, numerous studies have indicated that obesity and gestational weight gain are associated with an increase in the abundance of Firmicutes or an increase in the Firmicutes to Bacteriodetes ratio (Ley et al. 2006; Turnbaugh et al. 2009; Feng et al. 2015). It is noteworthy that the backfat thickness of sows did not differ greatly among the different stages of pregnancy in the present study. Due to the modulation of the diet, the sows did not gradually deposit more fat throughout the progression of pregnancy and the abundance of Firmicutes was maintained at a stable level in the gut. The Tenericutes phylum may provide some beneficial effects on the intestinal integrity because lower counts were detected in inflamed intestines, induced by dextran sodium sulphate (Nagalingam et al. 2011). The representatives of Fibrobacteres were characterized as having the potential to metabolize nonsoluble polysaccharides, such as cellulose, hemicellulose or pectin (Kubasova et al. 2017). Specifically, the genera Prevotella and CF231 (phylum: Bacteroidetes), Lactobacillus (phylum: Firmicutes) and Succinivibrio (phylum: Proteobacteria) increased, but the genera YRC22, Bacteroides and Parabacteroides (phylum: Bacteroidetes); and Desulfovibrio (phylum: Proteobacteria) decreased from day 30 of pregnancy until weaning. Most of the genera increased with the progression of pregnancy and decreased at weaning. These included the genera Prevotella and YRC22 and S24‐7 and p‐2534‐18B5 families within the phylum Bacteroidetes; the species Fibrobacter succinogenes within the phylum Fibrobacteres; the order Clostridiales and family Mogibacteriaceae within the phylum Firmicutes; the genera Succinivibrio and Desulfovibrio within the phylum Proteobacteria; the genus Treponema within the phylum Spirochaetes and the order RF39 within Tenericutes. Prevotella is more common in humans who consume a plant‐rich diet (Ley 2016). In addition to Prevotella, Succinivibrio is a known plant polysaccharide‐fermenting bacteria in the Prevotella‐type gut microbial community of native Africans (Ou et al. 2013). Bacteroides was characterized by a high expression of xylose isomerase. In addition to Bacteroides, Parabacteroides is a producer of propionic acid as a metabolic end‐product (MacFabe et al. 2007). Lactobacillus as a lactic acid‐producing bacterium could degrades lactose and other oligosaccharides into acetate and lactate (Walter 2008). Some members of Desulfovibrio can produce hydrogen sulphide by reducing sulphates (Heidelberg et al. 2004). By producing hydrogen sulphide, Desulfovibrio may, therefore, impact the metabolism of epithelial cells (Fite et al. 2004; Bisson‐Boutelliez et al. 2010). Fibrobacter succinogenes is an important degrader of lignocellulosic plant material (Bera‐Maillet et al. 2004). The order Clostridiales can digest protein, carbohydrate, sugar, amino acid, purine, pyrimidine and other organic compounds, to produce SCFAs (Zhang et al. 2016a). The genus Treponema comprises several uncultivable human and animal pathogens (Cejkova et al. 2015). As the LEfSe analysis showed in the present study, we also detected that the classes related to nutrient digestion increasing in the later stages of pregnancy included Fibrobacteraceae, Fibrobacterales, Bifidobacterium, Bifidobacteriales, Lactobacillus and Faecalibacterium. As previously mentioned, we observed numerous changes throughout the pregnancy of sows and most of the bacteria that increased in abundance with the progression of pregnancy are able to digest plant‐type materials, such as oligosaccharides, polysaccharides, lignocellulose and other cellulose‐type materials. This implies that undigestible or partly digestible dietary compounds can be metabolized by the intestinal microbiota for the potential benefit of the sows and their foetuses. The observed changes in the phylum and genera also revealed that most of the bacteria that showed increases during pregnancy, decreased at weaning. These findings showed that the changes in gut microbiota were transient at the critical period of pregnancy, but that the gut microbiota returned to a normal composition after this critical time. In addition to changes in the physiologies of animals, the changes in gut microbiota may also result from differences in dietary components between pregnancy and lactation stages. In a previous study, the diet of pregnant sows contained more wheat bran, which was demonstrated to influence pig gut microbial communities (Kraler et al. 2016).
As the major metabolites produced by gut microbiota, SCFAs have been suggested to be important regulators of energy balance, gut inflammation signalling and insulin sensitivity (Russell et al. 2011). Other compounds such as polyamines, other bioamines and indoles are produced by colonic bacteria from their respective amino acid derivities (Davila et al. 2013). According to the correlation analyses in the present study, a significant correlation was detected between the bacterial genera and metabolite concentrations. These results are consistent with those from a previous study that showed that bacteria with protein‐fermentation or AA‐fermentation capacities within the genera Bacteroides, Parabacteroides, Lactobacillus, Megasphaera, Roseburia and Ruminococcus (Davila et al. 2013; Dai et al. 2015; Zhang et al. 2016b) are positively correlated with bioamine concentrations. Some studies have shown that the genera Akkermansia, Fibrobacter and Desulfovibrio, i.e., are plant‐degradation bacteria (Fite et al. 2004; Hill and Spence 2017), as well as Sedimentibacter, Treponema and YRC22 are negatively correlated with bioamine concentrations. Higher concentrations of SCFAs were associated with carbohydrate‐degradation bacteria. Although some bacterial genera were correlated with SCFAs, it remains difficult to predict which bacteria are responsible for the production of specific SCFAs due to the complexity of microbial interactions, such as cross‐feeding (Rey et al. 2010) and resource competition (Mahowald et al. 2009). However, these bacteria probably play important roles in the gut fermentation of food in sows.
Collectively, our findings suggest that the progression of pregnancy is associated with changes in lipid metabolism and in the composition of the intestinal microbiota; notably of the bacteria involved in the degradation of carbohydrates, such as the genera Prevotella, Succinivibrio, Bacteroides, Parabacteroides and the order Clostridiales. The changes in gut microbiota diversity were transient and notably related to some critical processes at different stages during pregnancy. The present study mainly explored the associations between microbiota composition and metabolic parameters in sows during pregnancy until weaning, but did not determine the possible causal links between these parameters. Therefore, future studies should shed light on the ecological and functional mechanisms of the changes in the gut microbiota throughout pregnancy until weaning to determine such potential links between the metabolic activity of the microbiota and the metabolic changes in the hosts.
Author contributions
X.F.K., Y.J.J. and Y.L.Y. performed the experiments. Y.J.J., H.L., X.F.K. and F.B. wrote the manuscript. Y.J.J. and H.L. performed the statistical analyses. P.F.X., Z.H.L. and H.W.L. fed the animals. All authors reviewed the manuscript.
Conflict of Interest
The authors declare no competing financial interests.
Supporting information
Figure S1. The OTU‐level rarefaction curve of observed OTUs across all samples.
Acknowledgements
The present work was jointly supported by grants from the National Key R&D Program (2017YFD0500503), the National Nature Science Foundation of China (31572421, 31772613) and the Key Research Program of the Chinese Academy of Sciences (KFZD‐SW‐219‐2‐3). All authors have read and approved the final version of the manuscript and have declared that no competing interests exist. We would like to thank Editage (http://online.editage.cn/) for English language editing.
Y.J. Ji and H. Li contributed equally to this work.
References
- Bera‐Maillet, C. , Ribot, Y. and Forano, E. (2004) Fiber‐degrading systems of different strains of the genus Fibrobacter. Appl Environ Microbiol 70, 2172–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson‐Boutelliez, C. , Massin, F. , Dumas, D. , Miller, N. and Lozniewski, A. (2010) Desulfovibrio spp. survive within KB cells and modulate inflammatory responses. Mol Oral Microbiol 25, 226–235. [DOI] [PubMed] [Google Scholar]
- Blachier, F. , Beaumont, M. , Andriamihaja, M. , Davila, A.M. , Lan, A. , Grauso, M. , Armand, L. , Benamouzig, R. et al. (2017) Changes in the luminal environment of the colonic epithelial cells and physiopathological consequences. Am J Pathol 187, 476–486. [DOI] [PubMed] [Google Scholar]
- Caporaso, J.G. , Lauber, C.L. , Walters, W.A. , Berg‐Lyons, D. , Huntley, J. , Fierer, N. , Owens, S.M. , Betley, J. et al (2012) Ultra‐high‐throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo, C. , Hernandez, J. , Bravo, A. , Lopez‐Alonso, M. , Pereira, V. and Benedito, J.L. (2005) Oxidative status during late pregnancy and early lactation in dairy cows. Vet J 169, 286–292. [DOI] [PubMed] [Google Scholar]
- Cejkova, D. , Strouhal, M. , Norris, S.J. , Weinstock, G.M. and Smajs, D. (2015) A retrospective study on genetic heterogeneity within Treponema strains: subpopulations are genetically distinct in a limited number of positions. Plos Neglect Trop D 9, e0004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, S.F. , Murphy, E.F. , O'Sullivan, O. , Lucey, A.J. , Humphreys, M. , Hogan, A. , Hayes, P. , O'Reilly, M. et al (2014) Exercise and associated dietary extremes impact on gut microbial diversity. Gut 63, 1913–1920. [DOI] [PubMed] [Google Scholar]
- Dai, Z. , Wu, Z. , Hang, S. , Zhu, W. and Wu, G. (2015) Amino acid metabolism in intestinal bacteria and its potential implications for mammalian reproduction. Mol Hum Reprod 21, 389–409. [DOI] [PubMed] [Google Scholar]
- Davila, A.M. , Blachier, F. , Gotteland, M. , Andriamihaja, M. , Benetti, P.H. , Sanz, Y. and Tome, D. (2013) Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol Res 68, 95–107. [DOI] [PubMed] [Google Scholar]
- Debelius, J. , Song, S.J. , Vazquez‐Baeza, Y. , Xu, Z.Z. , Gonzalez, A. and Knight, R. (2016) Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol 17, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T.Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E.L. , Keller, K. , Huber, T. , Dalevi, D. et al (2006) Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen, L. and Relman, D.A. (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen, L. , McFall‐Ngai, M. and Relman, D.A. (2007) An ecological and evolutionary perspective on human‐microbe mutualism and disease. Nature 449, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiulio, D.B. , Callahan, B.J. , McMurdie, P.J. , Costello, E.K. , Lyell, D.J. , Robaczewska, A. , Sun, C.L. , Goltsman, D.S.A. et al. (2015) Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci USA 112, 11060–11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. , Haas, B.J. , Clemente, J.C. , Quince, C. and Knight, R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z.M. , Li, T.J. , Wu, L. , Xiao, D.F. , Blachier, F. and Yin, Y.L. (2015) Monosodium L‐glutamate and dietary fat differently modify the composition of the intestinal microbiota in growing pigs. Obes Facts 8, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fite, A. , Macfarlane, G.T. , Cummings, J.H. , Hopkins, M.J. , Kong, S.C. , Furrie, E. and Macfarlane, S. (2004) Identification and quantitation of mucosal and faecal desulfovibrios using real time polymerase chain reaction. Gut 53, 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friendship, R.M. (1992) Cardiovascular system, hematology, and clinical chemistry. Ames, IA: Iowa State University Press. [Google Scholar]
- Goff, J.P. and Horst, R.L. (1997) Physiological changes at parturition and their relationship to metabolic disorders. J Dairy Sci 80, 1260–1268. [DOI] [PubMed] [Google Scholar]
- Goodrich, J.K. , Waters, J.L. , Poole, A.C. , Sutter, J.L. , Koren, O. , Blekhman, R. , Beaumont, M. , Van Treuren, W. et al (2014) Human genetics shape the gut microbiome. Cell 159, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzeskowiak, L. , Collado, M.C. , Mangani, C. , Maleta, K. , Laitinen, K. , Ashorn, P. , Isolauri, E. and Salminen, S. (2012) Distinct gut microbiota in Southeastern African and Northern European infants. J Pediatr Gastroenterol Nutr 54, 812–816. [DOI] [PubMed] [Google Scholar]
- Heidelberg, J.F. , Seshadri, R. , Haveman, S.A. , Hemme, C.L. , Paulsen, I.T. , Kolonay, J.F. , Eisen, J.A. , Ward, N. et al. (2004) The genome sequence of the anaerobic, sulfate‐reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol 22, 554–559. [DOI] [PubMed] [Google Scholar]
- Hill, D.R. and Spence, J.R. (2017) Gastrointestinal organoids: understanding the molecular basis of the host–microbe interfacecannulated pigs. Cell Mol Gastroenterol Hepatol 3, 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Kong, X. , Li, H. , Zhu, Q. , Guo, Q.P. and Yin, Y.L. (2016) Effects of dietary nutrient levels on microbial community composition and diversity in the ileal contents of pregnant Huanjiang mini‐pigs. PLoS ONE 12, e0172086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Guo, Q. , Yin, Y. , Blachier, F. and Kong, X. (2018) Dietary proline supplementation alters colonic luminal microbiota and bacterial metabolite composition between days 45 and 70 of pregnancy in Huanjiang mini‐pigs. J Anim Sci Biotechnol 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost, T. , Lacroix, C. , Braegger, C. and Chassard, C. (2014) Stability of the maternal gut microbiota during late pregnancy and early lactation. Curr Microbiol 68, 419–427. [DOI] [PubMed] [Google Scholar]
- Kong, X.F. , Ji, Y.J. , Li, H.W. , Zhu, Q. , Blachier, F. , Geng, M.M. , Chen, W. and Yin, Y.L. (2016) Colonic luminal microbiota and bacterial metabolite composition in pregnant Huanjiang mini‐pigs: effects of food composition at different times of pregnancy. Sci Rep 6, 37224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, O. , Goodrich, J.K. , Cullender, T.C. , Spor, A. , Laitinen, K. , Backhed, H.K. , Gonzalez, A. , Werner, J.J. et al (2012) Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraler, M. , Ghanbari, M. , Domig, K.J. , Schedle, K. and Kneifel, W. (2016) The intestinal microbiota of piglets fed with wheat bran variants as characterised by 16S rRNA next‐generation amplicon sequencing. Arch Anim Nutr 70, 173–189. [DOI] [PubMed] [Google Scholar]
- Kubasova, T. , Davidova‐Gerzova, L. , Merlot, E. , Medvecky, M. , Polansky, O. , Gardan‐Salmon, D. , Quesnel, H. and Rychlik, I. (2017) Housing systems influence gut microbiota composition of sows but not of their piglets. PLoS ONE 12, e0170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman, A.A. and Koren, O. (2016) Antibiotic use during pregnancy: how bad is it? BMC Med 14, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R.E. (2016) Gut microbiota in 2015: Prevotella in the gut: choose carefully. Nat Rev Gastroenterol Hepatol 13, 69–70. [DOI] [PubMed] [Google Scholar]
- Ley, R.E. , Turnbaugh, P.J. , Klein, S. and Gordon, J.I. (2006) Microbial ecology ‐ human gut microbes associated with obesity. Nature 444, 1022–1023. [DOI] [PubMed] [Google Scholar]
- Li, W. and Godzik, A. (2006) Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li, H. , Li, T. , Yao, M. , Li, J. , Zhang, S. , Wirth, S. , Cao, W. , Lin, Q. et al (2016) Pika gut may select for rare but diverse environmental bacteria. Front Microbiol 7, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Li, T. , Berasategui, A. , Rui, J. , Zhang, X. , Li, C. , Xiao, Z. and Li, X. (2017) Gut region influences the diversity and interactions of bacterial communities in pikas (Ochotona curzoniae and Ochotona daurica). FEMS Microbiol Ecol 93, 13. [DOI] [PubMed] [Google Scholar]
- Li, H. , Qu, J. , Li, T. , Wirth, S. , Zhang, Y. , Zhao, X. and Li, X. (2018) Diet simplification selects for high gut microbial diversity and strong fermenting ability in high‐altitude pikas. Appl Microbiol Biotechnol 102, 6739–6751. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Hou, C. , Li, N. , Zhang, X. , Zhang, G. , Yang, F. , Zeng, X. , Liu, Z. et al. (2018) Microbial and metabolic alterations in gut microbiota of sows during pregnancy and lactation. FASEB J 33, 4490–4501. [DOI] [PubMed] [Google Scholar]
- Lozupone, C. and Knight, R. (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X.M. , Lu, P.Z. and Zhang, H. (2014) Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 98, 2657–2665. [DOI] [PubMed] [Google Scholar]
- MacFabe, D.F. , Cain, D.P. , Rodriguez‐Capote, K. , Franklin, A.E. , Hoffman, J.E. , Boon, F. , Taylor, A.R. , Kavaliers, M. et al. (2007) Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res 176, 149–169. [DOI] [PubMed] [Google Scholar]
- Machadoneto, R. , Graves, C.N. and Curtis, S.E. (1987) Immunoglobulins in piglets from sows heat‐stressed prepartum. J Anim Sci 65, 445–455. [DOI] [PubMed] [Google Scholar]
- Mahowald, M.A. , Rey, F.E. , Seedorf, H. , Turnbaugh, P.J. , Fulton, R.S. , Wollam, A. , Shah, N. , Wang, C. et al (2009) Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA 106, 5859–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, S. , Godfrey, K.M. , McDonald, D. , Treuren, W.V. , Bjørnholt, J.V. , Midtvedt, T. , Moen, B. , Rudi, K. et al (2016) Fat and vitamin intakes during pregnancy have stronger relations with a pro‐inflammatory maternal microbiota than does carbohydrate intake. Microbiome 4, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalef, R. (1963) On certain unifying principles in ecology. Am Nat 97, 357–374. [Google Scholar]
- McNaughton, S.J. (1977) Diversity and stability of ecological communities: a comment on the role of empiricism in ecology. Am Nat 111, 515–525. [Google Scholar]
- Nagalingam, N.A. , Kao, J.Y. and Young, V.B. (2011) Microbial ecology of the murine gut associated with the development of dextran sodium sulfate‐induced colitis. Inflamm Bowel Dis 17, 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, D.B. , Rockwell, L.C. , Prioleau, M.D. and Goetzl, L. (2016) The role of the bacterial microbiota on reproductive and pregnancy health. Anaerobe 42, 67–73. [DOI] [PubMed] [Google Scholar]
- Newbern, D. and Freemark, M. (2011) Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol 18, 409–416. [DOI] [PubMed] [Google Scholar]
- Ou, J.H. , Carbonero, F. , Zoetendal, E.G. , DeLany, J.P. , Wang, M. , Newton, K. , Gaskins, H.R. and O'Keefe, S.J.D. (2013) Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 98, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajarillo, E.A. , Chae, J.P. , Balolong, M.P. , Kim, H.B. , Seo, K.S. and Kang, D.K. (2015) Characterization of the fecal microbial communities of Duroc pigs using 16S rRNA gene pyrosequencing. Asian‐Australas J Anim Sci 28, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, J. , Barros, A.S. , Domingues, M.R.M. , Goodfellow, B.J. , Galhano, E. , Pita, C. , Almeida, M.D. , Carreira, I.M. et al (2015) Following healthy pregnancy by NMR metabolomics of plasma and correlation to urine. J Proteome Res 14, 1263–1274. [DOI] [PubMed] [Google Scholar]
- Rey, F.E. , Faith, J.J. , Bain, J. , Muehlbauer, M.J. , Stevens, R.D. , Newgard, C.B. and Gordon, J.I. (2010) Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem 285, 22082–22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura, V.K. , Faith, J.J. , Rey, F.E. , Cheng, J.Y. , Duncan, A.E. , Kau, A.L. , Griffin, N.W. , Lombard, V. , et al (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1079–U1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, W.R. , Gratz, S.W. , Duncan, S.H. , Holtrop, G. , Ince, J. , Scobbie, L. , Duncan, G. , Johnstone, A.M. et al (2011) High‐protein, reduced‐carbohydrate weight‐loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr 93, 1062–1072. [DOI] [PubMed] [Google Scholar]
- Saleri, R. , Sabbioni, A. , Cavalli, V. and Superchi, P. (2015) Monitoring blood plasma leptin and lactogenic hormones in pregnant sows. Animal 9, 629–634. [DOI] [PubMed] [Google Scholar]
- Shen, G.P. , Li, Z.S. , Zhang, Y. , Wu, H.F. and Feng, J.H. (2016) H‐1 NMR‐based metabolomics study on the physiological variations during the rat pregnancy process. Mol Cell Endocrinol 423, 40–50. [DOI] [PubMed] [Google Scholar]
- Tamaki, H. , Wright, C.L. , Li, X.Z. , Lin, Q.Y. , Hwang, C.C. , Wang, S.P. , Thimmapuram, J. , Kamagata, Y. et al (2011) Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next‐generation Titanium sequencing platform. PLoS ONE 6, e25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap, J. , Furet, J.P. , Bensaada, M. , Philippe, C. , Roth, H. , Rabot, S. , Lakhdari, O. , Lombard, V. et al (2015) Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ Microbiol 17, 4954–4964. [DOI] [PubMed] [Google Scholar]
- Tumbleson, M.E. , Burks, M.F. , Spate, M.P. , Hutcheson, D.P. and Middleton, C.C. (1970) Serum biochemical and hematological parameters of Sinclair(S‐1) miniature sows during gestation and lactation. Can J Comp Med 34, 312–319. [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P.J. , Hamady, M. , Yatsunenko, T. , Cantarel, B.L. , Duncan, A. , Ley, R.E. , Sogin, M.L. , Jones, W.J. et al (2009) A core gut microbiome in obese and lean twins. Nature 457, 480–U487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, J. (2008) Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl Environ Microb 74, 4985–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G.M. , Tiedje, J.M. and Cole, J.R. (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G.D. , Chen, J. , Hoffmann, C. , Bittinger, K. , Chen, Y.Y. , Keilbaugh, S.A. , Bewtra, M. , Knights, D. et al (2011) Linking long‐term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Fu, X. , Dai, X.H. , Chen, Y.G. and Dai, L.L. (2016a) A new biological process for short‐chain fatty acid generation from waste activated sludge improved by Clostridiales enhancement. Environ Sci Pollut Res Int 23, 23972–23982. [DOI] [PubMed] [Google Scholar]
- Zhang, J.Y. , Lv, C. , Tong, J. , Liu, J.W. , Liu, J.B. , Yu, D.W. , Wang, Y.W. , Chen, M.X. et al. (2016b) Optimization and microbial community analysis of anaerobic co‐digestion of food waste and sewage sludge based on microwave pretreatment. Bioresource Technol 200, 253–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The OTU‐level rarefaction curve of observed OTUs across all samples.