

Abstract
Essentials.
The utility of bleeding assessment tools regarding platelet function disorders is still elusive.
We studied consecutive patients in a prospective cohort study in a tertiary hospital.
Substantially higher scorings were observed in patients with platelet function disorders.
Bleeding assessment tools might provide a useful screening tool.
Background
Bleeding assessment tools (BATs) have been widely implemented in the evaluation of patients with suspected bleeding disorders. However, diagnostic BAT utility regarding platelet function disorders is still elusive.
Aim
We aimed to assess the diagnostic value of the International Society on Thrombosis and Haemostasis BAT (ISTH‐BAT) for platelet function disorders in clinical practice.
Methods
The clinical characteristics and laboratory data of all consecutive patients with a suspected bleeding disorder referred between January 2012 and March 2017 to an outpatient unit of a university hospital were prospectively collected. The diagnostic evaluation was performed according to current recommendations following a prespecified protocol and platelet function was tested using light transmission aggregometry as well as flow cytometry.
Results
Five hundred and fifty‐five patients were assessed; 66.9% were female, median age was 43.7 years (interquartile range [IQR] 29.3, 61.7). Confirmed platelet function disorder was diagnosed in 54 patients (9.7%), possible platelet function disorder in 64 patients (11.5%), and other disorders in 170 patients (30.6%). Median scoring of the ISTH‐BAT was 2 in patients without a bleeding disorder (IQR 1, 3), 4 in patients with a possible platelet function disorder (2, 7), and 7 in patients with confirmed platelet function disorder (5, 9). Area under the receiver operating characteristic curve (the area under the curve [AUC]) was 0.75 (95% CI 0.70, 0.80).
Conclusions
Presence of a platelet function disorder was associated with substantially higher BAT scorings compared to patients without. Our data suggest that the ISTH‐BAT provides a useful screening tool for patients with suspected platelet function disorders.
Keywords: blood platelet disorders/diagnosis, hemorrhagic disorders/diagnosis, platelet function tests, predictive value of tests, prognosis, surveys and questionnaires
1. INTRODUCTION
Platelet function disorders (PFDs) are among the most common hereditary bleeding disorders, exposing affected patients to the risk of bleeding, particularly in the context of trauma and medical interventions.1, 2 Diagnosing PFD is, however, cumbersome, and many patients are probably not identified.3 While severe PFD such as Glanzmann thrombasthenia, Bernard‐Soulier syndrome, or syndromic disorders are easier to recognize, diagnosis is much more elusive in the majority of PFD patients. The diagnostic evaluation requires laboratory assays that are technically challenging, time‐consuming, and difficult to interpret.4 These tests tend also to be available in specialized laboratories only.3 In addition, complex preanalytic requirements often necessitate blood drawing on site at the laboratory rather than having the sample shipped from farther afield.5, 6 It is therefore important to select the patients for referral adequately.
Structured BATs such as from the International Society on Thrombosis and Haemostasis (ISTH‐BAT) may be utilized as a screening instrument in specific patients and implemented in the diagnostic evaluation, thereby triggering a referral. The ISTH‐BAT is an instrument to record both the presence and the severity of bleeding symptoms covering 14 important sites of bleeding in patients.7, 8 It has been validated extensively in patients with suspected von Willebrand disease (VWD).9, 10, 11 The diagnostic utility of the ISTH‐BAT with regard to platelet function disorders is, however, still elusive.12 With respect to adult patients, it has never been studied within the appropriate target population, namely, consecutive patients referred for evaluation of a suspected bleeding disorder.
With the present study, we aimed to assess the diagnostic value of the ISTH‐BAT of the International Society on Thrombosis and Haemostasis for platelet function disorders in a representative cohort of patients.
2. METHODS
2.1. Study design, setting, and population
We included all patients older than 18 years of age referred with a suspected bleeding disorder to a specialized outpatient clinic of Inselspital University Hospital in Bern, Switzerland, between January 2012 and March 2017. Reasons for the referral were (a) a bleeding tendency, (b) a family history of bleeding disorders, or (c) abnormal laboratory test results. Inselspital Bern is a tertiary university hospital in Switzerland covering a region with 1.5 million inhabitants. The catchment area of the hospital is representative for Switzerland with regard to German‐speaking and French‐speaking residents, as well as patients living in either urban or rural areas. Inselspital University Hospital comprises the only specialized laboratory and its hematological outpatient clinic functions as a reference center for coagulation disorders.
Clinical data were prospectively recorded using an established in‐house questionnaire as well as the ISTH‐BAT sheet in the electronic hospital database. Laboratory data were stored accordingly. Patient files were coded using an in‐house identification system and data were retrieved by two investigators working in parallel (MA/JK, MN). All patients signed informed consent forms and the ethics committee approved the study protocol (No. 02289).
2.2. Evaluation of patients and determination of ISTH‐BAT
The evaluation of patients was done using a standardized protocol as proposed in previous recommendations.4, 7, 13, 14, 15 Before consultation, patients completed a 13‐item, in‐house questionnaire assessing the presence and severity of bleeding at specific organs, including the skin, nose, oral cavity, gastrointestinal and urogenital systems, joints and muscles, bleeding in association with minor injuries, dental procedures, surgery, transfusion requirements, bleeding after ingestion of drugs known to affect hemostasis, and family history.16 During the consultation, trained resident physicians took detailed history using a standardized form and applied the ISTH‐BAT.7, 8 The scores were applied prior to laboratory testing and physicians were not aware of any laboratory test results. The ISTH‐BAT scorings were challenged by a second experienced physician (attending). Patients were instructed to stop anticoagulant treatment, antiaggregant treatment, nonsteroidal antirheumatic drugs, and/or selective serotonin reuptake inhibitors 10 days prior to consultation. Potential bleeding stigmata including petechia, hematomas, as well as signs of amyloidosis, telangiectasia, or joint hyperflexibility, were documented after a thorough physical examination.
2.3. Handling of samples
To prevent preanalytical errors, a standardized protocol was implemented for blood drawing and preparation of blood samples.17 Blood was collected by standard venipuncture with a 21‐gauge needle in EDTA tubes (Monovette; Sarstedt, Nümbrecht, Germany) for blood cell count; in 0.106‐mol/L trisodium citrate (9:1, v/v) tubes (Monovette; Sarstedt) for standard coagulation testing and platelet flow cytometric assays; and buffered citrate (0.13 mol/L trisodium citrate, pH 5.5) (Monovette; Sarstedt) for platelet aggregation studies. Samples were transported manually to the laboratory.
2.4. Laboratory evaluation
Laboratory evaluation was done in a stepwise manner with the following initial tests conducted simultaneously: blood count with mean platelet volume, a blood smear analyzing platelet morphology, prothrombin time, activated partial thromboplastin time, thrombin time, fibrinogen concentration (Clauss's method); and coagulometric determination of factor II, factor V, factor VII, factor X, factor VIII, factor IX, factor XI, factor XIII, von Willebrand factor (VWF) antigen, VWF ristocetin cofactor activity, α2‐antiplasmin, and platelet function analyzer 100. Secondary tests included light transmission aggregometry (LTA) and platelet flow cytometry if the initial test results were normal. In selected cases, chromogenic factor VIII, VWF multimer analysis, and VWF factor VIII binding capacity were also performed. Lumiaggregometry and molecular diagnostics were conducted only in a few cases. Determination of LTA and platelet flow cytometry are described later; all other laboratory tests are reported in detail in the Supplementary Material.18
2.5. Determination of light transmission aggregometry and platelet flow cytometry
Light transmission aggregometry was done inline with current recommendations5, 19 and as previously described16 using the aggregometer APACT® 4004V (LABiTec GmbH, Ahrensburg, Germany). Platelet aggregation was induced by increasing concentrations of adenosine diphosphate (ADP) (Sigma‐Aldrich, St. Louis, MO; 4, 6, and 10 μmol/L for male patients and 3, 4, and 6 μmol/L for female patients); collagen (HORM®; Nycomed, Linz, Austria) at 1.5, 3, and 4 μg/mL; arachidonic acid at 2 mmol/L (Bio Data/Medonic Servotec AG, Interlaken, Switzerland); and ristocetin at 1.5 and 0.5 mg/mL (Socochim SA, Lausanne, Switzerland). Platelet‐rich plasma (PRP) was prepared (centrifuged at 150 g for 15 min) and platelet count was adjusted to 250 × 109/L. Then, 200 μL of PRP prewarmed at 37°C for 1 min was added to the aggregometer cuvette and run for an additional minute to exclude spontaneous aggregation; 20 μL of the agonist was added and the response was recorded. If the response to one agonist was outside the limits of the normal range, the test was repeated. The LTA was performed 1 h after collection of venous blood samples from the patient and was completed within 2.5 h. The in‐house reference values have been previously established.20 A sample from a healthy volunteer was analyzed as an internal control; LTA was not performed when the platelet count was <100 G/L.
Platelet flow cytometry was conducted as previously described.16 Surface glycoproteins (GPs) were analyzed using antihuman antibodies: Ibα (CD42b‐PE; Ibα; Dako), GPIIb/IIIa (CD41‐FITC, Becton Dickinson; CD61‐FITC, Becton Dickinson), baseline P‐selectin expression (CD62P‐PE, Becton‐Dickinson), and PAC‐1 binding (PAC1‐FITC, Becton Dickinson). FACSCanto® (Becton Dickinson, Heidelberg, Germany) flow cytometer was used. The dose response of platelet reactivity was investigated with ADP (0.5, 5.0, and 50 μmol/L), convulxin (5, 50, and 500 ng/mL), and thrombin (0.05, 0.5, and 5 nmol/L) with anti‐CD62P and PAC1. The surface expression of negatively charged phospholipids was investigated using Annexin V‐FITC (Roche, Rotkreuz, Switzerland) after incubation with either Ionophore A 23187 or the combination of convulxin (500 ng/mL) and thrombin (5 nmol/L). To evaluate the content and secretion of dense granules, platelets were loaded with mepacrine (0.17 as well as 1.7 μmol/L) and analyzed with thrombin. The in‐house reference values had been previously established.16 As a control, a sample from a healthy volunteer was analyzed in parallel with each run. Flow cytometric analysis was repeated once with different control platelets to confirm the results.
2.6. Definition of diagnoses
Bleeding disorders were diagnosed following current recommendations. Type 1 VWD was diagnosed with repeatable (two times) VWF:GPIbM levels of 0.05 to 0.4 U/mL and VWF:Ag of 0.05 to 0.4 U/mL, a VWF:GPIbM/VWF:Ag ratio of >0.7, a normal multimer pattern, and an appropriate bleeding history.21, 22, 23, 24, 25 The threshold of 0.4 U/mL was chosen rather than a 0.3 in order to simplify treatment decisions in clinical practice.26 Type 2 VWD was diagnosed according to ISTH criteria.23 Low VWF was diagnosed in patients with VWF:GPIbM or VWF:Ag below 0.5 U/mL, not meeting the criteria mentioned, and associated with blood group O.14 Hemophilia and other single‐factor deficiencies were diagnosed according to current definitions.27
Interpretation of LTA and flow cytometry was done according to previous recommendations and established in‐house reference ranges 16 by three experienced individuals; discrepancies were resolved by discussion.3, 4, 6, 28, 29, 30 Lumiaggregometry was additionally considered if available (in a few patients only). We categorized PFD into “confirmed platelet function disorder” in cases with repeated abnormal LTA and/or flow cytometry measurements in the absence of other disorders and “possible platelet function disorder” if only one measurement was available or there were inconclusive results, or concomitant disorders were present. Patients were categorized into one of the following PFD subgroups: (a) Glanzmann's thrombasthenia, defined as a defect in GPIIb/IIIa associated with a severely diminished aggregation of all agonists except ristocetin, reduced expression of GPIIb/IIIa, and/or markedly reduced activation of PAC1‐binding1, 3, 31, 32; (b) Gi‐like defects, defined as an accentuated deficiency in aggregation to the Gi‐coupled receptor antagonists ADP and adrenaline, associated with corresponding flow cytometry results1, 3, 32; (c) thromboxane A2 pathway defects, defined as an absent aggregation in response to arachidonic acid, and possibly associated with an impaired response to other agonists1, 3, 19, 31, 32; (d) dense granule secretion defects, defined as a defect in storage and/or secretion of mepacrine1, 3, 16, 31, 32; (e) collagen receptor defects, defined as an isolated reduction in aggregation and secretion after stimulation with collagen and convulxin1, 16, 32; (f) α‐granule disorders, defined as a reduced expression and/or secretion of P‐selectin, associated with varying impaired aggregation after stimulation with collagen and epinephrine 1, 16; (g) decreased generation of procoagulant (COAT) platelets, defined as an impaired binding of Annexin‐V after incubation with convulxin and thrombin16; (h) complex disorders, defined as defects in a number of agonists (LTA) and/or several flow cytometry results that cannot be attributed to any of the disorders mentioned.
We defined a “bleeder of undefined cause” as a patient with an abnormal ISTH‐BAT (male ≥4 points; female ≥6 points) if results of all other tests mentioned in the diagnostic evaluation were normal and no bleeding disorder was identified.33, 34, 35 Patients who did not have hemostatic disorders but did have systemic disorders associated with bleeding symptoms (e.g. hereditary telangiectasia and thrombocytopenia) were categorized as having “systemic disorders.”
2.7. Statistical analysis
Descriptive statistics were used to characterize the study population (numbers/percent or median/interquartile range as appropriate). To evaluate the ISTH‐BAT ability to discriminate between patients with or without a demonstrable platelet defect on platelet function testing, a receiver‐operating characteristic curve analysis was performed and simple logistic regression was computed with the Stata 14.2 statistics software package (StataCorp. 2014. Stata Statistical Software: Release 14 College Station, TX: StataCorp LP). Figures were created using Prism 6 (GraphPad Software, Inc., La Jolla, CA).
3. RESULTS
3.1. Patient characteristics
Between January 2012 and March 2017, 555 patients were referred with a suspected bleeding disorder and were included in the study cohort (Figure 1A). Patients were referred from general practitioners in 200 cases (36.0%), from gynecologists in 121 (21.8%), and from other medical specialists in 207 cases (37.2%). The reason for referral was a bleeding tendency in 453 cases (81.6%), abnormal coagulation tests in 35 cases (6.3%), family history in 42 cases (7.6%), and verification of a known hemostatic disorder (reevaluation) in 8 cases (1.5%). The median age was 42.9 years (interquartile range [IQR] 28.0, 64.8) and 371 patients were female (66.9%). Of all patients, 55 were being treated with antiplatelet drugs (9.9%; stopped 10 days before assessment) and 34 were receiving anticoagulant treatment (6.1%; predominantly vitamin K‐antagonists). The ISTH‐BAT was abnormal in 153 patients referred for a bleeding tendency (35%) and in 156 patients referred for any reason (28.1%). Detailed patient characteristics are displayed in Table 1.
Figure 1.
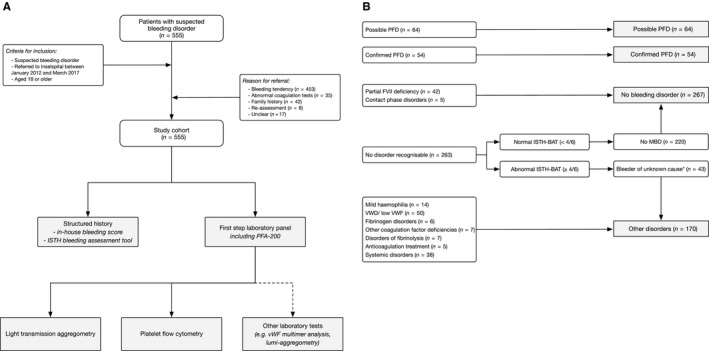
A, Flow of the patients. The clinical characteristics and laboratory data of all consecutive patients referred between January 2012 and March 2017 to an outpatient unit of a university hospital with a suspected bleeding disorder were collected prospectively. The diagnostic evaluation was performed according to current guidelines and platelet function was tested using light transmission aggregometry as well as flow cytometry. B, Classification of patients according to diagnosis. FVII, factor VII; MBD, mild bleeding disorder; PFD, platelet function disorder; VWD, von Willebrand disease; VWF, von Willebrand factor
Table 1.
Characteristics of patients referred with a suspected bleeding disorder (n = 555; 2012‐2017)
| Characteristics | Numbers (%) or median (IQR) as appropriate | Missing data | |||
|---|---|---|---|---|---|
| No bleeding disorder | Possible platelet function disorder[Link] | Confirmed platelet function disorder[Link] | Other bleeding disorders | ||
| Patients | 267 (48.0) | 64 (11.5) | 54 (9.7) | 170 (30.6) | 0 |
| Age (y) | 40.2 (27.3, 60.4) | 49.3 (34.9, 63.9) | 49.8 (33.5, 64.0) | 44.3 (30.7, 61.7) | 0 |
| Sex | |||||
| Female | 186 (69.7) | 52 (74.3) | 32 (66.7) | 101 (59.4) | 0 |
| Male | 81 (30.3) | 18 (25.7) | 16 (33.3) | 69 (40.6) | 0 |
| Reason for referral | |||||
| Bleeding tendency | 191 (75.5) | 63 (98.4) | 51 (94.4) | 148 (88.6) | 17 |
| Abnormal coagulation test results | 25 (9.9) | 1 (1.6) | 0 (0) | 9 (5.4) | |
| Family history | 33 (13.0) | 0 (0) | 3 (5.6) | 6 (3.6) | |
| Reevaluation | 4 (1.6) | 0 (0) | 0 (0) | 4 (2.4) | |
| Referring physician | |||||
| General practitioner | 90 (35.9) | 22 (32.8) | 20 (42.6) | 68 (41.7) | 27 |
| Gynecologist | 51 (20.3) | 18 (26.9) | 18 (38.3) | 34 (20.9) | |
| Other specialist | 110 (43.8) | 27 (40.3) | 9 (19.2) | 61 (37.4) | |
| Antiaggregant treatment | 23 (9.5) | 11 (17.2) | 3 (6.8) | 17 (10.8) | 47 |
| Anticoagulant treatment | 13 (4.9) | 3 (4.3) | 2 (4.2) | 16 (9.4) | 3 |
| SSRI treatment | 16 (6.0) | 8 (11.4) | 4 (8.3) | 7 (4.1) | 57 |
| VWF:C | 110 (82, 136) | 101 (79, 136) | 98 (79, 133) | 93 (57, 134) | 72 |
| VWF activity | 107 (81, 131) | 99 (72, 126) | 100 (75, 137) | 84 (55, 126) | 73 |
| Platelet count | 235 (200, 267) | 256 (223, 307) | 235 (186, 270) | 227 (184, 273) | 4 |
Abbreviations: IQR, interquartile range; SSRI, selective serotonin reuptake inhibitors; VWF:C, von Willebrand factor antigen.
Diagnosis of a platelet function disorder was made using light transmission aggregometry and platelet flow cytometry. “Confirmed platelet function disorder” was defined as abnormal results in repeated light transmission aggregometry/flow cytometry measurements in the absence of other disorders, “possible platelet function disorder” as an abnormal result in one measurement available, inconclusive results or presence of concomitant disorder.
3.2. Type of bleeding disorders
A bleeding disorder was diagnosed in 288 patients (51.9%; Figure 1; Table 1). The underlying type of bleeding disorder was a possible platelet function disorder in 64 cases (11.5%), confirmed platelet function disorder in 54 cases (9.7%), von Willebrand disease and low von Willebrand factor in 50 cases (9.1%), mild hemophilia in 5 cases (0.9%), deficiency of other coagulation factors in 7 cases (1.3%; including 6 patients with factor XI deficiency and 1 patient with factor X deficiency), fibrinogen disorders in 6 cases (1.1%), disorders of fibrinolysis in 7 cases (1.3%; including 1 patient with α2‐antiplasmin deficiency, 3 patients with PAI1 deficiency, and 3 patients with abnormal clot lysis time) anticoagulant treatment in 5 cases (0.9%), and a systemic disorder in 38 cases (6.9%).
3.3. Subgroups of PFD
Of 118 patients with a possible PFD, a confirmed subgroup could be established in 54 patients (46%). In contrast, diagnostic evaluation was not conclusive in 64 patients (54%). Distributions of PFD subgroups according to these two diagnostic groups are shown in Table 2. Gi‐like defects were found most frequently (32%), followed by complex disorders (28%), and diminished procoagulant COAT platelets (8%).
Table 2.
Platelet function disorder subgroups
| Disorder | Numbers (%) | |
|---|---|---|
| Possible PFD (n = 64) | Confirmed PFD (n = 54) | |
| Glanzmann's thrombasthenia[Link] | 0 | 4 (7.4) |
| Gi‐like defects[Link] | 20 (31.3) | 18 (33.3) |
| TxA2 pathway defects[Link] | 13 (20.3) | 1 (1.9) |
| Collagen receptor defects[Link] | 6 (9.4) | 2 (3.7) |
| Dense granule disorders[Link] | 1 (1.6) | 4 (7.4) |
| α‐granule disorders[Link] | 1 (1.6) | 5 (9.3) |
| Diminished procoagulant COAT platelets[Link] | 3 (4.7) | 7 (13.0) |
| Complex disorders[Link] | 20 (31.3) | 13 (24.1) |
Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; LTA. Light transmission aggregometry; PFD, platelet function disorder.
PFD subgroups were defined as follows: Defect in GPIIb/IIIa associated with a severely diminished aggregation of all agonists except ristocetin, reduced expression of GPIIb/IIIa, and/or markedly reduced activation of PAC1‐binding.1, 3, 31, 32
Accentuated deficiency in aggregation to the Gi‐coupled receptor antagonists ADP and adrenaline, associated with corresponding flow cytometry results.1, 3, 32
Absent aggregation in response to arachidonic acid, and possibly associated with an impaired response to other agonists.1, 3, 19, 31, 32
Isolated reduction in aggregation and secretion after stimulation with collagen and convulxin.1, 16, 32
Reduced expression and/or secretion of P‐selectin, associated with varying impaired aggregation after stimulation with collagen and epinephrine.1, 16
Impaired binding of Annexin‐V after incubation with convulxin and thrombin.16
Defects in a number of agonists (LTA) and/or several flow cytometry results that cannot be attributed to any of the disorders mentioned above.
3.4. Results of ISTH‐BAT scorings according to diagnosis
The median value of the ISTH‐BAT bleeding score in patients without a bleeding disorder was 2 (IQR 1, 4). In contrast, the ISTH‐BAT bleeding score was 4 (IQR 3, 7) in patients with a possible platelet function disorder and 7 (IQR, 5, 9) in patients with a confirmed platelet function disorder (Table 3; Figure 2). The score was 4 (IQR 2, 7) in patients with low VWF/VWD and 4 (IQR 1, 8) in patients with other coagulation disorders. Patients with systemic disorders associated with bleeding symptoms had a median score of 3 (IQR 1, 4).
Table 3.
ISTH‐BAT scorings according to disease in patients referred with a suspected bleeding disorder (n = 555)
| Disorder | Median score (IQR) | ||
|---|---|---|---|
| All patients | Female patients | Male patients | |
| No bleeding disorder | 2 (1, 3) | 2 (1, 4) | 2 (1, 3) |
| Possible platelet function disorder[Link] | 4 (2, 7) | 4 (2, 8) | 4 (2, 6) |
| Confirmed platelet function disordera | 7 (5, 9) | 7 (5, 11) | 6 (4, 8) |
| VWD/low VWF[Link] | 4 (2, 6) | 4 (3, 6) | 4 (2, 6) |
| Other disorder[Link] | 4 (3, 7) | 6 (3, 8) | 4 (2, 5) |
Abbreviations: IQR, interquartile range; ISTH‐BAT, International Society on Thrombosis and Haemostasis bleeding assessment tool; LTA, light transmission aggregometry; VWD, von Willebrand disease; VWF, von Willebrand factor.
Diagnosis of platelet function disorder was determined using light transmission aggregometry and platelet flow cytometry. “Confirmed platelet function disorder” was defined as abnormal results in repeated LTA/flow cytometry measurements in the absence of other disorders, “possible platelet function disorder” as abnormal result in one measurement available, inconclusive results or presence of concomitant disorders.
Low von Willebrand values associated with blood group 0; von Willebrand disease type 1 or type 2.
Systemic disorder associated with bleeding symptoms such as hereditary hemorrhagic telangiectasia, and thrombocytopenia.
Figure 2.
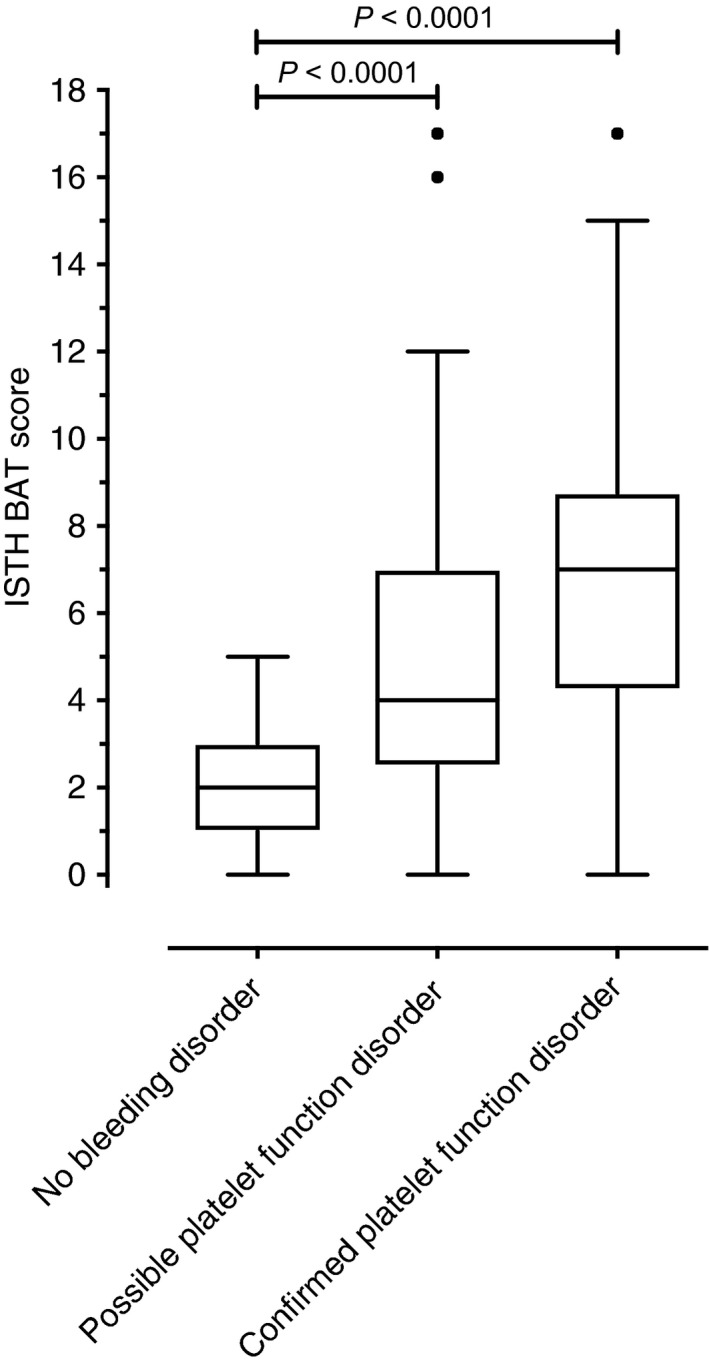
Results of the ISTH‐BAT according to the presence of a platelet function disorder. The ISTH‐BAT was conducted in the evaluation of 555 consecutive patients referred with a suspected bleeding disorder. Diagnosis of platelet function disorder was made using light transmission aggregometry and platelet flow cytometry. “Confirmed platelet function disorder” was defined as abnormal results in repeated LTA/flow cytometry measurements in the absence of other disorders, “possible platelet function disorder” as abnormal result in one measurement available, inconclusive results or presence of concomitant disorders. The Mann‐Whitney U test was applied. ISH‐BAT, International Society on Thrombosis and Haemostasis bleeding assessment tool; LTA, light transmission aggregomtery
3.5. Predictive value of ISTH‐BAT
The ROC curve analysis demonstrated an area under the curve (AUC) of 0.75 (95% CI 0.70, 0.80; Figure 3). In male patients only, AUC was 0.77 (0.68, 0.86); in female patients only, AUC was 0.74 (0.67, 0.80). At a threshold of 4 (males), sensitivity was 76.9% (95% CI 68.2, 84.2), and specificity was 62.4% (57.6, 67.0). At a threshold of 6 (females), sensitivity was 52.1% (42.7, 61.5), and specificity was 86.1% (82.4, 89.2).
Figure 3.
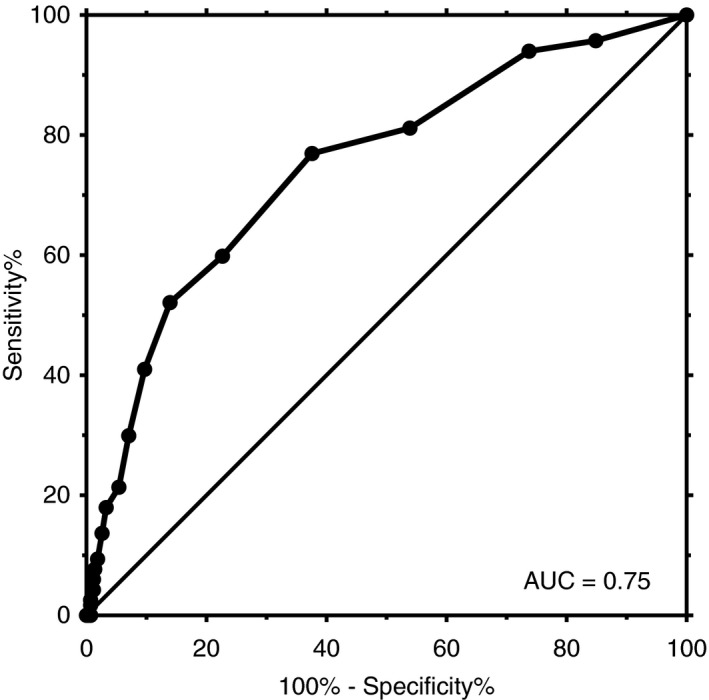
The receiver operating characteristic (ROC) curve of ISTH‐BAT for the presence of confirmed platelet function disorder. Area under the ROC curve (AUC) was 0.75 (95% CI 0.70, 0.80). At a threshold of 4, sensitivity was 76.9% (95% CI 68.2, 84.2) and specificity 62.4% (95% CI 57.6, 67.0). CI, confidence interval; IStH‐BAT, International Society on Thrombosis and Haemostasis bleeding assessment tool
[Correction added on May 31, 2019 after first online publication: In Figure 3, the percentages of sensitivity at a threshold of 4 have been amended from a previous version.]
4. DISCUSSION
In our cohort of consecutive patients assessed for a suspected bleeding disorder in clinical practice, we identified 54 patients with a confirmed PFD (9.7%) and 64 patients with a possible PFD (11.5%). Results of ISTH‐BAT were substantially higher in patients with possible and confirmed PFD compared to patients without a bleeding disorder. The predictive value of ISTH‐BAT for the presence of PFD was good.
Our results are essentially in line with previous investigations conducted in different settings and using other study designs. In a subgroup of patients studied in a pediatric setting, the ISTH score was higher in 5 patients with a possible PFD (median 3) compared to patients without a bleeding disorder (median 1).30 Rashid and colleagues compared ISTH‐BAT scorings between patients with suspected PFD and healthy volunteers in Pakistan and found a statistically significant difference (median 2 versus median 0).36 Lowe at al. studied 79 patients with excessive bleeding and 21 healthy volunteers and compared ISTH‐BAT according to lumiaggregometry results (adenosine triphosphate release). The ISTH‐BAT was significantly higher in patients with abnormal ATP release compared to patients without (median 12 versus median 0 points).37 Perez et al calculated ISTH‐BAT retrospectively in 61 patients with thrombocytopenia and/or suspected PFD and found an association between abnormal ISTH‐BAT and LTA results.38 Kaur and colleagues compared ISTH‐BAT scorings between 48 patients with Glanzmann's thrombasthenia or Bernard‐Soulier syndrome and healthy controls and found a significant difference.39
In contrast to previous investigations, we studied the ISTH‐BAT in a population that represents an unselected test target population and comprising all patients referred with a suspected bleeding disorder. Patients were consecutively included, preventing any selection bias. The number of patients studied was significantly higher than in previous investigations. We studied adult Caucasian patients and the evaluation was performed using a prespecified protocol including LTA as well as platelet flow cytometry. Our study does, however, have several limitations. First, the diagnostic evaluation was not finished appropriately in a number of patients resulting in a high proportion of patients with “suspected PFD.” Such patients were reluctant to appear several times in the outpatient unit and this has been encountered in clinical practice; we analyzed patients with suspected and confirmed PFD separately and a similar association was observed in both groups. In our setting, tests beyond LTA and flow cytometry were available in some cases only (e.g. molecular diagnostics, ATP release, expanded LTA agonist panel, fluorescence microscopy). Thus, the particular molecular defect in many patients could not be found. However, our results are in line with a previous study using ATP‐release37 and we do not anticipate that this introduced any bias.
Our results suggest that ISTH‐BAT is a useful screening tool in patients with suspected PFD. This is particularly supported by the high sensitivity. The ISTH‐BAT might be applied for triggering a referral of patients with any bleeding tendency at all (including patients with PFD as well as VWD), or for triggering platelet function studies in patients with normal first‐line test results. We do not believe that the ISTH‐BAT is able to discriminate different types of mild bleeding disorders, e.g. PFD from VWD, and we do not believe that the ISTH‐BAT might replace platelet function studies. Our study was not designed to demonstrate a respective difference and two observations suggest the opposite: (a) the specificity is moderate only, suggesting a relevant number of patients with abnormal ISTH‐BAT associated with other disorders than PFD and (b) a widely overlapping distribution of ISTH‐BAT scorings among patients with PFD, VWD, and other disorders. This study must be replicated in other settings and employing alternative platelet function assays as well.
In conclusion, in a large study of patients referred for suspected bleeding disorders in clinical practice, patients with PFD had substantially higher ISTH‐BAT scorings than patients without and the predictive value of ISTH‐BAT was good. Our results suggest that ISTH is a useful screening tool for PFD in clinical practice.
CONFLICT OF INTERESTS
M. Nagler reports receiving: grants from the Swiss National Science Foundation during the conduct of the study and research grants, lecture honoraria, or consultancy fees from Bayer, Pentapharm, Daiichi Sankyo, and Roche diagnostics, outside the submitted work. The other authors state that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Marcel Adler and Jonas Kaufmann collected the data, participated in data analysis, and wrote the manuscript. Lorenzo Alberio implemented the evaluation of patients, reviewed the data analysis, and intellectually reviewed the manuscript. Michael Nagler designed the study, conducted the analysis, and wrote the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Prof. J. A. Kremer Hovinga for her valuable comments on the design and results of the study. No particular funding was obtained.
Adler M, Kaufmann J, Alberio L, Nagler M. Diagnostic utility of the ISTH bleeding assessment tool in patients with suspected platelet function disorders. J Thromb Haemost. 2019;17:1104–1112. 10.1111/jth.14454
Manuscript handled by: David Lillicrap
Final decision: David Lillicrap, 11 April 2019
REFERENCES
- 1. Podda G, Femia EA, Pugliano M, Cattaneo M. Congenital defects of platelet function. Platelets. 2012;23:552–63. [DOI] [PubMed] [Google Scholar]
- 2. Nurden AT, Freson K, Seligsohn U. Inherited platelet disorders. Haemophilia. 2012;18(Suppl 4):154–60. [DOI] [PubMed] [Google Scholar]
- 3. Gresele P, Harrison P, Bury L, Falcinelli E, Gachet C, Hayward CP, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost. 2014;12:1562–1112. [DOI] [PubMed] [Google Scholar]
- 4. Gresele P; Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis . Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13:314–22. [DOI] [PubMed] [Google Scholar]
- 5. Cattaneo M, Cerletti C, Harrison P, Hayward CP, Kenny D, Nugent D, et al. Recommendations for the standardization of light transmission aggregometry: a consensus of the working party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost. 2013;11:1183–1112. [DOI] [PubMed] [Google Scholar]
- 6. Harrison P, Mackie I, Mumford A, Briggs C, Liesner R, Winter M, et al. British Committee for Standards in H. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br J Haematol. 2011;155:30–44. [DOI] [PubMed] [Google Scholar]
- 7. Tosetto A, Castaman G, Plug I, Rodeghiero F, Eikenboom J. Prospective evaluation of the clinical utility of quantitative bleeding severity assessment in patients referred for hemostatic evaluation. J Thromb Haemost. 2011;9:1143–8. [DOI] [PubMed] [Google Scholar]
- 8. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, et al.; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group . ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–5. [DOI] [PubMed] [Google Scholar]
- 9. Rodeghiero F, Castaman G, Tosetto A, Batlle J, Baudo F, Cappelletti A, et al. The discriminant power of bleeding history for the diagnosis of type 1 von Willebrand disease: an international, multicenter study. J Thromb Haemost. 2005;3:2619–26. [DOI] [PubMed] [Google Scholar]
- 10. Bowman M, Mundell G, Grabell J, Hopman WM, Rapson D, Lillicrap D, et al. Generation and validation of the Condensed MCMDM‐1VWD Bleeding Questionnaire for von Willebrand disease. J Thromb Haemost. 2008;6:2062–6. [DOI] [PubMed] [Google Scholar]
- 11. Biss TT, Blanchette VS, Clark DS, Bowman M, Wakefield CD, Silva M, et al. Quantitation of bleeding symptoms in children with von Willebrand disease: use of a standardized pediatric bleeding questionnaire. J Thromb Haemost. 2010;8:950–6. [DOI] [PubMed] [Google Scholar]
- 12. Tosetto A. Bleeding assessment tools: limits and advantages for the diagnosis and prognosis of inherited bleeding disorders. Semin Thromb Hemost. 2016;42:463–70. [DOI] [PubMed] [Google Scholar]
- 13. de Moerloose P, Levrat E, Fontana P, Boehlen F. Diagnosis of mild bleeding disorders. Swiss Med Wkly. 2009;139:327–332. [DOI] [PubMed] [Google Scholar]
- 14. Boender J, Kruip MJ, Leebeek FW. A diagnostic approach to mild bleeding disorders. J Thromb Haemost. 2016;14:1507–16. [DOI] [PubMed] [Google Scholar]
- 15. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost. 2007;5(Suppl 1):167–74. [DOI] [PubMed] [Google Scholar]
- 16. Daskalakis M, Colucci G, Keller P, Rochat S, Silzle T, Biasiutti FD, et al. Decreased generation of procoagulant platelets detected by flow cytometric analysis in patients with bleeding diathesis. Cytometry B Clin Cytom. 2014;86:397–409. [DOI] [PubMed] [Google Scholar]
- 17. CLSI . Collection, Transport, and Processing of Blood Specimens for Testing Plasma‐Based Coagulation Assays and Molecular Hemostasis Assays : Approved Guideline 8th ed. CLSI document H21‐A5 Wayne, PA, Clinical and Laboratory Standards Institute, 2008. [Google Scholar]
- 18. Zurcher M, Sulzer I, Barizzi G, Lammle B, Alberio L. Stability of coagulation assays performed in plasma from citrated whole blood transported at ambient temperature. Thromb Haemost. 2008;99:416–26. [DOI] [PubMed] [Google Scholar]
- 19. Hayward CP, Moffat KA, Raby A, Israels S, Plumhoff E, Flynn G, et al. Development of North American consensus guidelines for medical laboratories that perform and interpret platelet function testing using light transmission aggregometry. Am J Clin Pathol. 2010;134:955–63. [DOI] [PubMed] [Google Scholar]
- 20. Thommen D, Sulzer I, Buhrfeind E, Naef R, Furlan M, Lammle B. [Measurement of bleeding time and study of thrombocyte aggregation. Standardization of methods, normal values and results in patients with suspected hemorrhagic diathesis]. Schweiz Med Wochenschr. 1988;118:1559–67. [PubMed] [Google Scholar]
- 21. Laffan M, Brown SA, Collins PW, Cumming AM, Hill FG, Keeling D, et al. The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2004;10:199–217. [DOI] [PubMed] [Google Scholar]
- 22. Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101:2089–93. [DOI] [PubMed] [Google Scholar]
- 23. Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al.; Working Party on von Willebrand Disease Classification . Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. [DOI] [PubMed] [Google Scholar]
- 24. Sadler JE, Rodeghiero F; ISTH SSC Subcommittee on von Willebrand Factor . Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost. 2005;3:775–7. [DOI] [PubMed] [Google Scholar]
- 25. Quiroga T, Goycoolea M, Belmont S, Panes O, Aranda E, Zuniga P, et al. Quantitative impact of using different criteria for the laboratory diagnosis of type 1 von Willebrand disease. J Thromb Haemost. 2014;12:1238–43. [DOI] [PubMed] [Google Scholar]
- 26. Leebeek FW, Eikenboom JC. Von Willebrand's Disease. N Engl J Med. 2016;375:2067–80. [DOI] [PubMed] [Google Scholar]
- 27. White GC, 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J; Factor VIII and Factor IX Subcommittee . Definitions in hemophilia: recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85:560. [PubMed] [Google Scholar]
- 28. Knofler R, Eberl W, Schulze H, Bakchoul T, Bergmann F, Gehrisch S, et al. [Diagnosis of inherited diseases of platelet function. Interdisciplinary S2K guideline of the Permanent Paediatric Committee of the Society of Thrombosis and Haemostasis Research (GTH e. V.)]. Hamostaseologie. 2014;34:201–12. [DOI] [PubMed] [Google Scholar]
- 29. Kottke‐Marchant K, Corcoran G. The laboratory diagnosis of platelet disorders. Arch Pathol Lab Med. 2002;126:133–46. [DOI] [PubMed] [Google Scholar]
- 30. Bidlingmaier C, Grote V, Budde U, Olivieri M, Kurnik K. Prospective evaluation of a pediatric bleeding questionnaire and the ISTH bleeding assessment tool in children and parents in routine clinical practice. J Thromb Haemost. 2012;10:1335–41. [DOI] [PubMed] [Google Scholar]
- 31. Matthews DC. Inherited disorders of platelet function. Pediatr Clin North Am. 2013;60:1475–88. [DOI] [PubMed] [Google Scholar]
- 32. Dawood BB, Lowe GC, Lordkipanidze M, Bem D, Daly ME, Makris M, et al. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood. 2012;120:5041–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quiroga T, Mezzano D. Is my patient a bleeder? A diagnostic framework for mild bleeding disorders. Hematology Am Soc Hematol Educ Program. 2012;2012:466–74. [DOI] [PubMed] [Google Scholar]
- 34. Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, et al. Normal range of bleeding scores for the ISTH‐BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20:831–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quiroga T, Goycoolea M, Panes O, Aranda E, Martinez C, Belmont S, et al. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica. 2007;92:357–65. [DOI] [PubMed] [Google Scholar]
- 36. Rashid A, Moiz B, Karim F, Shaikh MS, Mansoori H, Raheem A. Use of ISTH bleeding assessment tool to predict inherited platelet dysfunction in resource constrained settings. Scand J Clin Lab Invest. 2016;76:373–8. [DOI] [PubMed] [Google Scholar]
- 37. Lowe GC, Lordkipanidze M, Watson SP; UK GAPP study group . Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11:1663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perez Botero J, Warad DM, He R, Uhl CB, Tian S, Otteson GE, et al. Comprehensive platelet phenotypic laboratory testing and bleeding history scoring for diagnosis of suspected hereditary platelet disorders: a single‐institution experience. Am J Clin Pathol. 2017;148:23–32. [DOI] [PubMed] [Google Scholar]
- 39. Kaur H, Borhany M, Azzam H, Costa‐Lima C, Ozelo M, Othman M. The utility of International Society on Thrombosis and Haemostasis‐Bleeding Assessment Tool and other bleeding questionnaires in assessing the bleeding phenotype in two platelet function defects. Blood Coagul Fibrinolysis. 2016;27:589–93. [DOI] [PubMed] [Google Scholar]