

Abstract
Background
Gaucher disease (GD) presents with a range of signs and symptoms. Physicians can fail to recognise the early stages of GD owing to a lack of disease awareness, which can lead to significant diagnostic delays and sometimes irreversible but avoidable morbidities.
Aim
The Gaucher Earlier Diagnosis Consensus (GED‐C) initiative aimed to identify signs and co‐variables considered most indicative of early type 1 and type 3 GD, to help non‐specialists identify ‘at‐risk’ patients who may benefit from diagnostic testing.
Methods
An anonymous, three‐round Delphi consensus process was deployed among a global panel of 22 specialists in GD (median experience 17.5 years, collectively managing almost 3000 patients). The rounds entailed data gathering, then importance ranking and establishment of consensus, using 5‐point Likert scales and scoring thresholds defined a priori.
Results
For type 1 disease, seven major signs (splenomegaly, thrombocytopenia, bone‐related manifestations, anaemia, hyperferritinaemia, hepatomegaly and gammopathy) and two major co‐variables (family history of GD and Ashkenazi‐Jewish ancestry) were identified. For type 3 disease, nine major signs (splenomegaly, oculomotor disturbances, thrombocytopenia, epilepsy, anaemia, hepatomegaly, bone pain, motor disturbances and kyphosis) and one major co‐variable (family history of GD) were identified. Lack of disease awareness, overlooking mild early signs and failure to consider GD as a diagnostic differential were considered major barriers to early diagnosis.
Conclusion
The signs and co‐variables identified in the GED‐C initiative as potentially indicative of early GD will help to guide non‐specialists and raise their index of suspicion in identifying patients potentially suitable for diagnostic testing for GD.
Keywords: lysosomal storage disease, metabolism, inborn error, splenomegaly, thrombocytopenia, algorithm
Introduction
Gaucher disease (GD) is an autosomal recessive, genetic disease that reduces levels of the enzyme glucocerebrosidase in the lysosome. Glucocerebrosidase is a key enzyme in the catabolism of the sphingolipid glucosylceramide, and deficiency of this enzyme results in accumulation of glucosylceramide in macrophages, which then accumulate in the bone marrow, liver, lungs, spleen and brain.1 At least 200 pathogenic variants of the glucocerebrosidase gene have been identified.2 Patients with GD may present with a broad range of signs and symptoms, and a continuum of disease severity is observed. The three disease phenotypes (GD types 1–3) are also diverse,3 ranging from cases that are lethal within a few months from birth,4 to mild or asymptomatic forms in which symptoms may not appear until as late as the eighth decade of life.3, 5 Most patients present with the non‐neuronopathic type 1 form (with a prevalence of 1 in 40 000–60 000 in the general population rising to approximately 1 in 850 among individuals with Ashkenazi Jewish ancestry),6, 7, 8 and may experience clinical problems, including bone pain, bone fracture, osteoporosis, anaemia, thrombocytopenia, reduced growth, thrombocytopenia, bleeding, anaemia and hepatosplenomegaly. Neurological complications are rarely seen in type 1 disease but are more common in types 2–3.
Given its rarity and heterogeneous nature, identifying patients for GD diagnostic testing remains a challenge in clinical practice (see case study in Fig. 1),9 and it is common for patients to experience substantial delays between symptom onset and diagnosis. In a UK‐based retrospective review (n = 86), the median time from first symptoms to diagnosis was 2 years (range: 0.5–26.0 years) and was 5.0 years or longer for almost one‐fifth of patients.10 Possible reasons for exclusion of GD from differential diagnoses may be poor disease awareness among clinicians or because investigation of other diagnostic possibilities associated with greater mortality takes precedence.9, 10, 11 Differential diagnoses may also be impeded by multiple referrals among medical specialists before a correct diagnosis is reached. Patients may visit up to eight (mean: 3.0) different specialists, including internists, paediatricians, haematologists, medical oncologists, gastroenterologists, geneticists, neurologists, obstetricians/gynaecologists, orthopaedists and rheumatologists.11
Figure 1.
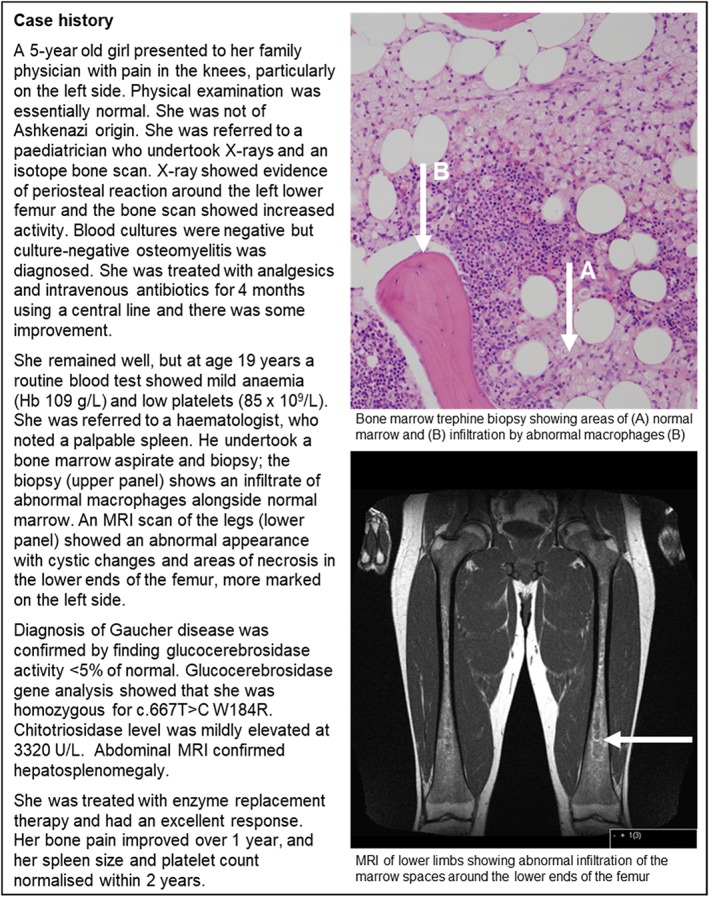
Type 1 Gaucher disease case study.
Diagnostic delays may lead to irreversible or preventable GD complications such as avascular necrosis, chronic bone pain, pathologic fractures, growth failure, life‐threatening sepsis, liver abnormalities and severe bleeding.11 In addition, patients often undergo unnecessary, invasive investigations (e.g. liver and bone marrow biopsies or splenectomy) before GD is diagnosed.11 The potential for such delays to impact on patient morbidity and quality of life is supported by the emergence of evidence on the benefits of early diagnosis.12 A GD diagnosis is confirmed with enzymatic and/or blood‐based genetic tests,1 but awareness among non‐specialists of available diagnostic services and regional differences in availability and in cost generally exclude GD testing from routine bloodwork. Algorithms have been developed to promote timely diagnosis of GD but they are typically aimed at specialists.13, 14 There is no consensus or guideline for identifying patients who may have early‐stage GD that is suited to non‐GD specialists.
The Gaucher Earlier Diagnosis Consensus (GED‐C) initiative is an ongoing global project that has applied Delphi methodology15, 16, 17 to build expert consensus on aspects relating to diagnosis and management of GD. Here, we summarise findings from the GED‐C initiative regarding consensus on signs and patient co‐variables suggestive of early type 1 or type 3 GD, on the most important barriers to early GD diagnosis, and on the likely impact of the initiative on patients and clinicians. We also propose some guidance for non‐specialists to help them both identify patients who may have GD and confirm or discount GD in the differential diagnosis.
Methods
The iterative Delphi process is summarised in Figure 2. Consensus is often reached within three rounds.15, 16, 17
Figure 2.
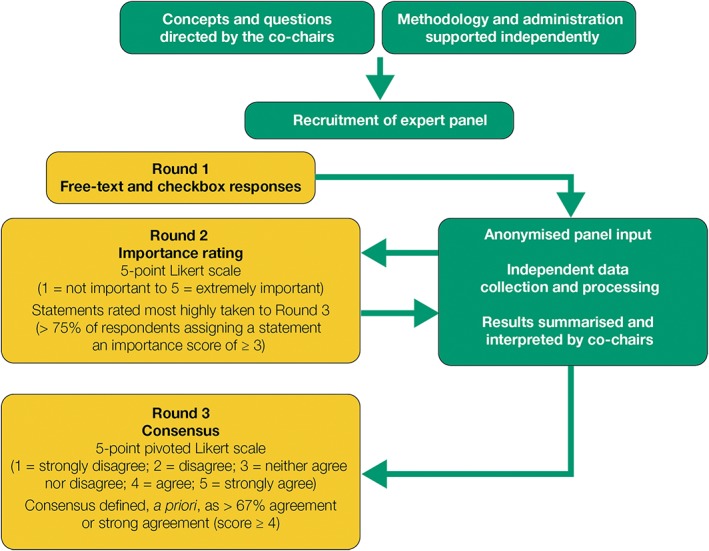
The Gaucher Earlier Diagnosis Consensus initiative Delphi methodology.
Selection of Chairs and expert panel
Three co‐Chairs were appointed: two clinical co‐Chairs recognised globally as leading GD experts, and one non‐clinical Chair with expertise in Delphi techniques who provided critical methodological input. The clinical co‐Chairs nominated individuals with established GD expertise (e.g. relevant research activities, participation in GD management initiatives or authorship of peer‐reviewed publications) to form the voting panel. Nominated individuals were recruited by an independent third‐party administrator (Oxford PharmaGenesis Ltd, Oxford, UK) and asked to respond independently to questions and remained anonymous to each other throughout the process. Based on published estimates that Delphi studies typically enrol 15–20 participants,17 it was agreed a priori that a panel of 22 experts was needed to provide adequate study power in case of dropouts.
Delphi process
All stages were overseen by the Chairs and conducted by the independent administrator. The administrator gathered the panel members’ responses using an online survey platform (SurveyMonkey, SurveyMonkey Europe, Dublin, Ireland) and blinded data before sharing with the co‐Chairs. In round 1 (Supporting Information Appendix S1), the expert panel provided free‐text answers to open questions about: clinical signs and co‐variables considered indicative of early type 1 or type 3 GD; barriers to diagnosis of GD; and the potential impact of the GED‐C initiative.
‘Early’ disease was defined as the time before symptoms impacted significantly on the quality of life. The administrator grouped responses from round 1 into similar themes, which the co‐Chairs checked, revised and consolidated into factors (for clinical signs and co‐variables) or statements (for ‘barriers to early GD diagnosis’ and ‘impact of the GED‐C initiative’). In round 2 (Appendix S2), the panel rated the importance of each factor or statement using a 5‐point Likert scale (1, not important; 2, slightly important; 3, important; 4, very important; 5, extremely important). Factors or statements awarded an importance score of 3 or more by more than 75% of respondents were taken to round 3. Factors not reaching this threshold were classified as ‘minor’; statements not reaching this threshold were excluded. In round 3 (Appendix S3), panel members indicated their level of agreement with each factor or statement using a 5‐point pivoted Likert scale (1, strongly disagree; 2, disagree; 3, neither agree nor disagree; 4, agree; 5, strongly agree). Consensus was defined a priori as more than 67% of the panel awarding an agreement score of 4 or more. Factors meeting these criteria were classified as major, otherwise as minor. Statements not meeting these criteria were excluded.
Statistical analyses
All data are reported descriptively owing to the exploratory (non‐hypothesis testing) nature of the study.
Literature review of presenting signs in GD at diagnosis
In parallel with the Delphi consensus, a comprehensive literature search was performed to compile evidence for the most common presenting signs at GD diagnosis. A detailed methodology is provided in Appendix S4.
Ethical approval
No patient data were required for conduct of this study, so no ethical approval was sought.
Results
Demographic and clinical practice information for the GED‐C expert panel
The expert panel comprised 22 practising physicians, representing 16 different countries (Argentina, Australia, Brazil, France, Germany, India, Ireland, Israel, Italy, Japan, The Netherlands, Russia, Spain, Sweden, UK and USA). All members had experience of managing type 1 GD, 19 also having managed type 3 GD. None had managed only type 3 GD, and experience of type 2 GD was not recorded. Collectively, the panel had more than 400 years of GD management experience (median, 17.5 years (range 6–40 years)) and had treated nearly 3000 patients with GD. Medical specialities of the expert panel represented those of physicians who would typically encounter patients with GD or who would confirm a diagnosis, including haematologists, paediatricians and experts in inborn errors of metabolism (Fig. 3). The panel members practised in public teaching/non‐teaching hospitals (n = 17), or in private hospitals/clinics/research centres (n = 5). For all questions, a response rate of 100% was achieved at each round of the consensus.
Figure 3.
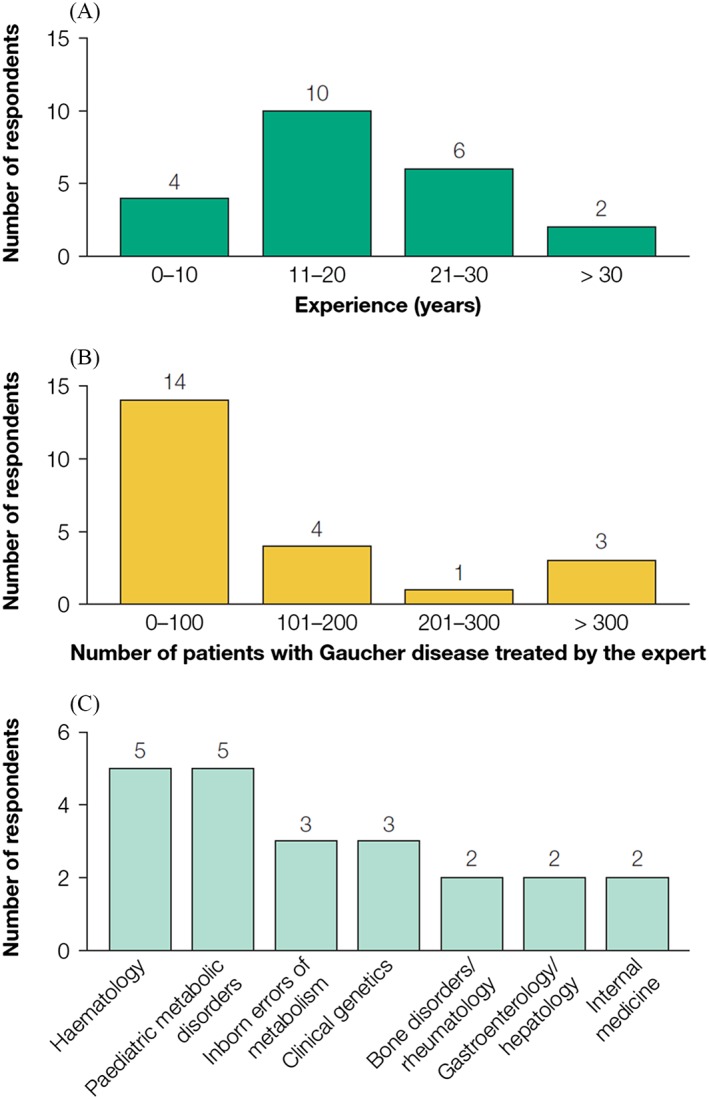
Characteristics of the experts involved in the Gaucher Earlier Diagnosis Consensus initiative (n = 22). (A) Years of experience in managing patients with Gaucher disease; (B) number of patients the expert has treated; (C) clinical specialty.
Presenting signs and patient co‐variables in early type 1 GD
For presenting signs in early type 1 GD, 84 individual free‐text phrases were supplied in round 1 and grouped into 29 themes. Seven themes (i.e. dental problems, depression, elevated β2‐microglobulin levels, elevated chitotriosidase levels, generalised pain, liver cirrhosis including abnormal liver function tests, and lymphadenopathy) were discarded as being either of low diagnostic value in GD or unlikely to be available in a patient's history. A further four themes were merged with others to yield 18 consolidated themes (termed ‘factors’) for assessment in round 2 (Appendix S5). Eight factors met the importance criteria in round 2 and were taken forward to round 3, in which consensus was reached on seven; these factors were classified as major signs. Of the remaining 11 factors from rounds 2 and 3, nine were classified as minor signs, and two were discounted as being of low relevance in early type 1 GD (Table 1).
Table 1.
Presenting clinical signs and co‐variables in early type 1 GD
| Round 2 | Round 3 | ||||||
|---|---|---|---|---|---|---|---|
| Importance score (Likert scale: 1–5)† | Agreement score (Likert scale: 1–5)‡ | ||||||
| Mean score | Median score | Respondents (%)§ | Mean score | Median score | Respondents (%)§ | ||
| Clinical signs | |||||||
| Major | Splenomegaly | 4.77 | 5 | 100 | 4.86 | 5 | 100 |
| Thrombocytopenia | 4.45 | 5 | 95 | 4.68 | 5 | 91 | |
| Bone issues including pain, crises, AVN and fractures | 4.00 | 4 | 91 | 4.45 | 5 | 91 | |
| Anaemia | 3.77 | 4 | 95 | 4.05 | 4 | 86 | |
| Hepatomegaly | 3.68 | 3.5 | 95 | 3.95 | 4 | 77 | |
| Elevated ferritin levels | 3.50 | 4 | 82 | 4.05 | 4 | 86 | |
| Gammopathy – monoclonal or polyclonal | 3.23 | 3 | 82 | 3.64 | 4 | 73 | |
| Minor | Bleeding, bruising or coagulopathy | 3.59 | 4 | 86 | 3.68 | 4 | 64 |
| Elevated serum angiotensin‐converting enzyme levels | 3.18 | 3.5 | 64 | — | — | — | |
| Growth retardation including low body weight | 3.00 | 3 | 73 | — | — | — | |
| Low bone mineral density | 2.95 | 3 | 59 | — | — | — | |
| Fatigue | 2.82 | 3 | 64 | — | — | — | |
| Asthenia | 2.59 | 2.5 | 50 | — | — | — | |
| Leukopenia | 2.50 | 3 | 55 | — | — | — | |
| Gallstones | 2.32 | 2 | 45 | — | — | — | |
| Dyslipidaemia | 2.27 | 2 | 36 | — | — | — | |
| Discounted | Neonatal cholestasis | 1.73 | 2 | 9 | — | — | — |
| Elevated bilirubin levels | 1.68 | 1.5 | 14 | — | — | — | |
| Co‐variables | |||||||
| Major | Family history of GD | 4.27 | 4.5 | 95 | 4.45 | 5 | 91 |
| Jewish ancestry | 3.91 | 4 | 86 | 4.18 | 4 | 86 | |
| Minor | Family history of PD | 3.14 | 3 | 73 | — | — | — |
Signs and co‐variables that did not meet the threshold for importance (a score of ≥3 by >75% of respondents) were classified as minor and not taken forward to round 3 for agreement rating. Signs and co‐variables that met the threshold for agreement in round 3 (a score of ≥4 by >67% of respondents) were classified as major; any that did not meet this threshold were classified as minor. Certain low‐scoring signs were discounted as being of little relevance in early type 1 GD.
The Likert scale used for importance rating in round 2 was: 1 = not important; 2 = slightly important; 3 = important; 4 = very important; 5 = extremely important.
The Likert scale used for agreement rating in round 3 was: 1 = strongly disagree; 2 = disagree; 3 = neither agree nor disagree; 4 = agree; 5 = strongly agree.
Percentage of respondents (n = 22) is the proportion awarding a Likert scale score that met the selection criteria in each round.
AVN, avascular necrosis; GD, Gaucher disease; PD, Parkinson disease.
Similarly, for patient co‐variables in early type 1 GD, 20 individual free‐text phrases were provided and grouped into nine themes. Two themes were discarded owing to their rarity (blood relative who died of foetal hydrops and/or with diagnosis of neonatal sepsis of uncertain aetiology; and child of consanguineous parents), and two were discarded for being of low diagnostic power in type 1 GD (age ≤18 years; age >18 years). Consolidation of the themes, including merging two themes with others, left three factors for consideration in round 2 (Appendix S6). Two factors met the importance criteria and were taken forward to round 3, and both met the consensus criteria. These two factors were classified as major co‐variables and the remaining factor as a minor co‐variable (Table 1).
Presenting signs and patient co‐variables in early type 3 GD
The panel provided 55 individual free‐text phrases relating to early type 3 GD signs, and these were grouped into 24 themes. Six themes were discarded as being of low diagnostic power (dysphagia, elevated serum acid phosphatase levels, gallstones, lymphadenopathy, paraesthesia and recurrent viral illness), leaving 18 factors for consideration in round 2 (Appendix S7). Of 13 factors that met the importance criteria in round 2, nine met the consensus criteria in round 3. These were classified as major signs, and the remaining nine factors from rounds 2 and 3 were classified as minor signs (Table 2).
Table 2.
Presenting clinical signs and co‐variables in early type 3 GD
| Round 2 | Round 3 | ||||||
|---|---|---|---|---|---|---|---|
| Importance score (Likert scale: 1–5)‡ | Agreement score (Likert scale: 1–5)‡ | ||||||
| Mean score | Median score | Respondents (%)§ | Mean score | Median score | Respondents (%)§ | ||
| Clinical signs | |||||||
| Major | Splenomegaly | 4.42 | 5 | 100 | 4.79 | 5 | 100 |
| Disturbed oculomotor function (slow horizontal saccades with unimpaired vision) | 4.58 | 5 | 100 | 4.74 | 5 | 100 | |
| Thrombocytopenia | 4.21 | 5 | 95 | 4.32 | 4 | 95 | |
| Myoclonus epilepsy | 3.95 | 4 | 95 | 4.00 | 4 | 79 | |
| Anaemia | 3.58 | 4 | 89 | 3.84 | 4 | 79 | |
| Hepatomegaly | 3.74 | 4 | 95 | 3.79 | 4 | 79 | |
| Bone pain, including fractures | 3.63 | 4 | 95 | 3.68 | 4 | 74 | |
| Disturbed motor function (impairment of primary motor development) | 3.11 | 3 | 79 | 3.68 | 4 | 79 | |
| Kyphosis | 3.79 | 4 | 84 | 3.58 | 4 | 68 | |
| Minor | Cardiac calcification | 3.32 | 3 | 79 | 3.63 | 4 | 53 |
| Growth retardation including low body weight | 3.47 | 3 | 95 | 3.58 | 4 | 53 | |
| Pulmonary infiltrates | 3.32 | 4 | 79 | 3.53 | 4 | 53 | |
| Cognitive deficit | 3.21 | 3 | 84 | 3.53 | 4 | 53 | |
| Elevated ferritin levels | 3.26 | 3 | 74 | — | — | — | |
| Bleeding, bruising or coagulopathy | 3.26 | 3 | 74 | — | — | — | |
| Gammopathy – monoclonal or polyclonal | 2.63 | 3 | 58 | — | — | — | |
| Elevated serum angiotensin‐converting enzyme levels | 3.11 | 3 | 63 | — | — | — | |
| Fatigue | 2.79 | 3 | 63 | — | — | — | |
| Co‐variables | |||||||
| Major | Family history of GD | 3.89 | 84 | 4.21 | 4 | 79 | |
| Minor | Age ≤18 years | 3.42 | 79 | 3.84 | 4 | 63 | |
| Blood relative who died of foetal hydrops and/or with diagnosis of neonatal sepsis of uncertain aetiology | 3.47 | 84 | 3.26 | 3 | 42 | ||
| Family history of PD | 2.79 | 63 | — | — | — | ||
| Ashkenazi Jewish ancestry | 2.47 | 37 | — | — | — | ||
Signs and co‐variables that did not meet the threshold for importance (a score of ≥3 by >75% of respondents) were classified as minor and not taken forward to round 3 for agreement rating. Signs and co‐variables that met the threshold for agreement in round 3 (a score of ≥4 by >67% of respondents) were classified as major; any that did not meet this threshold were classified as minor.
The Likert scale used for importance rating in round 2 was: 1 = not important; 2 = slightly important; 3 = important; 4 = very important; 5 = extremely important.
The Likert scale used for agreement rating in round 3 was: 1 = strongly disagree; 2 = disagree; 3 = neither agree nor disagree; 4 = agree; 5 = strongly agree.
Percentage of respondents (n = 19) is the proportion awarding a Likert scale score that met the selection criteria in each round.
GD, Gaucher disease; PD, Parkinson disease.
There were 15 individual free‐text phrases relating to co‐variables in type 3 GD, and these were grouped into 10 themes. Four themes were discounted owing to their rarity (Japanese or Taiwanese ancestry, Norbottnian Swedish ancestry, Palestinian Arabic ancestry and the L444P allele in Ashkenazi Jewish patients), and one theme was discounted as being of low clinical relevance (age >18 years). Of the five remaining factors assessed in round 2 (Appendix S8), three met the importance criteria, and consensus was reached for one of these in round 3. This was classified as a major co‐variable, and the other four factors from rounds 2 and 3 were classified as minor co‐variables (Table 2).
Levels of major signs considered consistent with a diagnosis of GD
During round 3, for major signs that were continuous rather than categorical variables (anaemia, hyperferritinaemia, hepatomegaly, splenomegaly and thrombocytopenia), most of the panel members believed that mild or moderate levels were consistent with early type 1 GD or early type 3 GD. However, except for splenomegaly, the panel tended to be divided about whether severe levels of each sign were consistent with early type 1 GD or type 3 GD (Fig. 4).
Figure 4.
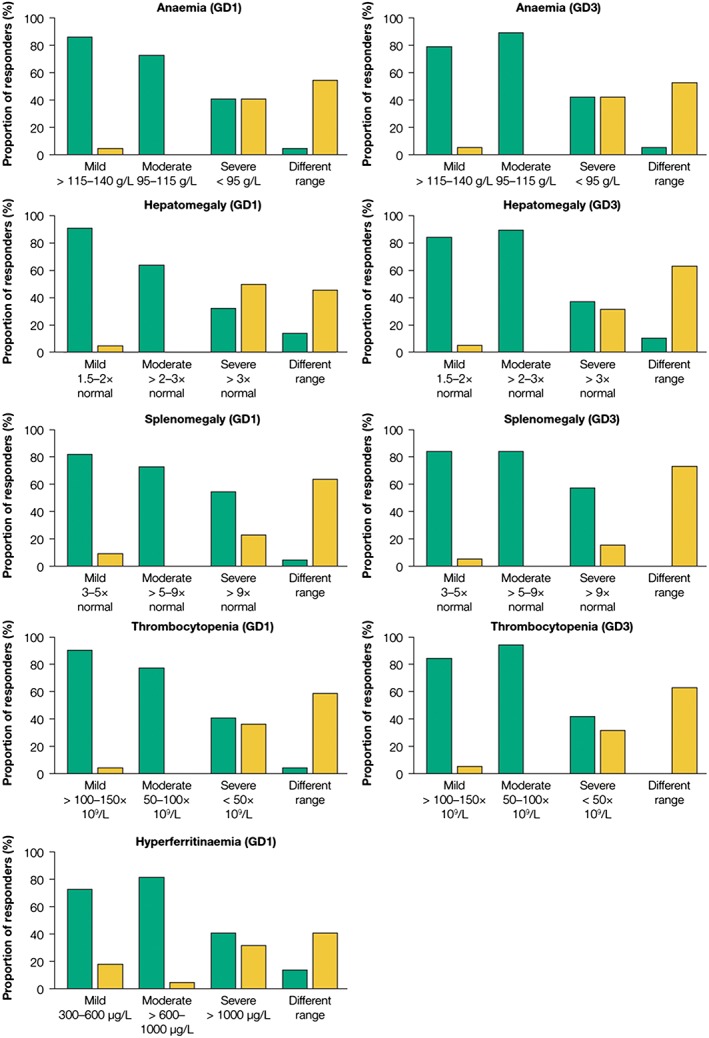
Major signs in Gaucher disease with continuous variables. Panel members were asked to indicate the levels of anaemia, hepatomegaly, splenomegaly and thrombocytopenia that were considered consistent with, or unlikely to indicate, early type 1 Gaucher disease (GD) (n = 22) or early type 3 GD (n = 19), and the levels of hyperferritinaemia consistent with, or unlikely to indicate, type 1 GD. Value ranges were proposed by the chairs and categorised as mild, moderate or severe; panel members could also nominate a different range. ( ), Consistent; (
), Consistent; ( ), unlikely.
), unlikely.
Consensus on the barriers to early GD diagnosis and the impact that the GED‐C initiative could have on clinical practice
Regarding barriers to early GD diagnosis, 47 individual phrases were provided in round 1, grouped into nine themes and consolidated into nine statements. In round 2, six statements met the importance criteria, and all met the consensus criteria in round 3. The highest‐scoring diagnostic‐barrier statement (mean score of 4.41 in round 3) was ‘Owing to its rarity, there is a lack of awareness of GD among healthcare professionals’ (Table 3).
Table 3.
Hypotheses on barriers to early diagnosis of patients with GD in clinical practice and on the impact that the GED‐C initiative could have on clinical practice as judged by an international panel of experts – GED‐C initiative Delphi rounds 2 and 3
| Round 2 | Round 3 | ||||||
|---|---|---|---|---|---|---|---|
| Importance score (Likert scale: 1–5)† | Agreement score (Likert scale: 1–5)‡ | ||||||
| Mean score | Median score | Respondents (%)§ | Mean score | Median score | Respondents (%)§ | ||
| Barrier statements | |||||||
| Importance criteria met/consensus achieved | Owing to its rarity, there is a lack of awareness of GD among healthcare professionals | 4.14 | 4 | 95 | 4.41 | 5 | 86 |
| Some early signs do not seem specific to GD, and their clinical presentation can be variable or heterogeneous | 3.91 | 4 | 100 | 4.27 | 4 | 91 | |
| Early signs are under‐recognised as characteristic of possible GD | 4.23 | 4 | 100 | 4.23 | 4 | 91 | |
| GD is not normally considered as a differential diagnosis | 4.09 | 4 | 100 | 4.18 | 4 | 86 | |
| Early signs can be mild and may be overlooked | 4.18 | 4 | 100 | 4.18 | 5 | 77 | |
| Access to diagnostic tests is poor in some countries and in some socioeconomic situations | 3.82 | 4 | 91 | 4.14 | 5 | 77 | |
| Importance criteria not met | Use of enzymatic diagnostic tests may be limited by cost or logistical barriers or by an HCP's unwillingness to request them | 3.18 | 3 | 64 | — | — | — |
| Lack of a diagnostic algorithm | 2.82 | 3 | 68 | — | — | — | |
| Geographic dispersion or socioeconomic division of families can reduce awareness of a family history of GD | 2.82 | 3 | 55 | — | — | — | |
| Impact statements | |||||||
| Importance criteria met/consensus achieved | Patients could be diagnosed earlier in the disease course, and monitored and managed appropriately to improve long‐term outcomes and quality of life | 4.18 | 4 | 95 | 4.55 | 5 | 100 |
| Earlier diagnosis would help to reduce serious or irreversible late‐onset complications, and comorbidities of the disease could be avoided or managed appropriately | 4.41 | 5 | 100 | 4.55 | 5 | 95 | |
| Healthcare professionals’ awareness of the disease might improve, and its inclusion as a differential diagnosis might avoid unnecessary invasive diagnostic procedures | 4.00 | 4 | 95 | 4.41 | 4 | 95 | |
| With earlier diagnosis, patients could be followed up from an earlier stage of disease, leading to a better understanding of disease phenotypes and progression | 3.86 | 4 | 86 | 4.36 | 5 | 86 | |
| Earlier diagnosis would allow family planning and genetic counselling to be offered earlier | 3.68 | 4 | 91 | 4.32 | 4 | 91 | |
| More rapid diagnosis would facilitate earlier decision‐making to support patients | 4.05 | 4 | 100 | 4.14 | 4 | 82 | |
| It might lead to wider use and availability of diagnostic testing | 3.73 | 4 | 86 | 3.86 | 4 | 68 | |
| Consensus not achieved | Goals for diagnosis would be clearer | 3.50 | 4 | 82 | 3.50 | 4 | 55 |
Statements that did not meet the threshold for importance (a score of ≥3 by >75% of respondents) were not taken forward to round 3 for agreement rating. Consensus was reached if statements met the threshold for agreement in round 3 (a score of ≥4 by >67% of respondents).
The Likert scale used for importance rating in round 2 was: 1 = not important; 2 = slightly important; 3 = important; 4 = very important; 5 = extremely important.
The Likert scale used for agreement rating in round 3 was: 1 = strongly disagree; 2 = disagree; 3 = neither agree nor disagree; 4 = agree; 5 = strongly agree.
Percentage of respondents (n = 22) is the proportion awarding a Likert scale score that met the selection criteria in each round.
GD, Gaucher disease; GED‐C, Gaucher Earlier Diagnosis Consensus; HCP, healthcare professional.
In response to questions about the potential impact of the initiative, 30 individual phrases were submitted and grouped into eight themes. These were consolidated into eight statements, all of which met the importance criteria in round 2. Consensus was reached for seven of the statements. The highest‐scoring impact statements (mean scores of 4.55 in round 3) were that ‘Patients could be diagnosed earlier in the disease course and monitored and managed appropriately to improve long‐term outcomes and quality of life’ and that ‘Earlier diagnosis would help to reduce serious or irreversible late‐onset complications, and comorbidities of the disease could be avoided or managed appropriately’ (Table 3).
Discussion
This international, multidisciplinary GED‐C initiative has identified a series of major and minor clinical signs and co‐variables that may facilitate the early diagnosis of GD among non‐specialist physicians and has provided important expert insights into the greatest barriers to early GD diagnosis. Collectively for type 1 and 3 GD, the GED‐C initiative identified 11 different major signs and two co‐variables indicative of early GD (Table 4). Based on these findings the authors suggest that the unexplained presence of at least two signs, or of one unexplained sign in conjunction with one of the co‐variables should cause physicians to consider GD in their differential diagnosis.
Table 4.
Summary of major signs and covariables of relevance in early Gaucher disease
| Major signs |
| Gastroenterological |
| Splenomegaly† |
| Hepatomegaly‡ |
| Orthopaedic |
| Bone pain§ |
| Kyphosis |
| General medical |
| Hyperferritinaemia‡ |
| Haematological |
| Anaemia‡ |
| Thrombocytopenia‡ |
| Gammopathy |
| Neurological¶ |
| Slow horizontal saccades with unimpaired vision |
| Impairment of primary motor development |
| Myoclonus epilepsy |
| Covariables |
| Jewish ancestry |
| Family history of GD |
GD should be included in differential diagnosis if two or more of these factors are present and unexplained, particularly if one factor is splenomegaly. The more signs and co‐variables that are present, the greater would be the suspicion of GD.
Typically this would be unexplained spleen enlargement of at least threefold, but spleen enlargement of less than threefold would not necessarily exclude GD.
Mild or moderate deviation from normal is most commonly seen with these signs in GD, but severe deviation would not exclude GD.
Bone pain is more common in early GD than are more severe bone issues such as avascular necrosis or fractures, but the presence of the latter may indicate that GD is already advanced.
Neurological signs generally only manifest in type 3 disease but are often preceded by the systemic signs listed.
GD, Gaucher disease.
The importance of considering GD increases with the number of unexplained signs and co‐variables, and investigation of the possibility of GD may require specialist referral. A specialist suspecting GD will test for low levels (typically 10–15% of normal) of lysosomal glucocerebrosidase, either in dried blot spots, which are relatively quick to prepare and submit for laboratory testing, or in total leukocytes, mononuclear cells or cultured fibroblasts, which provide a more reliable indication of deficiency.1 Diagnosis of GD is confirmed by DNA sequencing of the glucocerebrosidase gene. In the event of a positive diagnosis, specialist involvement is highly recommended to ensure that appropriate patient assessment and management plans are instigated as soon as possible.
Treatment of GD involves intravenous infusion of the deficient glucocerebrosidase enzyme every 2–4 weeks (‘enzyme‐replacement therapy’) or oral administration of medicine that reduces the amount of glucosylceramide substrate produced (‘substrate reduction’), thereby allowing any available enzyme to function more efficiently. Fortunately, not all patients with GD require disease‐modifying treatment and the optimum timing of treatment to prevent irreversible disease progression is unknown. However, delaying treatment can lead to worse outcomes, poorer prognoses18, 19 and the development of preventable late‐onset complications.11 Untreated patients with GD may have reduced quality of life relative to population norms,20 and the clinical benefits of enzyme‐replacement and substrate reduction therapies in treatment‐naïve patients with GD are well established.21, 22, 23, 24, 25, 26, 27, 28, 29
The diagnostic guidance in Table 4 is deliberately limited to major factors, but our wider findings could inform development of an algorithm that is highly selective for patients who may benefit from diagnostic GD testing. A point‐scoring system, such as that proposed in Table 5, could be developed in which weighted scores are assigned to each of the major signs and co‐variables, based on logistic regression analysis of retrospective data from patients already diagnosed with GD. Similar tools have been developed, validated prospectively and applied successfully in other disease areas.30 The arbitrary weighting values shown in Table 5 serve to illustrate the approach. Such an algorithm would expedite the appropriate use of diagnostic testing early in the disease course.
Table 5.
Prototype point‐scoring system as a screen for diagnostic testing in Gaucher disease
| Weighting | Clinical sign or co‐variable | |
|---|---|---|
| Major signs and co‐variables | 3 points | Splenomegaly (≥3× normal) |
| Disturbed oculomotor function (slow horizontal saccades with unimpaired vision) | ||
| 2 points | Thrombocytopenia, mild or moderate (platelet count, 50–150 × 109/L) | |
| Bone issues, including pain, crises, avascular necrosis and fractures | ||
| Family history of Gaucher disease | ||
| Anaemia, mild or moderate (haemoglobin, 95–140 g/L) | ||
| Hyperferritinaemia, mild or moderate (serum ferritin, 300–1000 μg/L) | ||
| Jewish ancestry | ||
| Disturbed motor function (impairment of primary motor development) | ||
| Hepatomegaly, mild or moderate (≤3× normal) | ||
| Myoclonus epilepsy | ||
| Kyphosis | ||
| Gammopathy – monoclonal or polyclonal | ||
| 1 point | Anaemia, severe (haemoglobin, <9.5 g/dL) | |
| Hyperferritinaemia, severe (serum ferritin, >1000 μg/L) | ||
| Hepatomegaly, severe (>3× normal) | ||
| Thrombocytopenia, severe (platelet count, <50 × 109/L) | ||
| Minor signs and co‐variables | 0.5 points | Gallstones |
| Bleeding, bruising or coagulopathy | ||
| Leukopenia | ||
| Cognitive deficit | ||
| Low bone mineral density | ||
| Growth retardation including low body weight | ||
| Asthenia | ||
| Cardiac calcification | ||
| Dyslipidaemia | ||
| Elevated angiotensin‐converting enzyme levels | ||
| Fatigue | ||
| Pulmonary infiltrates | ||
| Age ≤18 years | ||
| Family history of Parkinson disease | ||
| Blood relative who died of foetal hydrops and/or with diagnosis of neonatal sepsis of uncertain aetiology |
The weighting scores shown are arbitrary and will need to be validated. Major clinical signs and co‐variables were provisionally awarded a score of 2 points, and minor signs and co‐variables a score of 0.5 points. Given that 100% consensus was reached for the signs splenomegaly and disturbed oculomotor function (slow horizontal saccades with unimpaired vision) they were assigned a score of 3 points. Severe levels of the anaemia, hepatomegaly, hyperferritinaemia and thrombocytopenia were deemed more likely to indicate other pathologies than Gaucher disease but did not exclude it, so these were awarded a score of 1 point. Subject to validation in patient data, the scoring system will be used in the form of an online calculator to generate a total score based on a patient's presenting signs. The value of this total score will dictate whether diagnostic testing is recommended.
The signs identified during this initiative were consistent with those reported in the published literature. There was 100% consensus that splenomegaly was a major sign of both type 1 and type 3 GD, and splenomegaly was the single most‐often‐reported GD sign among abstracts identified in our literature search (Appendix S9). Among these was a longitudinal analysis of data from a large number of children and adolescents diagnosed with type 1 GD and registered in the International Collaborative Gaucher Group (ICGG) Gaucher Registry, which showed that splenomegaly was the most common presenting sign, reported in 95% of patients.31 Similarly, the five next most common presenting signs reported in our literature search aligned with those identified by the GED‐C expert panel, namely neurological abnormalities, bone abnormalities (including pain, fracture, crises and avascular necrosis), hepatomegaly, thrombocytopenia and anaemia. Thus, the available evidence strongly suggests that our consensus on major signs in early GD is robust, valid and representative of what is observed in clinical practice. In the ICGG analysis, the most common patient‐reported bone symptom was pain; skeletal abnormalities, such as fractures, and avascular necrosis were rare.31 Such signs were grouped as ‘bone issues’ in our survey because the presence of more severe signs of bone disease is consistent with GD, but ideally our consensus would identify patients before such signs were apparent. Some signs, such as gammopathy and hyperferritinaemia, were reported rarely or not at all in our literature search.
The barriers to earlier diagnosis of GD identified are also supported by the literature, one of the greatest challenges being a lack of awareness of GD among healthcare professionals, owing to its relative rarity.9, 32 Furthermore, the heterogeneous nature of GD (patients who are asymptomatic or who have only mild signs, disease progression at different rates in different tissues, or variation between disease phenotypes) also impedes its inclusion in differential diagnosis.9, 14 An international survey of haematologists and oncologists (n = 406) found that only 20% of those surveyed considered GD as a differential diagnosis in patients presenting with typical GD signs (e.g. bone pain, cytopenia, hepatomegaly and splenomegaly); leukaemia, lymphoma and multiple myeloma were considered the most likely diagnoses.11
Limitations
Delphi methodology is widely used for gathering and processing data from experts to achieve convergence of opinion on a specific real‐world issue, often in the absence of sufficient evidence to provide adequate guidance.15, 16, 17 The technique has been used to generate simple, robust, expert experience‐based consensus in a variety of disease settings to improve diagnosis.33, 34, 35, 36, 37, 38 An analysis of the relationship between the characteristics of Delphi surveys and variability in the consensus obtained found there was a little associated variation in consensus indices if the number of questions was in the range 6–40 (the maximum number in our survey was 26 in round 1); similarly, variability was modest if the number of respondents was in the range 6–50 (22 respondents to our survey).16 Although the Delphi technique is designed to minimise potential noise and data distortion often associated with conventional group interactions,15, 16, 17 it can have drawbacks. Regression to the mean is almost inevitable in a consensus exercise and could dilute insightful or nuanced responses offered by only a minority of participants. However, the technique eliminates individual bias, and a generalised approach is probably appropriate when creating non‐specialist guidance.
Conclusion
The GED‐C initiative has identified key clinical signs and co‐variables that are potentially indicative of type 1 GD and of type 3 GD in its early stages. Clinicians who are not specialists in GD can use this guidance to determine whether GD needs to be considered in the differential diagnosis of a patient and, when it is a possibility, arrange lysosomal glucocerebrosidase testing and specialist involvement as soon as possible. Facilitating early diagnosis of GD may ultimately lead to improved patient quality of life and outcomes.
Supporting information
Appendix S1. GED‐C Delphi process round 1 questionnaire.
Appendix S2. GED‐C Delphi process round 2 questionnaire.
Appendix S3. GED‐C Delphi process round 3 questionnaire.
Appendix S4. Literature review of presenting signs in GD at diagnosis.
Appendix S5. GED‐C Delphi round 1 results.
Appendix S6. GED‐C Delphi round 1 results.
Appendix S7. GED‐C Delphi round 1 results.
Appendix S8. GED‐C Delphi round 1 results.
Appendix S9. Literature review findings: GD signs at diagnosis.
Acknowledgements
The authors thank Dr Mark Rolfe, Oxford PharmaGenesis Ltd, Oxford, UK, for providing administrative support of the GED‐C initiative and for medical writing support in preparation of this manuscript. The authors chose to work with Oxford PharmaGenesis Ltd independently, without input from Shire International GmbH. This initiative was endorsed by the European Hematology Association Scientific Working Group ‘Quality of Life and Symptoms’.
Funding: This research was funded by unrestricted independent medical education grants (CME‐GBR‐12915 and IME‐GBR‐14397) from Shire International GmbH.
Conflict of interest: A. Mehta has received consulting fees from Amicus, Pfizer, Sanofi Genzyme and Shire, and has received research funding from Amicus, Protalix, Rigel, Sanofi Genzyme and Shire. D. J. Kuter has received consulting fees and research funding from Actelion, Genzyme, Protalix/Pfizer and Shire. S. S. Salek has received consulting fees and travel grants from Servier and Shire, and has received research funding from Bristol‐Myers Squibb, Novartis and Sanofi. N. Belmatoug has received fees for lectures, travel reimbursement and participation on advisory boards and scientific committees from Sanofi Genzyme and Shire. B. Bembi has received consulting fees and/or research support from Actelion, Genzyme and Shire. J. Bright was an employee of Oxford PharmaGenesis Ltd at the time this work was conducted. S. vom Dahl has received consulting fees, research funding and travel reimbursement from Actelion, Genzyme, Protalix/Pfizer and Shire. F. Deodato has received fees from Actelion, Sanofi Genzyme and Shire for lectures, participation on advisory boards and travel reimbursement. M. Di Rocco has received honoraria from Sanofi Genzyme and Shire. O. Göker‐Alpan has received consulting fees from Actelion, Genzyme, Pfizer and Shire, and research support from Alexion, Amicus, Genzyme, Pfizer and Shire. D. A. Hughes has received consultancy fees, honoraria and research support from Actellion, Amicus, Protalix, Sanofi Genzyme and Shire. E. A. Lukina has received honoraria from Sanofi Genzyme and Shire. M. Machaczka has received honoraria from Sanofi Genzyme and Shire. E. Mengel has received honoraria and/or consulting fees from Actelion, Alexion, BioMarin, Orphazyme, Sanofi Genzyme and Shire. A. Nagral has received travel reimbursement from Sanofi Genzyme. K. Nakamura has received honoraria from Shire and research funding from Sanofi Genzyme and Shire. A. Narita has received research funding from Actelion, Sanofi Genzyme and Shire and fees for attending advisory boards from Actelion, BioMarin, Sanofi Genzyme and Shire. B. Oliveri has received honoraria from GlaxoSmithKline and Shire. G. Pastores has received honoraria and/or consulting fees from BioMarin and Shire. J. Pérez‐López has received honoraria and/or consulting fees from BioMarin, Sanofi Genzyme and Shire. U. Ramaswami has received travel, research grants and/or honoraria from Amicus, Alexion, Genzyme, Protalix and Shire. I. Schwartz has received honoraria from Shire. J. Szer has received honoraria from Alexion and travel grants from Pfizer, Sanofi Genzyme and Shire. N. J. Weinreb has received honoraria and research support from Genzyme‐Sanofi, Pfizer and Shire. A. Zimran has received consulting fees and/or research support from Genzyme, Pfizer and Shire.
References
- 1. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017; 18: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pastores GM, Hughes DA. Gaucher disease In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, et al., eds. GeneReviews. Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]
- 3. Mistry PK, Belmatoug N, vom Dahl S, Giugliani R. Understanding the natural history of Gaucher disease. Am J Hematol 2015; 90(Suppl 1): S6–11. [DOI] [PubMed] [Google Scholar]
- 4. Mignot C, Gelot A, Bessieres B et al. Perinatal‐lethal Gaucher disease. Am J Med Genet A 2003; 120A: 338–44. [DOI] [PubMed] [Google Scholar]
- 5. Azuri J, Elstein D, Lahad A, Abrahamov A, Hadas‐Halpern I, Zimran A. Asymptomatic Gaucher disease implications for large‐scale screening. Genet Test 1998; 2: 297–9. [DOI] [PubMed] [Google Scholar]
- 6. Grabowski GA. Lysosomal storage diseases In: Braunwald E. and Fauci AS, eds. Harrison's Principles of Internal Medicine, 15th edn. New York, NY: McGraw‐Hill; 2001; 2276–81. [Google Scholar]
- 7. Morales LE. Gaucher's disease: a review. Ann Pharmacother 1996; 30: 381–8. [DOI] [PubMed] [Google Scholar]
- 8. Taddei TH, Kacena KA, Yang M, Yang R, Malhotra A, Boxer M et al. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hematol 2009; 84: 208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cassinerio E, Graziadei G, Poggiali E. Gaucher disease: a diagnostic challenge for internists. Eur J Intern Med 2014; 25: 117–24. [DOI] [PubMed] [Google Scholar]
- 10. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on‐going need for increased awareness amongst haematologists. Blood Cells Mol Dis 2013; 50: 212–17. [DOI] [PubMed] [Google Scholar]
- 11. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists‐oncologists and an opportunity for early diagnosis and intervention. Am J Hematol 2007; 82: 697–701. [DOI] [PubMed] [Google Scholar]
- 12. Andrade‐Campos M, Alfonso P, Irun P, Armstrong J, Calvo C, Dalmau J et al. Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national‐base study from the Spanish Registry of Gaucher disease. Orphanet J Rare Dis 2017; 12: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Rocco M, Andria G, Deodato F, Giona F, Micalizzi C, Pession A. Early diagnosis of Gaucher disease in pediatric patients: proposal for a diagnostic algorithm. Pediatr Blood Cancer 2014; 61: 1905–9. [DOI] [PubMed] [Google Scholar]
- 14. Mistry PK, Cappellini MD, Lukina E, Özsan H, Mach Pascual S, Rosenbaum H et al. A reappraisal of Gaucher disease – diagnosis and disease management algorithms. Am J Hematol 2011; 86: 110–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milholland AV, Wheeler SG, Heieck JJ. Medical assessment by a Delphi group opinion technic. N Engl J Med 1973; 288: 1272–5. [DOI] [PubMed] [Google Scholar]
- 16. Birko S, Dove ES, Ozdemir V. Evaluation of nine consensus indices in Delphi foresight research and their dependency on Delphi survey characteristics: a simulation study and debate on Delphi design and interpretation. PLoS One 2015; 10: e0135162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsu CC, Sandford BA. The Delphi technique: making sense of consensus. Pract Assess Res Eval 2007; 12: 1–8. [Google Scholar]
- 18. Arikan‐Ayyildiz Z, Yuce A, Emre S, Baysoy G, Saltik‐Temizel IN, Gurakan F. Outcome of enzyme replacement therapy in Turkish patients with Gaucher disease: does late intervention affect the response? Turk J Pediatr 2011; 53: 499–507. [PubMed] [Google Scholar]
- 19. Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol 2009; 147: 561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weinreb N, Barranger J, Packman S, Prakash‐Cheng A, Rosenbloom B, Sims K et al. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet 2007; 71: 576–88. [DOI] [PubMed] [Google Scholar]
- 21. Ibrahim J, Underhill LH, Taylor JS, Angell J, Peterschmitt MJ. Clinical response to eliglustat in treatment‐naive patients with Gaucher disease type 1: post‐hoc comparison to imiglucerase‐treated patients enrolled in the International Collaborative Gaucher Group Gaucher Registry. Mol Genet Metab Rep 2016; 8: 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith L, Rhead W, Charrow J, Shankar SP, Bavdekar A, Longo N et al. Long‐term velaglucerase alfa treatment in children with Gaucher disease type 1 naive to enzyme replacement therapy or previously treated with imiglucerase. Mol Genet Metab 2016; 117: 164–71. [DOI] [PubMed] [Google Scholar]
- 23. Zimran A, Duran G, Mehta A et al. Long‐term efficacy and safety results of taliglucerase alfa up to 36 months in adult treatment‐naive patients with Gaucher disease. Am J Hematol 2016; 91: 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ben Turkia H, Gonzalez DE, Barton NW et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol 2014; 88: 179–84. [DOI] [PubMed] [Google Scholar]
- 25. Gonzalez DE, Turkia HB, Lukina EA, Kisinovsky I, Dridi MFB, Elstein D et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: results from a randomized, double‐blind, multinational, phase 3 study. Am J Hematol 2013; 88: 166–71. [DOI] [PubMed] [Google Scholar]
- 26. Zimran A, Brill‐Almon E, Chertkoff R et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2012; 118: 5767–73. [DOI] [PubMed] [Google Scholar]
- 27. Kuter DJ, Mehta A, Hollak CE et al. Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells Mol Dis 2013; 51: 116–24. [DOI] [PubMed] [Google Scholar]
- 28. Zimran A, Wang N, Ogg C, Crombez E, Cohn GM, Elstein D. Seven‐year safety and efficacy with velaglucerase alfa for treatment‐naive adult patients with type 1 Gaucher disease. Am J Hematol 2015; 90: 577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sims KB, Pastores GM, Weinreb NJ, Barranger J, Rosenbloom BE, Packman S et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48‐month longitudinal cohort study. Clin Genet 2008; 73: 430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia‐Manero G, Sole F et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012; 120: 2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med 2006; 160: 603–8. [DOI] [PubMed] [Google Scholar]
- 32. Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: a Q&A expert roundtable discussion. Clin Adv Hematol Oncol 2012; 10(Suppl 8): 1–16. [PubMed] [Google Scholar]
- 33. Sherman PM, Hassall E, Fagundes‐Neto U, Gold BD, Kato S, Koletzko S et al. A global, evidence‐based consensus on the definition of gastroesophageal reflux disease in the pediatric population. Am J Gastroenterol 2009; 104: 1278–95. [DOI] [PubMed] [Google Scholar]
- 34. Lee JH, Park MS, Kwon H, Chung CY, Lee KM, Kim YJ et al. A guideline for differential diagnosis between septic arthritis and transient synovitis in the ED: a Delphi survey. Am J Emerg Med 2016; 34: 1631–6. [DOI] [PubMed] [Google Scholar]
- 35. Fox AR, Gordon LK, Heckenlively JR, Davis JL, Goldstein DA, Lowder CY et al. Consensus on the diagnosis and management of nonparaneoplastic autoimmune retinopathy using a modified Delphi approach. Am J Ophthalmol 2016; 168: 183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eubank BH, Mohtadi NG, Lafave MR, Wiley JP, Bois AJ, Boorman RS et al. Using the modified Delphi method to establish clinical consensus for the diagnosis and treatment of patients with rotator cuff pathology. BMC Med Res Methodol 2016; 16: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomkins‐Lane C, Melloh M, Lurie J, Smuck M, Battié MC, Freeman B et al. ISSLS prize winner: consensus on the clinical diagnosis of lumbar spinal stenosis: results of an international Delphi study. Spine (Phila Pa 1976) 2016; 41: 1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maher TM, Whyte MK, Hoyles RK et al. Development of a consensus statement for the definition, diagnosis, and treatment of acute exacerbations of idiopathic pulmonary fibrosis using the Delphi technique. Adv Ther 2015; 32: 929–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. GED‐C Delphi process round 1 questionnaire.
Appendix S2. GED‐C Delphi process round 2 questionnaire.
Appendix S3. GED‐C Delphi process round 3 questionnaire.
Appendix S4. Literature review of presenting signs in GD at diagnosis.
Appendix S5. GED‐C Delphi round 1 results.
Appendix S6. GED‐C Delphi round 1 results.
Appendix S7. GED‐C Delphi round 1 results.
Appendix S8. GED‐C Delphi round 1 results.
Appendix S9. Literature review findings: GD signs at diagnosis.