

Abstract
Based on the individual genetic profile, acute myeloid leukemia (AML) patients are classified into clinically meaningful molecular subtypes. However, the mutational profile within these groups is highly heterogeneous and multiple AML subclones may exist in a single patient in parallel. Distinct alterations of single cells may be key factors in providing the fitness to survive in this highly competitive environment. Although the majority of AML patients initially respond to induction chemotherapy and achieve a complete remission, most patients will eventually relapse. These points toward an evolutionary process transforming treatment‐sensitive cells into treatment‐resistant cells. As described by Charles Darwin, evolution by natural selection is the selection of individuals that are optimally adapted to their environment, based on the random acquisition of heritable changes. By changing their mutational profile, AML cell populations are able to adapt to the new environment defined by chemotherapy treatment, ultimately leading to cell survival and regrowth. In this review, we will summarize the current knowledge about clonal evolution in AML, describe different models of clonal evolution, and provide the methodological background that allows the detection of clonal evolution in individual AML patients. During the last years, numerous studies have focused on delineating the molecular patterns that are associated with AML relapse, each focusing on a particular genetic subgroup of AML. Finally, we will review the results of these studies in the light of Darwinian evolution and discuss open questions regarding the molecular background of relapse development.
Keywords: acute myeloid leukemia, clonal evolution, next generation sequencing, relapse, therapy resistance
1. INTRODUCTION
“According to Darwin's Origin of Species, it is not the most intellectual of the species that survives; it is not the strongest that survives; but the species that survives is the one that is able best to adapt and adjust to the changing environment in which it finds itself.” A 100 years after Charles Darwin's “On the Origin of Species”, Leon C. Megginson, business professor at Louisiana State University, shared this famous interpretation of Darwin's theory about the evolution by natural selection.1, 2 Today, this theory is not only applicable to the evolution of organisms, but also to the evolution of tumor diseases and the origin of distinct tumor populations in a human individual.
Darwinian evolution by natural selection is described as the result of a two‐step process: (a) the acquisition of heritable changes affecting an organism's physical or behavioral traits, that (b) allow the organism to better adapt to its environment, help it to survive and thereby increase the number of offspring. Evolution by natural selection is often referred to as “survival of the fittest,” whereas the effect on the fitness by a particular change always depends on the underlying selective pressure, which is given by the surroundings. Thereby, organisms as well as single cells might be perfectly adapted to a particular niche, whereas in a different environment, they would not be able to survive.
In the past, rules of Darwinian evolution have been applied to individual cells and cell populations during their progression to cancer,3, 4, 5 including the malignant transformation of hematopoietic progenitor cells to acute myeloid leukemia (AML).6, 7, 8, 9 The advent of next generation sequencing (NGS) techniques has greatly increased the detection of genetic events that are associated with tumor development and progression. In this review, we will summarize and discuss the results from recent NGS‐based studies of AML evolution from diagnosis to relapse and the development of therapy resistance. We will focus on different theoretical models of evolution, the dynamics of evolution, and the methodology to uncover evolutionary events including its strengths, drawbacks, and limitations.
2. MODELS OF CLONAL EVOLUTION OF AML
AML is the most common acute leukemia in adults and a highly aggressive disease with poor outcome. Although the prognosis of AML patients has improved over the last decades, only one of four patients survives 5 years or longer.10 In AML, immature blast cells reside in the bone marrow, peripheral blood, and extramedullary tissues, unable to differentiate. These cells are rapidly growing and continuously dividing, lacking maturation and function, thereby suppressing normal hematopoiesis. Even though two‐thirds of AML patients respond to induction chemotherapy and achieve complete remission, the majority of these patients will eventually relapse.11 AML induction chemotherapy has not changed profoundly over the last decades, and the molecular mechanisms mediating the transition of AML cells from therapy sensitive toward resistant are still not fully understood.12, 13 Although several studies have shown relapse‐specific gene mutations in individual cohorts of AML patients, no gene mutation was reported to be recurrently gained, while rarely or never lost at relapse across multiple studies. Mutations in DNMT3A, ASXL1, and RUNX1 as well as internal tandem duplications in the FLT3 gene (FLT3‐ITD) were frequently gained at relapse and associated with poor outcome if present at diagnosis, however, patients with these mutations may still respond to chemotherapy and achieve a complete remission. AML is a genetically heterogeneous disorder, which is characterized by mutations in a variety of genes, encoding for myeloid transcription factors, tumor suppressor genes, epigenetic modifiers, and splicing factors, altering normal hematopoietic function of myeloid progenitor cells. In total, more than 60 genes have been described to be recurrently mutated in AML and we are far from knowing AML genetics in detail as leukemias are highly heterogeneous not just between patients but also within patients.14, 15, 16 Moreover, the molecular profile of AML is changing during the disease, as multiple studies have described genetic and epigenetic evolution of AML from diagnosis to relapse. It is believed that AML originates from a single hematopoietic stem or progenitor cell (HSPC) acquiring somatic mutations over time that lead to a block of differentiation but also provide stem‐cell like properties of unrestricted self‐renewal, thus enabling mutated HSPCs to grow clonally.17, 18 Prior to leukemia‐initiating events, regulators of epigenetic marks (eg, DNMT3A, TET2, and ASXL1) commonly acquire mutations, which may provide a growth advantage, yet they are not sufficient to induce leukemia, and thus commonly described as preleukemic.19, 20, 21 Initiating mutations that provide leukemic potential are often found in the gene nucleophosmin (NPM1) and genes associated with signaling activation, for example, FLT3.14, 22 Each mutation adds to the genetic complexity and finally, increased clonal heterogeneity is associated with inferior outcome of AML.23, 24
During the disease, individual AML populations may follow distinct models of clonal evolution and the presence and the abundance of mutations at multiple time points paints a picture of dynamic changes. Linear evolution describes the stepwise acquisition of single mutations, whereas the eradication of the dominant clone, followed by outgrowth of a subclone is termed branching evolution (Figure 1). Without any change in the evolutionary pressure, cell populations are likely to follow a linear evolution, steadily increasing their fitness. Still, branching evolution may follow linear evolution, or vice versa, especially when the evolutionary pressure has changed profoundly (eg, at treatment start or change of therapy). During therapy, the AML cell population may evolve by either acquiring additional mutations mediating therapy resistance (linear evolution, Figure 1A), or by losing mutations that are for example, associated with sensitivity to the treatment (branching evolution, Figure 1B). In summary, AML cell populations at relapse may have evolved from either clonal or subclonal cell populations present at diagnosis, accompanied by potential acquisition of additional mutations. Following either linear or branching evolution, cell populations are steadily undergoing clonal evolution in order to the best adapt to their environment. As the majority of AML patients relapse after initial response to chemotherapy, in most patients a few AML cells find a niche to escape from therapy and eventually grow out again.
Figure 1.
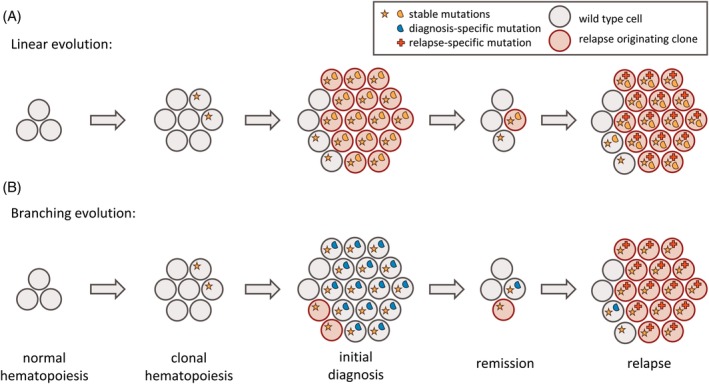
Models of clonal evolution of AML over time. Each circle corresponds to an individual cell, multiple identical cells correspond to cell clones, each defined by harboring the identical set of mutations. Cells without symbols refer to wild type cells without somatic mutations. Somatic mutations are represented by different symbols, colored based on their stability: orange (stable mutation), blue (diagnosis‐specific mutation), and red (relapse‐specific mutation). Cells of the relapse originating clone are highlighted in red. (A) Linear evolution describes the sequential acquisition of mutations. The relapse originating cell is part of the major clone at diagnosis. (B) Branching evolution describes the eradication of the major clone and subsequent outgrowth of a secondary clone. The relapse originating cell is part of a subclone at diagnosis. AML, acute myeloid leukemia
In an individual patient, the underlying model of clonal evolution can be assessed by comparing the abundance of each single mutation at multiple time points. In linear evolution, mutations of the major clone present at diagnosis are also present at relapse, accompanied by additional mutations (Figure 2A). Cells of the major clone were sensitive to the treatment, but residual cells have acquired new mutations that provide resistance resulting in therapy escape and development of relapse. New mutations can be both, driver mutations actively providing a growth advantage, or passenger mutations that were already present in a cell prior to the acquisition of a driver mutation. As during each step of linear evolution, a new clone evolves, both the ancestor and the descendent are sharing their particular mutational background except for the new mutations, including drivers and passengers. During linear evolution, mutations of the major clone are unlikely to get lost, as mutations reverting a mutated allele back to its wild type configuration are very rare events. In contrast, the loss of a mutation is a hallmark of branching evolution. The main clone at diagnosis disappears after treatment and a new clone that is resistant to the therapy is found at relapse. If no additional mutations occur at relapse, this clone likely was present at diagnosis at very low levels (Figure 2B). If additional mutations are detected at relapse, the clone has evolved from a common ancestor that was present at diagnosis (Figure 2C). In neither of the two cases, the relapse has evolved from the dominant clone at diagnosis itself. While initial and relapsed disease may share mutations, branched evolution is characterized by a loss of mutations at relapse.
Figure 2.
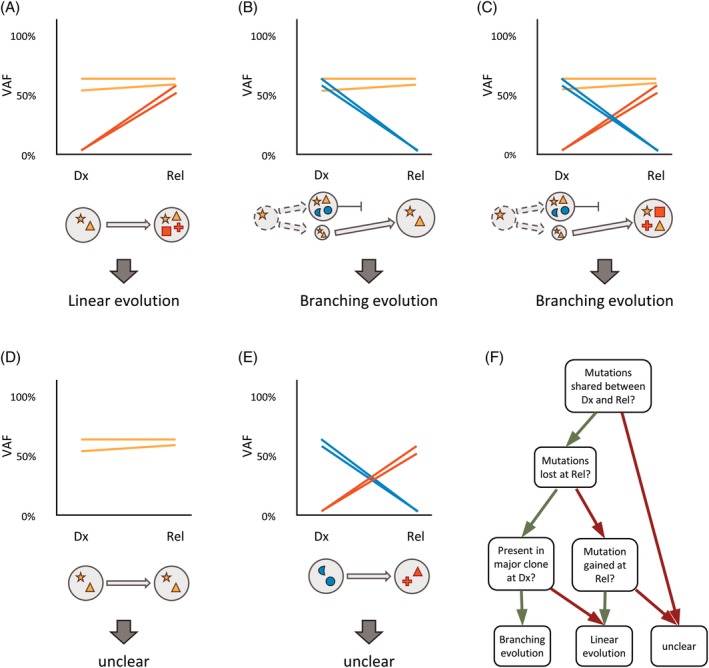
Examples of clonal evolution between diagnosis and relapse in a single patient. Each line corresponds to an individual mutation and illustrates the presence of the mutation at both time points, colored by its stability. Each circle corresponds to an individual cell clone, defined by harboring the identical set of mutations. Somatic mutations are represented by different symbols, colored based on their stability. Cells without symbol refer to wild type cells without somatic mutations. VAF, variant allele frequency; Dx, diagnosis; Rel, relapse; orange, stable at Dx and Rel; blue, lost at Rel; red, gained at Rel. (A) Genetic evolution with stable mutations and gained mutations. (B) Genetic evolution with stable mutations and lost mutations. (C) Genetic evolution with stable mutations, gained mutations, and lost mutations. (D) Genetic evolution with stable mutations only. (E) Genetic evolution with gained mutations and lost mutations only. (F) Decision tree to define the underlying model of genetic evolution
It is important to distinguish between clonal (present in the majority of cells, ie, the dominant clone) and subclonal mutations (present in a minority of cells). In contrast to the loss of a clonal mutation, the loss of a subclonal mutation does not necessarily imply branching evolution. In this case, a subclone, harboring a particular mutation, is eradicated at a later time point. On the other hand, the major clone is still present and evolves linearly. As linear and branching evolution are defined by the clonal origin of the relapse cell population (clonal or subclonal), it may not be possible to decipher the underlying model of evolution if multiple clones with similar size coexist at diagnosis. The classification of a mutation as clonal or subclonal is derived from the variant allele frequency (VAF), as it provides information about how many cells in a sample carry a particular variant. The VAF is defined as the ratio of sequence reads carrying the mutation to the total number of reads at a given nucleotide position.
As genetic evolution is defined by changes in the mutational profile between diagnosis and relapse, the model of evolution remains unclear in AML patients without detectable differences. If mutations are shared at both time points, relapsed disease is related to the initial disease and has evolved from it (Figure 2D). In contrast, if the mutational profiles at diagnosis and at relapse are completely different, it is unclear if relapse has evolved from the original disease, or if it is an independent, secondary AML (Figure 2E). However, a single shared mutation might not be sufficient to draw any conclusions about genetic evolution, as identical mutations can be found in different, independent malignancies (including solid tumors) such as mutations in KRAS (hotspot at residues G12 and G13), NRAS (hotspots G12, G13, and Q61), and BRAF (hotspot V600). In summary, simple rules can be applied in order to describe clonal evolution from diagnosis to relapse in a particular AML patient (Figure 2F). The presence of common mutations defines the relationship of initial and relapsed AML and the loss of a clonal mutation defines branching evolution, while mutation gains are possible in both, linear and branching evolution. As the loss of a clonal mutation is the hallmark of branching evolution, it is necessary to include as many genomic target regions as possible in order to describe the evolutionary model appropriately.
Finally, clonal evolution occurs at multiple levels. Relapsed AML must not necessarily differ genetically from initial AML. Clonal evolution may also take place on the epigenetic level, when different epigenetic profiles confer therapy resistance. Changes in epigenetic profiles may be detected between diagnosis and relapse using appropriate methods. A recent study suggests that both genetic and epigenetic evolution may complement each other, and thus a lack of genetic evolution can be compensated by increased epigenetic evolution.25
3. AML RELAPSE AFTER CHEMOTHERAPY
Since the first AML genomes were sequenced about 10 years ago,26, 27 numerous researchers have made efforts to catalogue the genetic lesions in AML introducing a huge number of genetic subclasses of AML with distinct clinical characteristics such as response to therapy and risk of relapse.15, 16, 22 By comparing diagnostic and relapse samples, differences in the mutational landscape can be detected and recurrent changes are very likely to contribute to the development of resistance. In about 30%‐40% of AML cases, cytogenetic alterations at relapse not present at initial diagnosis were described as potential mechanism of AML relapse.28, 29, 30, 31 In the majority of these cases, a more complex karyotype was detected at relapse, which is generally associated with inferior survival. However, in <10% of AML cases, the karyotype found at relapse was described to be less complex. Trisomies 8 and 21, as well as deletions affecting the long arm of chromosome 9 are recurrently gained at relapse, still their association with therapy resistance remains unclear, as these alterations are not described as prognostically relevant if present at diagnosis.32, 33, 34 In contrast, deletions of the long arm of chromosomes 5 or 7 are both reported to be recurrently gained at relapse and associated with poor outcome at diagnosis.16, 35 Moreover, NGS enabled the investigation of AML relapse at single nucleotide resolution. Based on whole genome sequencing (WGS) of paired diagnosis/relapse samples, AML relapse has been described to evolve either from the initial founding clone or from a subclone present at diagnosis.36 Over the past years, numerous studies have delineated the molecular patterns associated with AML relapse (Table 1).
Table 1.
| Publication | AML subtype | N total patients | N matched Dx/Rel | Sequencing strategy | Models of evolution | Dx associated mutations | Rel associated mutations |
|---|---|---|---|---|---|---|---|
| Garg et al.,37 Blood 2015 | AML with FLT3‐ITD | 80 |
WES: 13 (+CR), GPS: 37 (+CR) |
Initial WES, extended GPS | Linear (n = 2), branching (n = 5) | NPM1, CEBPA, GATA2, SRCAP | SETD1A, ASXL1 |
| Madan et al.,38 Leukemia 2016 | APL | 212 |
WES: 8 (+CR), GPS:22 (‐CR) |
Initial WES, extended GPS | Linear (n = 5), branching (n = 1) | FLT3‐SNV, NRAS, KRAS | PML, RARA, RUNX1, ARID1B |
| Sood et al.,39 Leukemia 2016 | AML with inv(16) or t(8;21) | 13 |
10 (+CR), 3 (‐CR) |
WES | Linear (n = 7), branching (n = 4) | NRAS | GATA2 deletions‐ |
| Sun et al.,40 Leukemia 2017 | AML with MLL‐PTD | 85 |
WES: 5 (+CR), GPS: 8 (‐CR) |
Initial WES, extended GPS | NA | STAG2 | FAT1 |
|
Greif et al.,41 Clin Can Res 2018 |
CN‐AML | 50 | 50 (+CR) | WES, validated by GPS | Linear (n= 33), branching (n = 11) | NPM1, FLT3‐SNV, PTPN11, NRAS | FLT3‐ITD, IDH1, WT1, KDM6A |
|
Höllein et al.,43 Blood Adv 2018 |
AML with NPM1 mut | 104 | 11 (‐CR) | GPS | NA | NPM1, PTPN11 | RUNX1, TP53 |
| Christen et al.,44 Blood 2019 | AML with t(8;21) | 331 | 19 (+CR) | GPS | NA | ASXL2 | G2E3 |
| Höllein et al.,45 Hemasphere 2019 | AML with RUNX1‐RUNX1T1 | 94 | 17 (‐CR) | GPS | NA | ASXL1, ASXL2, NRAS | KIT |
|
Cocciardi et al.,42 Nat Commun 2019 |
AML with NPM1 mut | 129 |
WES: 20 (+CR), GPS: 109 (‐CR) |
Initial GPS, selected WES | Linear (n = 5), branching (n = 15) | NPM1, NRAS, FLT3‐SNV | FLT3‐ITD, MLL‐PTD, RUNX1 |
Note: Gene symbols in bold represent common events reported at least in two independent cohorts.
Abbreviations: Dx, diagnosis; Rel, relapse; WES, whole exome sequencing; GPS, gene panel sequencing; CR, complete remission.
These studies have focused mostly on well‐defined and clinically meaningful molecular subclasses of AML. The number of genes analyzed ranged from as little as five by targeted gene panel sequencing (GPS) up to >20 000 by whole exome sequencing (WES). The number of paired patient samples collected at time points of initial diagnosis and relapse after chemotherapy ranged from 11 to 129 (GPS) and 5 to 50 (WES), respectively (Table 1). WES is commonly applied to a limited number of matched diagnosis/relapse patient samples, while targeted GPS is usually performed in larger cohorts in order to estimate the frequency of gene mutations. To detect the frequency of individual mutations at diagnosis and/or relapse, it might be sufficient to collect any diagnosis and relapse samples, even if they do not stem from the same individuals. However, to draw meaningful conclusions about the clonal evolution from diagnosis to relapse, it is imperative to sequence longitudinal samples from the same patients. So far, most studies focused on the detection of mutation frequencies rather than the longitudinal analysis of individual mutations. Moreover, as GPS studies are focusing on mutations that are well‐known to be acquired somatically in AML, germline control samples are usually not included. In WES studies, the larger number of detected mutations, including passenger mutations, helps to assess clonal evolution more precisely. Thus, in this review, the estimation of linear vs branching evolution in individual cohorts was limited to WES cases only. However, to assign the somatic status of mutations detected by WES, it is necessary to include a germline control sample, for example, a sample at complete remission. Due to limited sample availability and increased sequencing costs, WES studies commonly include a limited number of patients only.
Several studies have applied WES in order to genetically characterize AML at diagnosis and relapse (Table 1). In 2015, the genetic profiles of 13 matched diagnosis/remission/relapse triplets of AML patients carrying an FLT3‐ITD were described based on WES analysis. Further, an additional 67 patients including 37 diagnosis/relapse pairs were analyzed using GPS, covering a total of 50 paired diagnosis/relapse sample pairs.37 In another study from the same group, 12 relapsed acute promyelocytic leukemia (APL) patients were analyzed using WES (including eight diagnosis/remission/relapse triplets), followed by the comparison of 153 relapsed APL cases with 69 non‐relapsed APL cases using GPS (including 30 diagnosis/relapse pairs).38 Sood and colleagues described the mutational landscape of relapsed core binding factor (CBF) leukemia, defined by either inv(16) or t(8;21) and a total of 13 patients was characterized by WES, including 10 diagnosis/remission/relapse triplets and three diagnosis/relapse pairs.39 AML relapse with partial tandem duplication of the MLL gene (MLL‐PTD) was evaluated by sequencing samples from 85 patients. Five diagnosis/remission/relapse triplets were analyzed by WES and eight diagnosis/relapse pairs were analyzed by GPS.40 Relapse of the largest cytogenetic subgroup of AML, patients with normal karyotype, was characterized by 50 matched diagnosis/remission/relapse triplets using WES, followed by GPS for variant validation.41 Very recently, Cocciardi et al. described the mutational spectrum at diagnosis and relapse of NPM1 mutated AML in 129 cases using GPS of five commonly mutated genes (DNMT3A, FLT3, NRAS, IDH1, and IDH2). In addition, they performed WES of 10 cases positive for NPM1 mutation at diagnosis with loss of the NPM1 mutation at relapse (here referred to as NPM1 +/NPM1 − AML) and 10 cases with stable NPM1 mutation.42
In general, larger cohorts of relapsed AML patients have been characterized using GPS only. Another cohort of 104 NPM1 mutated AML patients at diagnosis was described, however, at relapse only 11 matched samples were included, all with loss of NPM1 mutation (NPM1 +/NPM1 − AML).43 A study on CBF leukemia, focusing on t(8;21) positive AML only, covered a total of 331 patients, including 19 diagnosis/remission/relapse triplets.44 CBF leukemia defined by RUNX1‐RUNX1T1 fusion was characterized in 94 patients, including 17 diagnosis/relapse pairs.45
In summary, the major limitation of most studies describing the genetic evolution from AML diagnosis to relapse is either the focus on a small set of genes only, or the limited number of paired samples. Only few studies included a reasonable number of patients and comprehensive genetic profiling by WES.
In all of these studies, the vast majority of patients share at least one mutation between diagnosis and relapse. This suggests that, in general, initial and relapsed AML have a common origin, defined by these founding mutations, and relapse has not developed as an independent leukemia. Both evolutionary models, linear and branching evolution, were found across all cytogenetic subgroups (Table 1). On average, the number of mutations was similar at diagnosis and relapse in all cohorts, although in FLT3‐ITD positive AML, a lower mutational load at relapse was reported,37 while in the CN‐AML cohort, the mutation load increased.41 The frequency of transversions (C > A and C > G changes) was commonly higher among relapse‐specific mutations,37, 41, 42, 44 which is in line with previous reports and may be due to the mutagenic effect of cytarabine treatment.36, 46 Of note, this was not observed in APL, however, APL treatment does not include cytarabine.38 The most frequently mutated genes, NPM1, FLT3, and DNMT3A, show variable evolutionary patterns in different AML cohorts. Mutations in NPM1 are rather stable in all cohorts, lost only in 2/36 CN‐AML patients41 and 4/21 FLT3‐ITD positive patients,37 which is in line with the reported frequency of NPM1 mutation loss at relapse in general.42, 43, 47 Similarly, FLT3‐ITDs have a rather stable mutation pattern in the CN‐AML cohort, while they are gained at relapse in 6/25 patients,41 which is in line with the ELN classification that associates FLT3‐ITD with poor prognosis.12 Interestingly in NPM1 +/NPM1 − AML patients, FLT3‐ITDs were not detected at relapse although present in 18/61 NPM1 + diagnostic samples, pointing towards the development of an independent AML at relapse.42, 43 FLT3 point mutations are often lost in the CN‐AML cohort41 and NPM1 + patients,42, 43 while gained in patients of the FLT3‐ITD AML cohort.37 DNMT3A is stably mutated in most of the patients without any loss in the CN‐AML cohort,41 still recurrently lost in 2/18 FLT3‐ITD patients37 and 6/91 NPM1 + patients.42, 43 Additional examples for stable mutations detected in multiple cohorts include IDH1 and IDH2, although these mutations are also gained in several patients.37, 40, 41, 43 Genes specifically mutated at diagnosis include NRAS, KIT, and PTPN11.38, 39, 41, 43 In contrast, FLT3‐ITDs and mutations in WT1, KDM6A, and RUNX1 are often gained at relapse.38, 41, 42, 43 Interestingly, mutations in ASXL1 were found to be relapse‐specific in FLT3‐ITD AML,37 but stable in CN‐AML41 and diagnosis‐specific in NPM1 +/NPM1 − AML.43 Similarly, TET2 mutations were stable in the CN‐AML cohort41 but frequently gained and lost in APL.38
In summary, mutations in NPM1 and in signaling genes (eg, FLT3, NRAS, KIT, and PTPN11) are often found in AML at diagnosis but are frequently lost at relapse. This is true for CBF leukemia as well as for CN‐AML, although these two subtypes of AML generally have distinct mutational profiles. Interestingly, FLT3 point mutations are recurrently lost at relapse, while FLT3‐ITDs are recurrently gained. In contrast, epigenetic regulators show a variety of evolutionary patterns. While regulators of DNA methylation (eg, DNMT3A, TET1/2, and IDH1/2) are rather stable but also recurrently gained at relapse, regulators of chromatin remodeling and histone modifiers (eg, ASXL1/2, ARID1A/B, KDM6A, and MLL2/3) may be recurrently gained but also lost at relapse. Taken together, while deregulation of signaling pathways is critical for AML initiation, epigenetic regulation might play an essential role in the evolution towards relapse. In particular, reprogramming the DNA methylation landscape during chemotherapy treatment might be the key for AML cells to find their niche, develop resistance, and escape therapy.
4. MOLECULAR MECHANISMS OF CHEMOTHERAPY RESISTANCE
Several mechanisms of resistance development have been proposed, based on the deregulation of individual genes in relapsed AML, including SAMHD1, EZH2, and KDM6A. SAMHD1 encodes for a deoxynucleoside triphosphate triphosphohydrolase and is commonly mutated in T cell lymphomas, chronic lymphocytic leukemia, and colon cancer.48, 49, 50 Although SAMHD1 has been shown to sensitize cancer cells towards chemotherapy,51 it was reported that in AML, expression of SAMHD1 is inversely correlated to cytarabine response in vitro and in vivo.52 EZH2 encodes a histone methyltransferase and is commonly mutated at AML diagnosis and at relapse. Inactivation of EZH2 has been associated with poor prognosis in AML as well as in other hematologic malignancies.53, 54, 55, 56 Loss of EZH2 is commonly found in AML relapse and induces resistance towards multiple drugs, including tyrosine kinase inhibitors. Different from its role in AML, EZH2 mutations are associated with better outcome of patients with follicular lymphomas.57 The histone H3 lysine 27 demethylase KDM6A is commonly inactivated in several types of cancer, including leukemia.58, 59, 60 In AML, mutations in KDM6A are recurrently gained at relapse. In vitro knock out of KDM6A results in increased resistance to cytarabine and daunorubicin, while re‐expression of KDM6A sensitizes cells to treatment.61
5. AML RELAPSE AFTER ALLOGENEIC STEM CELL TRANSPLANTATION
Recently, it was reported that in AML cells after allogeneic stem cell transplantation (SCT), no relapse‐specific mutational patterns were observed.62 In contrast, other studies found an enrichment of WT1 mutations in AML relapse after chemotherapy as well as after SCT.41, 63 Although the gain of WT1 mutations were not significant in any of the studies, combined analysis of two independent cohorts showed a significant association of WT1 mutations with relapse after SCT suggesting a common evolutionary pattern (Figure 3). While WT1 mutations are not relapse‐specific, they are recurrently gained at relapse pointing towards a role in disease progression. WT1 constitutes an important epitope for immune response to leukemia, mediating the graft‐vs‐leukemia effect and providing the rationale for vaccination strategies.64, 65 However, somatic mutations may disrupt the immunogenic potential of WT1, thereby contributing to immune escape after allogeneic transplantation.
Figure 3.
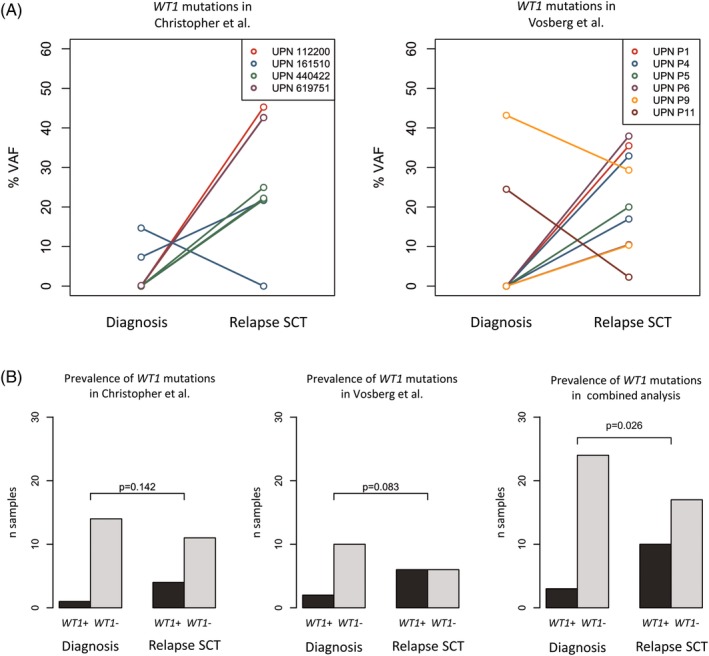
The frequency of WT1 mutations increase upon relapse after allogeneic stem cell transplantation (relapse SCT) in studies of Christopher et al. and Vosberg et al. (A) Variant allele frequencies (VAF) of WT1 mutations before and after relapse SCT in single data sets. (B) Prevalence of WT1 mutations in the single data sets and in the combined analysis
This exemplary meta‐analysis demonstrates the potential to obtain more conclusive results upon integrative analysis of independent studies. So far, individual studies of AML evolution have not been very conclusive, especially if they are limited to few patients only.
6. SUMMARY, DISCUSSION, OUTLOOK
Over the last years, several studies have focused on deciphering the genetic differences of AML at diagnosis and at relapse. By comparing these studies, several common features can be described. In the vast majority of cases, AML relapse evolved from the initially diagnosed disease, as the paired samples share common founding mutations. This process can follow either linear or branching evolution. Mutations in genes associated with signaling activation are commonly found at diagnosis but frequently lost at relapse, suggesting a major role in the development of leukemia but a minor role in escape from chemotherapy. Mutations in epigenetic regulators, especially regulators of DNA methylation, are generally stable and commonly gained at relapse, which points towards an important role of epigenetic reprogramming in therapy resistance. Nevertheless, the overall number of matched samples at both time points was limited in most studies, and thus the detection of true relapse drivers for all patients may not be possible. As AML samples are genetically highly heterogeneous, common features are rare and the picture of how AML clones evolve over time under the pressure of chemotherapy is still incomplete. Additional studies with increasing patient numbers will be necessary in order to bridge this gap.
In order to improve clonality analysis in AML patients, it is necessary to maximize the sequencing coverage, the number of target genes, and the number of longitudinal samples from a single patient. Although GPS commonly offers higher sensitivity to detect subclonal mutations as compared to WES, in most cases we are still not able to distinguish between the outgrowth of a very small subclone present at diagnosis and the de‐novo acquisition of a mutation in a single cell that survived chemotherapy using either of these techniques. Thus, additional and more detailed studies with increased sequencing coverage are necessary in the future. Current NGS techniques using unique molecular identifiers offer the possibility to precisely detect a single mutation in more than 10 000 cells. However, they are still limited to very few target regions only.66, 67 Ultimately, single cell studies may provide the sensitivity required to precisely track individual mutations.68 Regarding the genes of interest, most studies focus on a limited target region, either all genes by WES or a subset of previously described genes using GPS, as high coverage WGS is still expensive. Of note, in any kind of targeted sequencing, the selection of the enrichment method is critical for estimating the underlying clonality. As during each step of polymerase chain reaction (PCR), which is necessary for target enrichment, sequencing artifacts may be introduced and subsequently amplified. As a consequence, these PCR artifacts bias the computation of the VAF, resulting in incorrect estimation of the clonality. Using hybridization capture‐based enrichment methods only few PCR steps are involved and moreover, artifacts introduced by PCR can be detected more readily allowing for their removal from further analysis. In contrast, correcting for PCR artifacts is not possible when using amplicon‐based enrichment strategies as here the sequence of interest is intended to be enriched by PCR amplification. Thus, during subsequent data analysis it is not possible to distinguish desired PCR amplification from PCR artifacts. Finally, in order to correctly estimate clonality, it is critical to set the VAFs of individual mutations in relation to each other. Even in high‐depth sequencing data, the VAF may not perfectly represent the number of cells carrying a particular mutation or the VAFs of multiple mutations may not be distinguishable from each other in order to assign the mutations to separate clones and multiple constellations of clonality are possible. Tracking these mutations over time in as many longitudinal samples as possible increases the power to correctly assign common mutations shared by the same individual clone. In each sample, the VAFs may be corrected for the individual tumor burden. However, the number of tumor cells may be unknown and may also not be deduced from mutation analysis because preleukemic variants are also present in non‐leukemic blast cells to an unknown extent and the founding mutation, present in all leukemic cells, might be unknown.
The model of evolution is playing a major role not just biologically but also clinically. The evolution from treatment‐sensitive AML toward treatment‐resistant AML is based on losing a particular feature that provides sensitivity and/or gaining a feature that provides resistance. AML cells that followed linear evolution have been in the need of acquiring a mutation that provides resistance, which was not yet present in the main clone at diagnosis. This process, involving erroneous DNA replication during multiple cell divisions, may need some time, while the cells are under the pressure of chemotherapy. If residual leukemia cells are not able to gain the mutation quickly enough, they may be eradicated by the therapy. Thus, mutation clearance is directly correlated to the risk of AML relapse and its dynamics.69, 70, 71, 72 On the other hand, as the subclonal diversity of AML at diagnosis is commonly high, individual cells capable of giving rise to relapse may already exist at diagnosis.73, 74 If a particular mutation that provides resistance is already present in a single cell, this cell has the strongest fitness of all cells under the new selective pressure of chemotherapy. Within this niche, the adapted cell survives and may grow out, while all its competitors are eradicated by the therapy, thereby resulting in early relapse. In contrast, if AML cells at diagnosis harbor a feature that provides sensitivity, these cells may either lose it by back‐mutation, or an ancestral clone which is not carrying this mutation may grow out under therapy. Whether a resistance‐mediating mutation is needed, or a sensitivity‐mediating mutation must get lost in order to find a niche which allows for leukemia cell survival, the presence of a corresponding subclone at diagnosis leads to rapid development of relapse. Generally, the model of selective outgrowth in branching evolution may follow faster dynamics compared to the acquisition of de‐novo mutations in linear evolution, as this usually takes time and the number of tumor cells that may possibly acquire such mutation has been reduced to a minimum by the therapy. Indeed, when correlating clinical outcome with evolutionary models, early relapsing AML is often associated with a loss of mutations, the major hallmark of branching evolution.41 Although linear evolution can also be detected in AML patients with early relapse, branching evolution is a very rare event in patients with late relapse.
Within the model of branching evolution, it is important to differ between the outgrowth of a common ancestor of initial and relapsed AML, and the evolution of an independent leukemia due to pre‐existent and probably preleukemic clonal hematopoiesis. In clonal hematopoiesis, very early mutations, commonly in epigenetic regulators, are present before the onset of the disease, sometimes decades before the diagnosis of leukemia. Although clonal hematopoiesis is associated with an increased lifetime risk of developing hematologic malignancies, these mutations are not sufficient to induce leukemogenesis. Thus, relapsed AML sharing only preleukemic mutations with diagnostic AML should not be viewed as an example of branching evolution but rather as independent AML. Further, to clearly describe different mechanisms of AML evolution, it is also important to distinguish between preleukemic mutations, founding mutations, driver mutations and passenger mutations when looking at stable mutations between diagnosis and relapse.
Although previous studies have shown that a transformation from therapy‐sensitive towards therapy‐resistant AML may be mediated by a change in the mutational profile, still in about 25% of AML patients no mutational gain at relapse was detected. Moreover, these patients relapsed significantly earlier, indicating an even more aggressive disease. Thus, it is important to search for drivers of AML relapse not only at the level of genetics, but also at additional biological layers like epigenetics, especially as mutations in epigenetic regulators are relatively stable over time and moreover frequently gained at relapse.
Although every study cohort of relapsed AML helps to identify common features and mechanisms, they also add to the complexity of AML genetics. When looking deep enough, each AML (epi) genome is as individual as the patient itself and moreover, each evolutionary process from diagnosis to relapse is defined by multiple patient‐ and treatment‐specific features. Meta‐analyses of multiple studies will help to detect common mechanisms of therapy resistance. Finally, we are facing a paradigm shift from classifying AML into common genetic subgroups towards treating AML as a genetically individual disease that evolves over time.
ACKNOWLEDGMENTS
P.A.G. acknowledges support by the Deutsche Forschungsgemeinschaft (DFG), Collaborative Research Centre (SFB) 1243 “Cancer Evolution,” Project A08.
Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer. 2019;58:839–849. 10.1002/gcc.22806
REFERENCES
- 1. Darwin C. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. London: John Murray; 1859. [PMC free article] [PubMed] [Google Scholar]
- 2. Megginson LC. Lessons from Europe for American business. Southwest Soc Sci Q. 1963;44(1):3‐13. [Google Scholar]
- 3. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338‐345. [DOI] [PubMed] [Google Scholar]
- 4. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924‐935. [DOI] [PubMed] [Google Scholar]
- 5. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23‐28. [DOI] [PubMed] [Google Scholar]
- 6. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sykes SM, Kokkaliaris KD, Milsom MD, Levine RL, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells in acute myeloid leukemia. Exp Hematol. 2015;43(12):989‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howlader NNA, Krapcho M, Miller D, et al. SEER Cancer Statistics Review, 1975–2013. Bethesda, MD: National Cancer Institute; 2015. [Google Scholar]
- 11. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136‐1152. [DOI] [PubMed] [Google Scholar]
- 12. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453‐474. [DOI] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Metzeler KH, Herold T, Rothenberg‐Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686‐698. [DOI] [PubMed] [Google Scholar]
- 16. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid Leukemia. N Engl J Med. 2016;374(23):2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dick JE, Lapidot T. Biology of normal and acute myeloid leukemia stem cells. Int J Hematol. 2005;82(5):389‐396. [DOI] [PubMed] [Google Scholar]
- 18. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet. 2018;392(10147):593‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corces‐Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548‐2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bochtler T, Stolzel F, Heilig CE, et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J Clin Oncol. 2013;31(31):3898‐3905. [DOI] [PubMed] [Google Scholar]
- 24. Medeiros BC, Othus M, Fang M, Appelbaum FR, Erba HP. Cytogenetic heterogeneity negatively impacts outcomes in patients with acute myeloid leukemia. Haematologica. 2015;100(3):331‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li S, Garrett‐Bakelman FE, Chung SS, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22(7):792‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garson OM, Hagemeijer A, Sakurai M, et al. Cytogenetic studies of 103 patients with acute myelogenous leukemia in relapse. Cancer Genet Cytogenet. 1989;40(2):187‐202. [DOI] [PubMed] [Google Scholar]
- 29. Kern W, Haferlach T, Schnittger S, Ludwig WD, Hiddemann W, Schoch C. Karyotype instability between diagnosis and relapse in 117 patients with acute myeloid leukemia: implications for resistance against therapy. Leukemia. 2002;16(10):2084‐2091. [DOI] [PubMed] [Google Scholar]
- 30. Kim Y, Jang J, Hyun SY, et al. Karyotypic change between diagnosis and relapse as a predictor of salvage therapy outcome in AML patients. Blood Res. 2013;48(1):24‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Testa JR, Mintz U, Rowley JD, Vardiman JW, Golomb HM. Evolution of karyotypes in acute nonlymphocytic leukemia. Cancer Res. 1979;39(9):3619‐3627. [PubMed] [Google Scholar]
- 32. Alpermann T, Haferlach C, Eder C, et al. AML with gain of chromosome 8 as the sole chromosomal abnormality (+8sole) is associated with a specific molecular mutation pattern including ASXL1 mutations in 46.8% of the patients. Leuk Res. 2015;39(3):265‐272. [DOI] [PubMed] [Google Scholar]
- 33. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peniket A, Wainscoat J, Side L, et al. Del (9q) AML: clinical and cytological characteristics and prognostic implications. Br J Haematol. 2005;129(2):210‐220. [DOI] [PubMed] [Google Scholar]
- 35. Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354‐365. [DOI] [PubMed] [Google Scholar]
- 36. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature. 2012;481(7382):506‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garg M, Nagata Y, Kanojia D, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3‐ITD at diagnosis and relapse. Blood. 2015;126(22):2491‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madan V, Shyamsunder P, Han L, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. 2016;30(12):2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sood R, Hansen NF, Donovan FX, et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse leukemia. Leukemia. 2016;30(2):501‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun QY, Ding LW, Tan KT, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL‐PTD). Leukemia. 2017;31(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greif PA, Hartmann L, Vosberg S, et al. Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: an exome sequencing study of 50 patients. Clin Cancer Res. 2018;24:1716‐1726. [DOI] [PubMed] [Google Scholar]
- 42. Cocciardi S, Dolnik A, Kapp‐Schwoerer S, et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat Commun. 2019;10(1):2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hollein A, Meggendorfer M, Dicker F, et al. NPM1 mutated AML can relapse with wild‐type NPM1: persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018;2(22):3118‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Christen F, Hoyer K, Yoshida K, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood. 2019;133(10):1140‐1151. [DOI] [PubMed] [Google Scholar]
- 45. Hollein A, Nadarajah N, Meggendorfer M, et al. Molecular characterization of AML with RUNX1‐RUNX1T1 at diagnosis and relapse reveals net loss of co‐mutations. HemaSphere. 2019;3(1):e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fordham SE, Cole M, Irving JA, Allan JM. Cytarabine preferentially induces mutation at specific sequences in the genome which are identifiable in relapsed acute myeloid leukaemia. Leukemia. 2015;29(2):491‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1‐mutated acute myeloid leukemia. Blood. 2013;122(1):100‐108. [DOI] [PubMed] [Google Scholar]
- 48. Clifford R, Louis T, Robbe P, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123(7):1021‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Merati M, Buethe DJ, Cooper KD, Honda KS, Wang H, Gerstenblith MR. Aggressive CD8(+) epidermotropic cutaneous T‐cell lymphoma associated with homozygous mutation in SAMHD1. JAAD Case Rep. 2015;1(4):227‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rentoft M, Lindell K, Tran P, et al. Heterozygous colon cancer‐associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci U S A. 2016;113(17):4723‐4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015;14:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schneider C, Oellerich T, Baldauf HM, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med. 2017;23(2):250‐255. [DOI] [PubMed] [Google Scholar]
- 53. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722‐726. [DOI] [PubMed] [Google Scholar]
- 54. Gollner S, Oellerich T, Agrawal‐Singh S, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med. 2017;23(1):69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118(19):5227‐5234. [DOI] [PubMed] [Google Scholar]
- 56. Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665‐667. [DOI] [PubMed] [Google Scholar]
- 57. Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first‐line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population‐based registry. Lancet Oncol. 2015;16(9):1111‐1122. [DOI] [PubMed] [Google Scholar]
- 58. Nickerson ML, Dancik GM, Im KM, et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin Cancer Res. 2014;20(18):4935‐4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ntziachristos P, Tsirigos A, Welstead GG, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514(7523):513‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van Haaften G, Dalgliesh GL, Davies H, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stief SM, Hanneforth A‐L, Weser S, et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia. 2019. 10.1038/s41375-019-0497-6. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Christopher MJ, Petti AA, Rettig MP, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. 2018;379(24):2330‐2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vosberg S, Hartmann L, Metzeler KH, et al. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation is associated with gain of WT1 alterations and high mutation load. Haematologica. 2018;103:e581‐e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ishikawa T, Fujii N, Imada M, et al. Graft‐versus‐leukemia effect with a WT1‐specific T‐cell response induced by azacitidine and donor lymphocyte infusions after allogeneic hematopoietic stem cell transplantation. Cytotherapy. 2017;19(4):514‐520. [DOI] [PubMed] [Google Scholar]
- 65. Rezvani K, Yong AS, Mielke S, et al. Leukemia‐associated antigen‐specific T‐cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111(1):236‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kou R, Lam H, Duan H, et al. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS ONE. 2016;11(1):e0146638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stahlberg A, Krzyzanowski PM, Egyud M, Filges S, Stein L, Godfrey TE. Simple multiplexed PCR‐based barcoding of DNA for ultrasensitive mutation detection by next‐generation sequencing. Nat Protoc. 2017;12(4):664‐682. [DOI] [PubMed] [Google Scholar]
- 68. Potter N, Miraki‐Moud F, Ermini L, et al. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia. 2019;33(5):1113‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hirsch P, Tang R, Abermil N, et al. Precision and prognostic value of clone‐specific minimal residual disease in acute myeloid leukemia. Haematologica. 2017;102(7):1227‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Morita K, Kantarjian HM, Wang F, et al. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid Leukemia. J Clin Oncol. 2018;36(18):1788‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rothenberg‐Thurley M, Amler S, Goerlich D, et al. Persistence of pre‐leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia. 2018;32(7):1598‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yilmaz M, Wang F, Loghavi S, et al. Late relapse in acute myeloid leukemia (AML): clonal evolution or therapy‐related leukemia? Blood Cancer J. 2019;9(2):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bhola PD, Mar BG, Lindsley RC, et al. Functionally identifiable apoptosis‐insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J Clin Invest. 2016;126(10):3827‐3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]