

Abstract
Introduction
Although the clinical manifestations of severe haemophilia A (HA) are well studied, the challenges, if any, of living with mild HA are not clearly delineated to date.
Aim
To assess available evidence of clinical risks and societal/economic impacts of disease in adult patients with mild HA using a systematic literature review.
Methods
Prespecified study selection criteria were applied in a comprehensive literature search. Included studies varied in design and reported outcomes of interest for adults (≥13 years of age) with mild HA.
Results
Seventeen studies with a total of 3213 patients met eligibility criteria (published or presented in English, 1966‐2017). Most studies were observational, and the outcomes reported were too sparse and dissimilar to support a formal meta‐analysis. Mean annual bleeding rates ranged from 0.44 to 4.5 episodes per patient per year. Quality of life (QoL; SF‐36 General Health) was impacted compared to healthy controls. Health care costs and productivity were seldom assessed and no robust comparisons to healthy controls were available.
Conclusion
Quantifying outcomes for adult patients with mild HA remains challenging, with estimates of key QoL and cost data often based on small data sets and without comparison to population norms. Therefore, the clinical impact of mild haemophilia may be under‐represented and unmet needs may remain unaddressed. As paradigm‐changing therapies for HA emerge, stronger knowledge of mild HA can guide the development of care options that minimize burden and enhance the QoL for this segment of the haemophilia community, and for the haemophilia community in totality.
Keywords: acute bleeding, disease burden, haemophilia, quality of life
1. INTRODUCTION
Haemophilia A (HA) is an X‐linked bleeding disorder caused by a deficiency of blood coagulation factor VIII (FVIII), occurring in approximately 1 out of every 5000 male live births.1 HA can be a life‐threatening condition that requires lifelong monitoring and/or treatment. Management strategies include the use of plasma‐derived or recombinant factor concentrates and can vary between treatment centres and disease severity.2
Factor VIII replacement therapy, however, is not without risk (eg the development of inhibitors) and is expensive, ranging from $50 000 to nearly $300 000 annually in a recent US study, depending on disease severity and treatment approach.3 Severity levels for HA have generally been defined by baseline FVIII activity: severe <1% of normal or <1 IU/dL, moderate 1% to <5% and mild 5%‐40%.4 The clinical manifestations and cost impacts of severe HA are well studied, particularly with regard to the clinical benefit, but high cost, of ongoing FVIII prophylaxis.2, 5, 6
In contrast, the clinical burden, societal impact and economic impact of mild HA are not as clearly understood. Patients with mild HA often require and receive less intense therapy and medical attention, but may still experience limitations in daily activities and impacts of the disease on their morbidity, quality of life (QoL) and health care utilization.7, 8 It is also unclear to what extent variations in treatment strategies, patient characteristics and geographic location are associated with clinical outcomes and potential delay in diagnosis and treatment. In this study, we sought out published estimates of the burden of mild HA from clinical, patient, payer and/or societal perspectives.
2. MATERIALS AND METHODS
This systematic literature review was performed using recommended best practices, including a prespecified protocol defining the search strategy, inclusion criteria and statistical analysis plan addressing the burden of disease and clinical risks associated with mild HA in adults. This review was conducted and reported according to PRISMA guidelines.9 The protocol for the review was agreed to in advance by all authors but was not submitted to an external registry.
The population of interest was adults (defined as being 13 years of age or older) with mild HA. We were unable to find any recent summary of the literature in this population, and the clinical picture may be distinct from both moderate/severe HA and mild haemophilia B. Because we anticipated small numbers of studies focused exclusively on mild HA adults over 18 years of age, we included studies of mild HA individuals 13 years of age and older. Studies of any design (interventional or observational, controlled or uncontrolled) were eligible so long as they reported data for at least 10 patients in the population of interest. This lower limit of 10 patients was prospectively defined in order to exclude studies of insufficient sample size, for which outcomes may not be generalizable as they may focus on reports of unusual cases. Outcomes of interest included bleeding events (any event as defined or described by authors), QoL, joint pain, function/disability, health care utilization and cost (direct health care cost and/or indirect societal cost). As our goal was to assess the burden of disease, no specific intervention or comparison was required in the included studies.
2.1. Data sources and search strategies
A comprehensive search of English‐language biomedical literature was conducted. We searched electronic repositories including PubMed/MEDLINE, the Cochrane Library and EMBASE for all available dates up to the search cut‐off date of 31 December 2017. The search terms used for each database are detailed in Appendix S1. In addition to published journal articles, recent haematology meetings (2016‐2017) were searched for abstract presentations of eligible studies which may not yet have been published in full. Finally, we hand‐searched the reference lists of eligible articles and recent reviews.
2.2. Study selection
Two levels of screening were performed on all non‐duplicate citations downloaded from the search and each level of screening involved up to two reviewers. Level I screening was conducted on the title and abstract of each citation to identify potentially eligible studies, including haemophilia populations with mixed severity. Level I screening was performed by a single reviewer, with any questions resolved by consultation with a second reviewer. Level II screening was conducted as defined in the protocol, by reviewing the full text of each article to identify eligible studies that fit with study selection criteria. Level II screening was conducted by one reviewer and verified by a second reviewer. A published study would be included in our analysis and moved forward for data extraction only if both reviewers in the Level II screening agreed that study selection criteria were met.
Per predefined exclusion criteria, meeting abstracts or presentations prior to 2016, white papers, publications with no primary data, animal or in vitro studies, studies exclusively in FVIII inhibitor patients or paediatric patients (age <13 years), and studies of mixed/unspecified haemophilia populations, with data not separable by disease severity, were excluded.
2.3. Data collection and outcomes
For the purposes of this assessment, mild HA is defined by FVIII levels of 5%‐40% and adults are defined as being 13 years of age and older. Where data were available for a subset of patients meeting this definition, we included the article and extracted data only for the subset of interest. Studies without at least one outcome separately reported for at least 10 patients of interest were excluded. The primary endpoint was the rate of bleeding events, on a mean episodes per year basis or any other units reported by authors. We summarized all events reported by authors, regardless of whether bleeds required treatment, and captured the definition of bleeding events if reported. As mild HA patients tend to bleed less frequently and typically only after trauma, to fully characterize the clinical, patient‐reported and societal burden of mild HA, additional outcomes were captured. Secondary outcomes included QoL; joint pain; function/disability, including productivity and employment; other morbidity or mortality attributed to HA; and health care utilization and cost.
Dual review was used to collect data from each eligible study using a standardized template. Any discrepancies in interpretation between the two reviewers were resolved through a discussion of the text of the original articles. Each included study was appraised for quality and risk of bias using the Oxford Center for Evidence‐Based Medicine (Oxford CEBM) Levels of Evidence.10 Industry sponsorship was captured for each included study, based on author disclosures or affiliations.
2.4. Synthesis of results
We planned to conduct a meta‐analysis if sufficient comparable data were found for a primary or secondary outcome. Differences in author definitions of the primary endpoint, the rate of bleeding events, were available. These were captured and reviewed for comparability (eg only bleeds requiring treatment, only spontaneous bleeds unrelated to surgical or dental procedures, any bleeding event). Where data were too sparse or too heterogeneous to be combined across studies, descriptive statistics were performed in order to qualitatively summarize available evidence. The risk of bias across studies was assessed informally based on study design, level of evidence, size, and representativeness of the population of interest, and comparability of estimates from different studies.
3. RESULTS
3.1. Study selection
The search identified 1845 unique citations across all sources. After screening of the titles and abstracts, 139 potentially eligible studies were obtained in full text for review (Figure 1). Application of eligibility criteria resulted in 17 included studies, comprising 20 publications due to separate reports on the same or overlapping study populations.3, 8, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 The primary reasons for study exclusion were that patients with haemophilia were not separated by severity (or only moderate/severe HA patients were included), or study populations were mixed with regard to type of haemophilia (A/B/other coagulopathies) and/or age (adults and children). In addition, 14 studies did not report an outcome of interest and 20 studies were reviews, case reports or studies not containing at least 10 mild HA patients.
Figure 1.
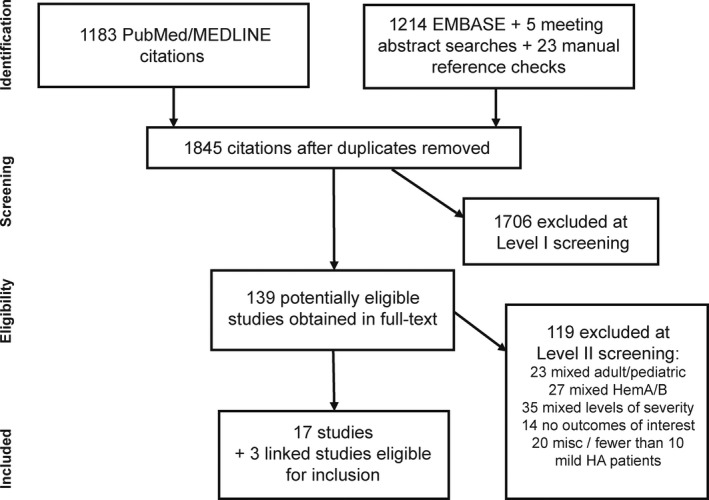
Study attrition
3.2. Study characteristics
The 17 studies included data for 3213 mild HA patients aged 13 years and older. These patients were a subset of the overall study population in all cases. The total number of haemophilia patients (all ages, severities and types) in the 17 studies was 20 587. Eligible studies were conducted in Europe (59%), North America (35%) and Japan (6%) (Table 1). Most studies were observational in nature, and few details on typical treatment protocols were available. A variety of treatments were described by authors, including FVIII concentrates, recombinant FVIII and DDAVP. Fewer than 1% of patients had prophylactic use of FVIII,11, 13, 27 except in one study reported as a meeting abstract, for which long‐term use of FVIII concentrate was required.18
Table 1.
Characteristics of included studies
| Number of studies | Number of mild HA adults | |
|---|---|---|
| Total | 17 | 3213 |
| Country/sites | ||
| Europe | 10 | 1045 |
| North America | 6 | 2049 |
| Japan | 1 | 119 |
| Patient population of entire studya | ||
| Mild HA adults and age‐matched healthy controls | 1 | 47 |
| Mild/moderate HA, mild HA/HB | 3 | 217 |
| HA, any severity | 5 | 122 |
| HA or HB | 5 | 2044 |
| Various coagulopathies | 3 | 783 |
| Year of publication | ||
| 1996‐2007b | 2 | 56 |
| 2008‐2017 | 15 | 3157 |
| Level of evidence/study design | ||
| IB (Prospective cohort or registry) | 6 | 1517 |
| IIB (Retrospective cohort) | 6 | 316 |
| IIC (Outcomes research, survey data) | 5 | 1380 |
Data were extracted for the subset of patients of interest (mild HA adults) in this review.
Search dates extended back to 1966 (the initiation of MEDLINE database), but the earliest study meeting eligibility criteria was published in 1996.
3.3. Risk of bias
Most evidence was Oxford Level of Evidence IB or IIB, consisting of prospective or retrospective cohort studies, in many cases from a single centre (Table 1). No randomized controlled trials meeting inclusion criteria were identified. The risk of selective reporting/outcome availability bias was high, as mild HA adults were typically a subset of the entire study population, and not all outcomes were available for this subset. Seven studies were industry‐sponsored. No differences in outcomes were apparent between industry‐sponsored and non‐industry‐sponsored studies; no formal meta‐analysis or assessment of publication bias was conducted.
3.4. Clinical burden: bleeding events
Clinical burden of mild HA was reported in a variety of formats in the included studies. We sought data on annual bleeding rates (ABR) or other characterizations of bleeding risk. Most often, bleeding events were reported as a percentage of patients with any bleeding episode over a period of time (cumulative incidence), with both follow‐up duration and the definition of events varying between studies (Table 2). One multicentre, prospective study from the US reported a mean ABR of 4.5 ± 10.0 episodes/year in 23 mild HA adult patients3; however, other estimates were lower by an order of magnitude.8, 25 Most studies meeting inclusion criteria did not report mean ABR.
Table 2.
Bleeding episodes reported in mild HA patients
| Study | Population (FVIII level/age) | Follow‐up period | Bleeding events | |
|---|---|---|---|---|
| % (n/N) of patients with bleeding | episodes/pt year (SD) | |||
| Prospective cohort (US, Canada)12 |
6%‐40% (median NR) Age NR |
5 y (2002‐2006) | 36% (4/11) had bleeding scorea >0, indicating at least one bleeding episode other than surgery, dental procedures and major accidents | NR |
| Cross‐sectional survey (Finland)22 |
5%‐40% (median NR) Age ≥16 |
1 y (years NR; publication 1996) | 40% (8/20) had at least one episode of moderate bleedingb; none had severe bleeding | NR |
| Retrospective cohort (Italy)8 |
0.6‐0.33 IU/mL (median 0.15 IU/mL) Median age 35 (range 3‐88)c |
10 y (range 1‐39; 1985‐2010) | 91% (68/75) had at least one bleed during follow‐upc: 27% joint, 35% muscle, 73% muco‐cutaneous, 13% postoperative, 21% after dental work |
0.56 (0.67) Includes all bleeds regardless of treatment |
| Retrospective cohort (Slovenia)25 |
5%‐40% (mean 8.5%) Age 13‐54 |
6 y (2007‐2014) |
57% (8/14) Includes surgery, trauma, dental procedures |
0.44d Includes all bleeds regardless of treatment |
| Retrospective cohort (Canada)26 e |
5%‐40% (mean 0.15 IU/mL) Adults ≥18 (mean age 46) |
5 y (year NR; publication 2008) | 17% (8/46) with 2 or more bleeds requiring medical assessment or therapy in past 5 y | NR |
| Prospective cohort (Italy)27 |
NR (median NR) Adults ≥18 |
Duration NR (2011‐2013) | 5.5% (1/18) had evidence of cerebral microbleedf | NR |
| Prospective cohort (US)3, 20, 28, g |
6%‐30% (median NR) Adults ≥18 |
2 y (2005‐2007) | NR (23 patients with data) |
4.5 (10.0) Definition of bleeding episodes not available |
Abbreviation: NR, not reported.
Bleeding score calculated from 5 y mean scores of haemarthrosis and soft tissue haematoma.
Bleeding histories were divided into severity based on symptom clustering across the whole cohort of coagulation disorders (n = 224); most episodes were joint and soft tissue bleeds.
Outcomes may include some patients under 13 y; study was retained due to long follow‐up duration and detailed reporting. Three patients had FVIII inhibitors.
Eleven soft tissue bleeds occurred in one patient who developed FVIII inhibitors.
Subjects are from large kindred with specific mutation (VAL2016ala).
Patients without prior symptomatic brain bleeding were assessed for evidence of asymptomatic bleeding using cerebral MRI.
This analysis of HUGS‐Va included only participants with complete follow‐up data.
Bleeding events in mild HA, such as joint bleeds or cerebral bleeds, may not be readily apparent. One study used cerebral MRI to assess mild HA adults with no prior symptomatic cerebral bleeding; 5.5% (1/18) of patients had evidence of a previous cerebral microbleed.27
3.5. Other morbidity
Joint pain and damage resulting from subclinical joint bleeds may also be problematic in mild HA. Joint score data, like data for bleeding events, were not reported in a standard way across studies (Table 3). In a small cross‐sectional survey study from 1996, 20% of mild HA patients age 16 or older experienced moderate or severe pain in the previous year, with pain and disability increasing with age.22 Another study comparing mild HA adults with age‐matched controls found the maximum joint score to be significantly higher (worse) in mild HA.26 Development of inhibitors to FVIII in mild HA reached a cumulative incidence of 4.0%‐7.8% over 10 years in two retrospective cohort studies with long‐term follow‐up.8, 12
Table 3.
Joint score and pain reported in mild HA patients
| Study | Population (FVIII level/age) | Follow‐up period | Number of mild HA patients | Mean (SD) or Median (range) | |
|---|---|---|---|---|---|
| Joint score (name of scale) | Pain or overall severity score (name of scale) | ||||
| Prospective cohort (US)3, 20, 28, a |
6%‐30% (median NR) Age ≥18 |
2 y (2005‐2007) | 23 | Joint range of motion limitation (AAOS): 5.4 ± 4.5 | NR |
| Cross‐sectional survey (Finland)22 |
5%‐40% (median NR) Age ≥16 |
1 y (years NR, publication 1996) | 20 | NR | Pain in previous year: 5% (1/20) severe, 15% (3/20) moderateb |
| Retrospective cohort (Italy)8 |
0.06‐0.33 IU/mL (median 0.15 IU/mL) Median age 35 (range 3‐88)c |
10 y (range 1‐39) (1985‐2010) | 75 | Mean physical joint score 0.88 ± 1.78 (scale NR) | NR |
| Retrospective cohort (Canada)26, d |
5%‐40% (mean 0.15 IU/mL) Age ≥18 (mean age 46) |
5 y (year NR, publication 2008) | 47 | Max joint score by Colorado PE0.5: 5.3 mild haemophilia vs 2.8 control (P < 0.05) | HAQ pain score: 0.51 haemophilia vs 0.61 controls (no significant difference) |
Abbreviations: AAOS, American Academy of Orthopaedic Surgeons; Colorado PE0.5: Scale based on modified World Federation of Haemophilia (WFH) Physical Joint Examination instrument.19; NR, not reported.
This analysis of HUGS‐Va included only participants with complete follow‐up data.
Pain and disability increased with age.
Outcomes may include some patients under 13 y; study was retained due to long follow‐up duration and detailed outcome reporting.
Subjects are from large kindred with specific mutation (VAL2016ala). Normative data are from age‐matched healthy controls (n = 32).
3.6. Quality of life
Patient‐reported outcomes were available in three studies, all of which were published in the past 10 years (Figure 2). Each study used a different QoL instrument: the SF‐36,26 SF‐123 and HAEMO‐QoL‐A.18 SF‐36 general health was lower for mild HA vs age‐matched healthy controls in a Canadian cohort (58.1 vs 70.8, P < 0.05, n = 47).26 No other studies reported QoL in mild HA adults compared to healthy controls or population norms.
Figure 2.
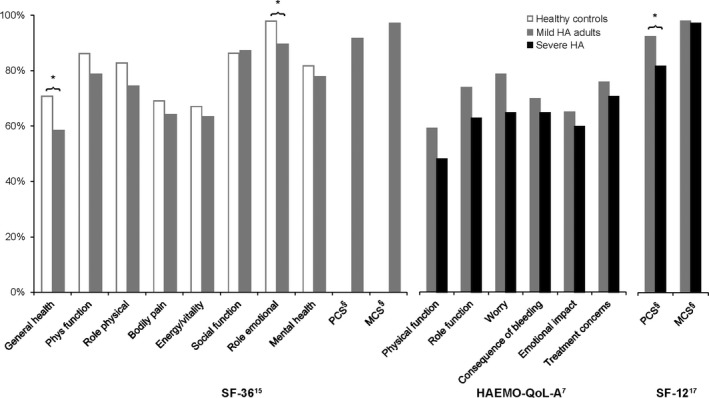
Quality of life scores in mild HA (as % of maximum score). § Physical component summary (PCS) and mental component summary (MCS) for SF‐36 and SF‐12 use norm‐referenced scoring (not available as transformed/scaled scores) and are shown as % of the closest available age and country norms. For HUGS‐Va, normative data were not reported within the study, so SF‐12 norms for Utah, age >18 were used.21 * P < 0.05 vs comparison
In a prospective US study, SF‐12 physical component summary was higher (better QoL) for mild HA compared to severe HA (P = 0.014, n = 42), while mental component summary was not significantly different between mild and severe HA.3 No differences between severe and mild HA were found using the Haemo‐QOL‐A, in a small UK survey study reported to date only as a meeting abstract.18 It is notable that this study required long‐term use of FVIII concentrate, meaning that mild HA patients in this study were probably not representative of the overall mild HA population.
3.7. Societal impacts
Societal impacts, which should include productivity loss, disability and time lost from work/school, were mentioned in five studies, but were not often quantified in ways that would allow comparison to populations without HA. One study estimated 5 disability‐adjusted life years (DALYs) lost per case of mild HA, using a modelling approach to assess lifetime burden of disease.16 Employment of mild HA patients was 60% (27/45) compared to 68% (21/31) for healthy controls in a Canadian cohort; the difference did not reach statistical significance.26 In a multicentre US study, mild HA patients missed an average of 6.2 work days per year, of which 4.7 days (76%) were due to HA.3 Finally, a retrospective study in Italy noted 3.4 missed days of work per year, with no benchmark provided for patients without HA.23 Table 4 summarizes the data available for health care cost and utilization and societal impacts (direct and indirect costs of mild HA).
Table 4.
Healthcare cost and indirect cost/societal impact for mild HA patients
| Study | Population (FVIII level/age) | Follow‐up period | Number of mild HA patients | Cost outcomes | |
|---|---|---|---|---|---|
| Annual direct cost (mean or median, with currency and year) | Indirect cost/productivity | ||||
| Retrospective cohort (US)14 |
6%‐50% (median NR) Adults |
12 mo | 36 |
22 182 USD (1995; mean) total cost 18 017 USD factor costa |
NR |
| Retrospective cohort (Portugal)21, b |
5%‐40% (median NR) Age ≥18c |
12 mo | 16 |
€793 (year of currency NR; mean or median not specified) €506 hospital costs €+ 287 clotting factor |
NR |
| Cost model (Belgium)16 |
6%‐40% Adults |
Lifetime (simulated 2011 birth cohort, projected to future) | N/A (simulated cohort) | NR |
Disability weight vs norms: 0.054 Lifetime DALYs per mild haemophilia case: 5 (95% CrI 2‐10)d SF‐36D data used for disability weights |
| Prospective cohort (US)3, 20, 28, e |
6%‐30% (median NR) Age ≥18 |
2 y (2005‐2007) | 23 | NR |
6.2 d lost from work per year (total) 4.7 per year due to HA |
| Retrospective cohort (Italy)8, e |
0.06‐0.33 IU/mL (median 0.15 IU/mL) Median age 35 (range 3‐88) |
10 y (range 1‐39) (1985‐2010) | 75 | NR | 64% employed/in training (mild HA adults) |
| Prospective cohort (Italy)23, e |
>5% (median NR) Age NR (assumed 17‐65, based on reporting of employment status) |
12 mo (2005) | 109 | NR | 3.4 d lost from work per year |
| Retrospective cohort (Canada)26, f |
5%‐40% (mean 0.15 IU/mL) Age ≥18 (mean age 46) |
5 y (years NR, publication 2008) | 47 | NR |
60% (27/45) employed mild HA vs 68% (21/31) controls. HAQ standard disability score: 0.24 vs 0.12 controls |
Abbreviations: 95% CrI, 95% credible interval; DALY, disability‐adjusted life year; NR, not reported; SD, standard deviation.
Costing by CPT code using Medicare fee schedule. A substantial portion of the study population was HIV+.
Data on outpatient visits, hospital admissions and surgeries are not available by severity.
Costs are not reported separately for adults and children; authors stated no statistical difference.
Sensitivity analyses under different discounting assumptions: 1‐2 lifetime DALYs after discounting. No excess mortality was assumed for mild or moderate haemophilia patients.
No normative/ healthy control data available.
Subjects are from large kindred with specific mutation (VAL2016ala). No significant difference between mild HA and age‐matched healthy controls.
3.8. Health care cost and utilization
We sought details on health care utilization (clinic visits, hospitalization days, etc) and health care costs (direct costs) in adults with mild HA; while such costs are a topic of discussion in recent literature, we found they were generally reported for HA overall. Mild HA health care costs were reported in 2 studies, with rather different health care settings: a 2015 study from Portugal reported mean costs of 793€/year, 36% of which was the cost of clotting factor.21 In contrast, a US‐based study conducted in 1995 estimated annual costs of $22 182 for mild HA, 81% of which was clotting factor.15 Health care utilization was not reported for adults with mild HA in any of the studies in our review.
4. DISCUSSION
Limited data were available on ABR in mild HA adults (three studies meeting our eligibility criteria, reporting between 0.44 and 4.5 mean bleeding episodes/year).8, 25, 28 Because data were scarce and estimates varied, we consulted studies not meeting inclusion criteria to provide context for ABR. Two studies were found with an ABR estimate for mild HA patients of all ages (including children); mean ABR centred on 0.5‐0.6 episodes per year.29, 30 Mild HA patients in our review also experienced other morbidity, QoL impacts and economic impacts.
Quantifying outcomes for adult patients with mild HA remain challenging, with estimates of key QoL and cost data often based on small subsets of a single study. One complicating factor is the definition of mild HA based on FVIII levels, which can fluctuate. In general, the mild HA population is defined as patients with FVIII levels >5% (or 5 IU/dL) and up to 40% (or 40 IU/dL). However, there has been occasional variation in this range, as shown in Table 2, as well as in a recent communication from the Scientific Subcommittee (SSC) of the International Society of Thrombosis and Haemostasis.31 Few studies put mild HA outcomes in the context of population norms; therefore, the clinical impact of mild HA may be under‐represented and unmet needs remain unaddressed. While mean ABRs were relatively low, mild HA had measurable impact on QoL and represents an area of therapeutic concern requiring further study. Until recently, treatment goals in haemophilia were FVIII trough levels of >1%, to prevent major bleeding. With the advent of extended half‐life FVIII concentrates and novel non‐replacement therapies, and the potential for gene therapies, understanding around target trough levels is evolving to suggest there may be additional benefit in targeting non‐haemophilia levels (ie >40%), as opposed to levels within the mild HA range. Such targets may be achievable through recent therapeutic advances such as those in adeno‐associated viral vector‐mediated gene therapy, which may enable sustained expression of FVIII activity following a one‐time infusion.32 We would need to consider currently unmeasured, cumulative benefits to those living with mild HA. This review should be followed by further prospective studies to understand the need of all patients with mild haemophilia.
Strengths of this systematic review include the comprehensive search and rigorous methodology including a prospective plan, using current best practices for systematic literature review. Limitations relate primarily to the availability and comparability of published evidence. Data on mild HA adults are frequently aggregated with moderate HA, mild haemophilia B and/or mild HA paediatric patients in the published literature. Despite our efforts to extract clinical and humanistic outcomes data for comparable populations (mild HA adults), meta‐analysis was ultimately not feasible due to variability in reporting. Studies used different measures, scales and time points, with too few studies using consistent formats. The body of evidence in mild HA is also limited by missing data and selective availability of outcomes, making it difficult to gain a comprehensive picture of disease burden across all outcomes of interest.
This review has revealed a lack of evidence in the specific population of mild HA adults; evidence is low quality in part because data had to be separated out for the population of interest. Considering the limitations of the current body of evidence, higher quality studies in this area are needed. Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild HA adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QoL and other patient‐reported outcomes. A recent multi‐stakeholder effort to define core outcomes for trials in haemophilia highlighted the importance of mental health outcomes; this is an area where existing published evidence for mild HA adults is lacking.33
5. CONCLUSION
To the best of our knowledge, this is the first systematic review to assess available evidence of clinical risks and societal/economic impacts of disease in adult patients with mild HA. Based on our review, data from adult patients with mild HA are frequently aggregated with other types of haemophilia in the published literature. Few studies to date have focused on this population alone, nor have many authors described mild HA outcomes in the context of population norms. While mean ABRs were relatively low, mild HA had measurable impact on QoL and productivity and represents an area of therapeutic need requiring further study.
DISCLOSURES
FP has received honoraria for participating as a speaker at satellite symposia and educational meetings organized by Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex and CSL Behring; moreover, she is a member of the scientific advisory board of Sanofi. JQ, BK, AL, MCL, and WYW are employees and stockholders of BioMarin. FT was an employee of BioMarin at the time of the study. DF has received consulting fees from BioMarin. FP defined the publication need and scope. DF and ER managed data collection and curation. DF drove manuscript drafting and revisions. All authors made intellectual contributions to protocol content and study inception; aided in the interpretation of results; and reviewed and approved the final manuscript.
Supporting information
ACKNOWLEDGEMENTS
Erica Roulier and Fathima Mubarack assisted with literature searches and data extraction for the systematic review. Yuqing Yang provided statistical guidance. Gillian Clague assisted with preparation of graphics and coordination of author review. Kendra Bolt provided editorial support and assistance with manuscript finalization.
Peyvandi F, Tavakkoli F, Frame D, et al. Burden of mild haemophilia A: Systematic literature review. Haemophilia. 2019;25:755–763. 10.1111/hae.13777
REFERENCES
- 1. Beckman MG, Hulihan MM, Byams VR, et al. Public health surveillance of nonmalignant blood disorders. Am J Prev Med. 2014;47:664‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berntorp E, Dolan G, Hay C, et al. European retrospective study of real‐life haemophilia treatment. Haemophilia. 2017;23:105‐114. [DOI] [PubMed] [Google Scholar]
- 3. Zhou Z‐Y, Koerper MA, Johnson KA, et al. Burden of illness: direct and indirect costs among persons with hemophilia A in the United States. J Med Econ. 2015;18:457‐465. [DOI] [PubMed] [Google Scholar]
- 4. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐47. [DOI] [PubMed] [Google Scholar]
- 5. O'Hara J, Hughes D, Camp C, Burke T, Carroll L, Diego DG. The cost of severe haemophilia in Europe: the CHESS study. Orphanet J Rare Dis. 2017;12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van den Berg HM, Fischer K. Prophylaxis for severe hemophilia: experience from Europe and the United States. Semin Thromb Hemost. 2003;29:49‐54. [DOI] [PubMed] [Google Scholar]
- 7. Osooli M, Lövdahl S, Steen Carlsson K, et al. Comparative burden of arthropathy in mild haemophilia: a register‐based study in Sweden. Haemophilia. 2017;23:e79‐e88. [DOI] [PubMed] [Google Scholar]
- 8. Tagliaferri A, Di Perna C, Riccardi F, Pattacini C, Rivolta GF, Franchini M. The natural history of mild haemophilia: a 30‐year single centre experience. Haemophilia. 2012;18:166‐174. [DOI] [PubMed] [Google Scholar]
- 9. Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howick J, Phillips B, Ball C, et al. Oxford Centre for Evidence‐based Medicine: Levels of evidence (March 2009). Oxford, UK: Centre for Evidence‐Based Medicine, Oxford University; 2009. [Google Scholar]
- 11. Biss TT, Chan AK, Blanchette VS, Iwenofu LN, McLimont M, Carcao MD. The use of prophylaxis in 2663 children and adults with haemophilia: results of the 2006 Canadian national haemophilia prophylaxis survey. Haemophilia. 2008;14:923‐930. [DOI] [PubMed] [Google Scholar]
- 12. Brummel‐Ziedins KE, Whelihan MF, Gissel M, Mann KG, Rivard GE. Thrombin generation and bleeding in haemophilia A. Haemophilia. 2009;15:1118‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eckhardt CL, Menke LA, van Ommen CH, et al. Intensive peri‐operative use of factor VIII and the Arg593–>Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009;7:930‐937. [DOI] [PubMed] [Google Scholar]
- 14. Globe DR, Cunningham WE, Anderson RM, et al. The Hemophilia Utilization Group Study: cost of out patient, in patient and pharmaceutical care. Int J Ped Hematol Oncol. 2001;7:87‐100. [Google Scholar]
- 15. Globe DR, Curtis RG, Koerper MA. Utilization of care in haemophilia: a resource‐based method for cost analysis from the Haemophilia Utilization Group Study (HUGS). Haemophilia. 2004;10(Suppl 1):63‐70. [DOI] [PubMed] [Google Scholar]
- 16. Henrard S, Devleesschauwer B, Beutels P, et al. The health and economic burden of haemophilia in Belgium: a rare, expensive and challenging disease. Orphanet J Rare Dis. 2014;9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iorio A, Oliovecchio E, Morfini M, Mannucci PM. Italian Registry of Haemophilia and Allied Disorders. Objectives, methodology and data analysis. Haemophilia. 2008;14:444‐453. [DOI] [PubMed] [Google Scholar]
- 18. Luo PL, Rangarajan S, Austin S. Quality of life and activities in patients treated with long term plasma derived FVIII concentrate (Fanhdi). European Association for Haemophilia and Allied Disorders (EAHAD). 2017; abstract P150.
- 19. Manco‐Johnson M, Soucie JM, Sharathkumar A, et al. Characteristics of males with hemophilia in the United States Hemophilia Treatment Center Network (USHTCN) with new‐onset elevated inhibitor titers, December 15, 2013 – December 31, 2016. American Society of Hematology 2017; abstract 3408.
- 20. Poon J‐L, Zhou Z‐Y, Doctor JN, et al. Quality of life in haemophilia A: Hemophilia Utilization Group Study Va (HUGS‐Va). Haemophilia. 2012;18:699‐707. [DOI] [PubMed] [Google Scholar]
- 21. Rocha P, Carvalho M, Lopes M, Araujo F. Costs and utilization of treatment in patients with hemophilia. BMC Health Serv Res. 2015;15:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santavirta N, Bjorvell H, Solovieva S, Santavirta S, Hurskainen K, Konttinen YT. Empirically derived classification of coagulation disorders in 224 patients. Haematologica. 1996;81:316‐323. [PubMed] [Google Scholar]
- 23. Tagliaferri A, Rivolta GF, Biasoli C, et al. A web‐based registry of inherited bleeding disorders in the region of Emilia‐Romagna: results at three and a half years. Haemophilia. 2008;14:343‐354. [DOI] [PubMed] [Google Scholar]
- 24. Taki M, Shirahata A. Current situation of regular replacement therapy (prophylaxis) for haemophilia in Japan. Haemophilia. 2009;15:78‐82. [DOI] [PubMed] [Google Scholar]
- 25. Trampus Bakija A, Debeljak M, Preloznik Zupan I, Benedik Dolnicar M, Kovac J, Jazbec J. Specific and global coagulation tests in patients with mild haemophilia A with a double mutation (Glu113Asp, Arg593Cys). Blood Transfus. 2015;13:622‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walsh M, Macgregor D, Stuckless S, Barrett B, Kawaja M, Scully MF. Health‐related quality of life in a cohort of adult patients with mild hemophilia A. J Thromb Haemost. 2008;6:755‐761. [DOI] [PubMed] [Google Scholar]
- 27. Zanon E, Manara R, Milan M, et al. Cognitive dysfunctions and cerebral microbleeds in adult patients with haemophilia A: a clinical and MRI pilot‐study. Thromb Res. 2014;134:851‐855. [DOI] [PubMed] [Google Scholar]
- 28. Zhou Z‐Y, Wu J, Baker J, et al. Haemophilia utilization group study ‐ Part Va (HUGS Va): design, methods and baseline data. Haemophilia. 2011;17:729‐736. [DOI] [PubMed] [Google Scholar]
- 29. Aznar JA, Lucía F, Abad‐franch L, et al. Haemophilia in Spain. Haemophilia. 2009;15:665‐675. [DOI] [PubMed] [Google Scholar]
- 30. Ng HJ, Lam J, Koh PL, et al. A comprehensive study of current haemophilia care and outcomes in Singapore. Haemophilia. 2015;21:e428‐e431. [DOI] [PubMed] [Google Scholar]
- 31. Makris M, Oldenburg J, Mauser‐Bunschoten EP, et al. The definition, diagnosis and management of mild hemophilia A: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16:2530‐2533. [DOI] [PubMed] [Google Scholar]
- 32. Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377:2519‐2530. [DOI] [PubMed] [Google Scholar]
- 33. Iorio A, Skinner MW, Clearfield E, et al. Core outcome set for gene therapy in haemophilia: Results of the coreHEM multistakeholder project. Haemophilia. 2018;24:e167‐e172. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials