

Abstract
Macrophages are important innate immune cells that respond to microbial insults. In response to multi-bacterial infection, the macrophage activation state may change upon exposure to nascent mediators, which results in different bacterial killing mechanism(s). In this study, we utilized two respiratory bacterial pathogens, Mycobacterium bovis (Bacillus Calmette Guẻrin, BCG) and Francisella tularensis live vaccine strain (LVS) with different phagocyte evasion mechanisms, as model microbes to assess the influence of initial bacterial infection on the macrophage response to secondary infection. Non-activated (M0) macrophages or activated M2-polarized cells (J774 cells transfected with the mouse IL-4 gene) were first infected with BCG for 24–48 h, subsequently challenged with LVS, and the results of inhibition of LVS replication in the macrophages was assessed. BCG infection in M0 macrophages activated TLR2-MyD88 and Mincle-CARD9 signaling pathways, stimulating nitric oxide (NO) production and enhanced killing of LVS. BCG infection had little effect on LVS escape from phagosomes into the cytosol in M0 macrophages. In contrast, M2-polarized macrophages exhibited enhanced endosomal acidification, as well as inhibiting LVS replication. Pre-infection with BCG did not induce NO production and thus did not further reduce LVS replication. This study provides a model for studies of the complexity of macrophage activation in response to multi-bacterial infection.
Keywords: Francisella, BCG, co-infection, macrophage, IL-4
Introduction
Our understanding of host–pathogen interaction is largely based on the host response to a single pathogen. This approach has provided valuable information about the action(s) taken by the host against invasion by specific microbes and the mechanisms employed to subvert the host immune system.1 However, many human infections are now known to be polymicrobial in nature (see the review by Bakaletz2). In such an environment, immune responses triggered by one pathogen may influence a host reaction to co-infecting pathogens.1
During the initial stage of a host–pathogen interaction, the pathogen encounters strategically placed phagocytic cells, e.g., macrophages that target and eliminate infectious agents. Because macrophages may encounter a variety of microbes, they are exposed to a multitude of stimulatory and suppressive signals that may have a profound effect on pathogen control. For example, persistent infection with respiratory syncytial virus in macrophages was shown to reduce the phagocytic capacity to engulf Haemophilus influenzae.3 Moreover, interaction with acidified Coxiella burnetii phagolysosomes resulted in reduced growth of Mycobacterium tuberculosis, which normally inhibits phagolysosomal fusion.4 Additionally, resident M0 macrophages can be activated classically by Th1 cytokines (e.g., IFN-γ) or alternatively by Th2 cytokines (e.g., IL-4 and IL-13) to become phenotypically and functionally distinct (M1 and M2, respectively) in response to different pathogens and environmental insults, and such M1 and M2 macrophage populations may vary under different health conditions. For example, a significant increase of M2 macrophages has been reported in allergen-exposed mouse lungs,5 the lungs of asthmatic patients,6 and of individuals exposed to cigarette smoke.7
Thus, this paper seeks to provide a model to address the question of whether the outcome of multi-bacterial infection is different in M0 and M2 dominant microenvironments. For this purpose, we used two well-characterized intracellular bacteria, M. bovis BCG (Bacillus Calmette Guẻrin) and Francisella tularensis live vaccine strain (LVS) as model organisms because of different outcomes in the phagosome, i.e. BCG mainly resides in membrane-bound phagosomes while LVS rapidly escapes to the cytosol.8 Both Mycobacterium and Francisella can cause pulmonary infection and cervical lymphadenopathy. Although not the purpose of this study, a recent survey of 1170 tuberculosis cervical lymphadenitis patients in Turkey (a country with emergent endemic tularemia and epidemic tuberculosis) by Karabay et al. found that ≈7% of these patients were seropositive for F. tularensis.9
Previously, we have shown a novel IL-4 dependent pathway that aids in reducing F. tularensis replication in macrophages.10,11 IL-4-activated M2 macrophages, subsequently infected with F. tularensis, exhibited increased ATP levels and an associated increase in phagosomal acidification that led to a decrease in F. tularensis phagosomal escape and reduced replication.10,11 In contrast, Mycobacterium infection results in a dominant M1 polarization of alveolar macrophages during the first 3 wk of infection.12 Taking advantage of this established LVS-infected macrophage system, and the well-characterized inhibitory mechanisms in M0 and M2 macrophages, we studied the influence of BCG on LVS infection in these two distinctly different macrophages. While both BCG and LVS target and replicate inside macrophages, they possess different survival mechanisms. F. tularensis is a facultative, intracellular, Gram-negative bacterium that causes the human disease tularemia.13 The LVS is a human attenuated strain derived from F. tularensis subsp. holarctica14 that is often used as an experimental alternative in lieu of the more virulent subsp. tularensis15 and which, like F. tularensis, escapes from phagosomes and replicates in high numbers in the cytosol.16 In contrast, BCG is an acid fast bacterium and vaccine strain derived from multiple in vitro passages of M. bovis, which arrests phagosomal maturation and fusion with lysosomes and replicates within the phagosome.17 This phagosomal maturation arrest is crucial for the persistence and replication of mycobacteria in macrophages.18
Considering that LVS replicates much faster than BCG and can kill infected macrophages between 48 and 72 h post-infection, we focused on studying the effects of pre-BCG infection-induced host modulation on the subsequent endocytic trafficking and replication of LVS. Distinct LVS killing mechanisms mediated by BCG pre-infection in non-polarized and M2 polarized macrophages are discussed and modeled.
Materials and methods
Mice
All animal experiments were performed in compliance with the Animal Welfare Act, the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the ‘Guide for the Care and Use of Laboratory Animals’ published by the National Research Council. All animal work was done in accordance with the guidelines set forth by the University of Texas at San Antonio Institutional Animal Care and Use Committee, which specifically approved this study under protocol IS00000029. Animals were euthanized in a closed chamber with CO2 followed by cervical dislocation and all tissues were collected post-mortem. Specific pathogen-free 4–8-wk-old mice were used for all procedures. C57BL/6 mice were purchased from the National Cancer Institute. TLR2−/−19 and TLR4−/−20 mice were provided by Dr M. T. Berton (UT Health San Antonio).
Bacteria
F. tularensis LVS (obtained from Dr R. Lyons, University of New Mexico and Dr Karen Elkins, Food and Drug Administration) and F. tularensis Schu S4 (obtained from the Centers for Disease Control, CDC) were grown in tryptic soy broth supplemented with L-cysteine.21 Experiments using Schu S4 were conducted in a CDC-registered and annually certified Animal Biosafety Level 3 (ABSL-3) facility. M. bovis BCG was obtained from Heartland National TB Center and grown in Middlebrook 7H9 broth supplemented with Middlebrook Albumin Dextrose Complex enrichment and 0.05% Tween 80.
Generation of IL-4-expressing J774 cells
Standard molecular cloning methods were employed to insert the mouse IL-4-encoding nucleotide sequence into a pRetroX-Tight-Pur vector using the Retro-XTM Tet-On® Advanced Expression System, according to the manufacturer’s recommendation (Clontech). The vector containing the IL-4 gene or vector alone was used to transfect J774A.1 cells, and the resulting stable cell lines containing either the IL-4 gene or vector alone were designated as J774.IL4 and J774.vec, respectively. IL-4 production in J774.IL4 cells is minimal but can be induced with the addition of doxycycline. To determine optimal induction, J774.IL4 cells were exposed to increasing concentrations (25–100 ng/ml) of doxycycline for 4–12 h (data not shown). Based on the kinetics of IL-4 production, we observed that a minimum of 50 ng/ml doxycycline was required for maximal IL-4 induction by 12 h, and thus this induction condition was used throughout the study.
Cell culture and generation of primary cells for infection
Cells (J774, J774.vec and J774.IL4) were cultured at 37℃ in complete DMEM with 4.5 g/l Gluc, L-glutamine, sodium pyruvate and 10% FBS. Primary macrophages were derived from C57BL/6 wild type (WT), TLR2−/− and TLR4−/− mouse bone marrow as previously described.10 Mincle−/−22, caspase recruitment domain family member 9-deficient (CARD9−/−23) and MyD88−/− bone marrow were gifts from Dr Garry Cole, University of Texas at San Antonio. For infection, J774, J774.vec and J774.IL4 cells were counted and seeded (5 × 105/well in 24-well plates) in the presence of 50 ng/ml doxycycline for a period of 12 h. After 12 h, cells were infected with 10 MOI BCG for 48 h. At the end of the BCG infection period, supernatants were collected and filtered. BCG-infected macrophages were washed with DMEM and co-infected with 10 MOI LVS suspended in the filtered supernatants. LVS uptake and replication were then measured at 3 and 24 h post-LVS inoculation, by lysing infected macrophages with 0.2% deoxycholate and determining the number of viable LVS by serial dilution in sterile PBS and plating on supplemented Tryptic Soy Agar plates.
Quantification of cytokines, NO and arginase activity
Cell supernatants were collected at the indicated time points for assessment of IL-4 concentrations by ELISA, according to the manufacturer’s recommendations (BD Bioscience). Cells were lysed with 0.2% deoxycholate in the presence of protease inhibitor (Roche Diagnostics) for arginase activity measurement using the QuantiChrom Arginase Assay Kit (Gentaur) and reported as U/g (Units per gram of cell lysate protein). NO was detected in culture supernatants using Griess reagent.24 In some experiments, the NO inhibitor NG-monomethyl-L-arginine acetate salt (L-NMMA, 1.0 mM) was added at the time of doxycycline addition and throughout the infection.25
Quantitation of cytosolic and total intracellular LVS
In order to assess escape of LVS from phagosomes to the cytosol, we employed a differential membrane permeabilization method using digitonin and saponin.16,26 Briefly, macrophages were seeded (5 × 105cells/well) onto cover slips, induced with 50 ng/ml doxycycline for 12 h and subsequently infected with 5 × 106 CFU BCG, followed 48 h later by infection with LVS (5 × 106 CFU). Cover slips were fixed with 2% paraformaldehyde at indicated time points and treated with 50 µg/ml digitonin for 5 min at room temperature (approx. 25℃) or with 2% saponin for 30 min at room temperature. Cover slips without detergent treatment were used to determine surface LVS. Cover slips were subsequently blocked using 1% BSA, 0.3 M glycine in PBS for 30 min at room temperature and then incubated with rat anti-LVS primary Ab (generated in our lab) for 2 h. Digitonin permeabilizes the plasma membrane and allows Ab binding to the LVS only in the cytosol. In contrast, saponin permeabilizes all the membranes, including phagosomal membranes, allowing access to all intracellular LVS. Alexa Fluor 488-conjugated goat anti-rat IgG (H + L) (Life Technologies) was used as a secondary Ab to label LVS for 1 h. Cover slips were mounted using FluroSave reagent (Calbiochem) and the presence of the Alexa Fluor 488-labeled LVS was visualized using a Zeiss LSM 510 confocal microscope. Total intracellular LVS was calculated by subtracting surface LVS counts (sum of 25 randomly selected macrophages) from saponin-treated samples (sum of 25 randomly selected macrophages). Similarly, cytosolic LVS was calculated by subtracting surface LVS from LVS counts of digitonin-treated samples.
Statistics
Data were analyzed by Student’s t–test between the two examined groups. A P value of 0.05 or less was considered statistically significant. Data are representative of experiments repeated at least twice.
Results
LVS replication is reduced in alternatively activated (M2) J774 cells
Previously, we have demonstrated that mast cells inhibit F. tularensis replication in macrophages via IL-4 secretion.10,11 In order to study this bacterial inhibition independent of mast cells, we generated the J774.IL4 macrophage cell line, which is capable of producing IL-4 upon doxycycline induction. As shown in Figure 1a, IL-4 was produced in the J774.IL4 cells after 12 h doxycycline (50 ng/ml) induction but not in its absence. In contrast, doxycycline did not induce IL-4 production in J774 or J774.vec (J774 transfected with an empty vector) cells. Increased arginase activity in J774.IL4 cells following doxycycline induction (Figure 1b) further suggested that these cells behaved in a similar fashion to that of an activated M2 phenotype.27 Although there was no marked difference in cell morphology among uninfected doxycycline-treated J774, J774.vec and J774.IL4 cells (Figure 1c, upper panels), J774 and J774.vec cells exhibited a more (38 and 13%, respectively) amoeboid-like shape with multiple pseudopodia following 24 h LVS infection (Figure 1c, lower panels) compared to infected J774.IL4 cells (less than 1% elongated cells), which were found to be more spherical in shape, resembling non-infected macrophages. Similar observations were reported in LVS-infected bone marrow-derived macrophages (BMMØ), which exhibited an increase in surface area, loss of sphericity, elongation and decrease of volume due to apoptosis;10,11 while the presence of IL-4 (addition of exogenous rIL-4 or co-culture with mast cells) led to increased intramacrophage killing of LVS with restoration of spherical morphology. To assess the inhibitory effect of M2 J774.IL4 cells on LVS replication, macrophages were grown for 12 h in the presence of doxycycline, culture medium removed, and the cells infected with LVS (10 MOI) for 3 h (bacterial uptake) and 24 h (replication) without doxycycline. As shown in Figure 2a, bacterial uptake was comparable among the three macrophage types; however, LVS replication was significantly reduced (approximately 1.5 log) in the J774.IL4 compared to J774 and J774.vec cells. Enhanced LVS inhibition in J774.IL4 cells correlated with increased IL-4 production (Figure 2b) and arginase activity (Figure 2c), consistent with the M2 macrophage activation phenotype. Similarly, reduced growth of the human virulent strain F. tularensis Schu S4, associated with increased IL-4 levels, was observed in M2 J774.IL4 macrophages (Figure 2d and e).
Figure 1.

Alternative activation of J774.IL-4 macrophages. J774, J774.vec or J774.IL4 cells were grown for 12 h in the presence (50 ng/ml) or absence of DOX. IL-4 secretion (a) in culture media was measured by ELISA, cellular arginase activity (b) was determined using the QuantiChrom Arginase Assay Kit, and cellular morphology (c) was visualized (400× phase contrast microscopy) 24 h after LVS inoculation (10 MOI). *P < 0.05 between indicated groups. ǂP < 0.05 between the indicated J774.IL4 and respective J774 and J774.vec groups.
BCG: Bacillus Calmette Guẻrin; DOX: doxycycline; LVS: live vaccine strain.
Figure 2.

F. tularensis replication in J774 cell lines. J774, J774.vec and J774.IL4 cells (5 × 105 per well) were incubated with doxycycline (50 ng/ml) for 12 h and subsequently infected with 5 × 106 LVS ((a) to (c)) or Schu S4 ((d) and (e)). Bacterial uptake and replication were measured 3 h and 24 h post-inoculation ((a) and (d)). Additionally, the IL-4 concentration in culture media was measured ((b) and (e)) and the cellular arginase activity was determined (C). ǂP < 0.05 between the indicated J774.IL4 and respective J774 and J774.vec groups.
LVS: live vaccine strain.
BCG-mediated control of intramacrophage LVS replication
In order to study how bacterial superinfection affects LVS replication, macrophages were infected with BCG 48 h prior to LVS infection. Using a GFP-expressing BCG28,29 and an mCherry-expressing LVS30 for image flow cytometry assays (the Imagestream MKII, Amnis, EMD Millipore),31 we estimated that 62% of J774 cells were co-infected with BCG and LVS at 4 h post-LVS inoculation. In addition, 4 and 23% of J774 cells harbored LVS and BCG alone, respectively (Figure 3a to d). These results suggest that > 90% of LVS-infected cells were pre-infected by BCG. It also was noted that uptake of LVS by J774 cells, with or without pre-BCG infection, was comparable (Figure 3e). Similarly, comparable rates (> 90%) of LVS infection were associated with BCG pre-infection among doxycycline-treated J774, J774.vec and J774.IL4 cells. Representative images from flow cytometric assays of these BCG–LVS co-infected cells are shown in Figure 3f. A twofold increase in cellular GFP intensity was observed in BCG-infected J774.IL4, compared to J774 (and J774.vec), cells, suggesting that M2 macrophages are more susceptible to BCG infection, which is consistent with other reports.32,33
Figure 3.

Uptake of BCG and LVS by J774 macrophage-like cells. Untreated J774 cells ((a) to (e)) or 50 ng/ml doxycycline-treated J774, J774.vec and J774.IL4 cells (f) were seeded (5 × 105/well in 24-well plates) for 12 h followed by infection with GFP-expressing BCG (10 MOI) for 48 h, and subsequent inoculation with mCherry-expressing LVS (10 MOI). After 4 h incubation, co-infection with BCG and LVS was visualized and frequency analyzed using the Imagestream MKII (Amnis, EMD Millipore). (a) The dot-plot depicts the GFP and mCherry intensity of each cell and three gated cell populations: (b) mCherryhiGFPlow, (c) mCherryhiGFPhi and (d) mCherrylowGFPhi. The representative cell images of these three gated populations are shown in (b) LVS-infected J774 cells, (c) BCG–LVS co-infected cells (cellular localization of bacteria within this population was 93% using the Internalization Index analysis, IDEAS®) and (d) BCG-infected cells. (e) Uptake of mCherry LVS by J774 cells was comparable between LVS alone (pink area) and BCG + LVS co-infection (green line). (F) Shown are representative J774, J774.vec and J774.IL4 cells co-infected with BCG and LVS.
BCG: Bacillus Calmette Guẻrin; BF: Bright Field; LVS: live vaccine strain.
It has been well documented that LVS escapes from phagosomes within 30–60 min following phagocytosis with resulting replication in the cytosol.13 Our data are consistent with these observations, indicating that in both J774 and J774.vec cells the cytosolic LVS numbers (macrophages permeabilized with digitonin) were similar to the total intracellular LVS counts (macrophages permeabilized with saponin) following 1 and 4 h of LVS infection, respectively, suggesting that most LVS had escaped from phagosomes by 1 h (Figure 4a). In contrast, approximately 50% of LVS remained in the phagosomes of doxycycline-activated J774.IL4 (M2) cells at 4 h post-infection (Figure 4a). Interestingly, pre-BCG infection (48 h) did not prevent LVS escape from phagosomes in J774 and J774.vec cells within 4 h (Figure 4b), but pre-infection with BCG did result in the reduction of LVS replication at 24 h in macrophages compared to LVS infection alone (Figure 4c). This BCG-mediated LVS inhibition appears to be associated with NO production (Figure 4d, J774 and J774.vec). Infection with BCG 12 h prior to LVS infection (time 0, Figure 4d, J774 and J774.vec) induced a significant amount of NO, which had minimal effect on uptake of LVS (Figure 4c, 3 h). However, increased NO production (Figure 4d, comparing BCG + LVS to LVS alone) correlated with decreased LVS replication (Figure 4c, 24 h). In contrast, in M2 polarized J774.IL4 cells, pre-BCG infection did not reduce arginase production (data not shown) nor induce NO production (Figure 4d), and had little effect on LVS escape (Figure 4a and b) and replication (Figure 4c, LVS vs BCG + LVS).
Figure 4.

BCG and LVS co-infection in J774 cell lines. J774 cell lines (5 × 105) were seeded in wells on cover slips and IL-4 expression induced with 50 ng/ml doxycycline for 12 h. Cells were then infected with either (a) LVS (5 × 106) or (b) BCG (5 × 106) for 24 h, followed by LVS (5 × 106). At 1 and 4 h post-LVS inoculation, cells were fixed, treated with either Dig or Sap, and stained for LVS. The cytosolic (LVS-Dig and BCG + LVS-Dig) and total intracellular (LVS-Sap and BCG + LVS-Sap) LVS quantifications were determined as described in the Materials and methods. Concurrently, J774 cell lines (5 × 105 per well) were induced with doxycycline, infected with BCG or mock treated with medium for 48 h, and infected with LVS. Uptake and replication of LVS were measured at 3 and 24 h post-LVS inoculation (c), and NO levels in culture medium were measured prior to and 24 h after LVS inoculation using the Griess reagent (d). *P < 0.05 between indicated groups. ǂP < 0.05 between the indicated J774.IL4 and respective J774 and J774.vec groups.
BCG: Bacillus Calmette Guẻrin; Dig: digoxin; LVS: live vaccine strain; Sap: saponin.
Inhibition of intramacrophage LVS replication by BCG was associated with NO production
To confirm the essential role of NO in BCG-mediated LVS inhibition, macrophages were treated with the NO inhibitor L-NMMA. As shown in Figure 5a, BCG infection significantly elevated NO production in J774 and J774.vec cells as well as in BMMØs 48 h post-inoculation, while addition of L-NMMA significantly abrogated NO production. We further assessed the role of NO in suppression of LVS intramacrophage replication with LVS infection alone and pre-infection with BCG. We observed that LVS infection alone induced minimal NO in all three J774 cell lines and in BMMØs (Figure 5b, upper panels). Consistent with previous observations, LVS replication was significantly reduced in the M2 J774.IL4 macrophages in contrast to M0 J774 and J774.vec cells, and addition of the NO inhibitor L-NMMA had no effect on LVS replication in all three cell lines or in BMMØs (Figure 5b, lower panels). However, when pre-infected with BCG, LVS replication was inhibited in J774, J774.vec and BMMØs (Figure 5b, lower panels), and this inhibition correlated with marked NO production (Figure 5b, upper panels).
Figure 5.

NO-mediated control of LVS replication. J774, J774.vec and J774.IL-4 cells (1 × 106 per well) were induced with doxycycline (50 ng/ml, 12 h). BMMØs derived from C57BL/6 mice were infected with BCG (1 × 107) in the presence or absence of NO inhibitor L-NMMA, and NO levels in culture media measured 24 h post-inoculation (a). In a similar study, BMMØs and doxycycline-induced J774 cell lines were infected with BCG, or mock treated with medium in the presence or absence of L-NMMA for 48 h, and infected with LVS. NO concentration ((b), upper panel) and LVS replication ((b), lower panel) were measured 24 h post-LVS inoculation. *P < 0.05 between indicated groups. ǂP < 0.05 between the indicated J774.IL4 and respective J774 and J774.vec groups.
BCG: Bacillus Calmette Guẻrin; BMMØ: bone marrow-derived macrophages; L-NMMA: NG-monomethyl-L-arginine acetate salt; LVS: live vaccine strain.
Additionally, inhibition of LVS was abrogated in the presence of the NO inhibitor L-NMMA. These results strongly suggest that BCG-mediated LVS inhibition is NO-dependent in M0 macrophages. However, BCG and NO have a minimal role in the control of LVS replication in M2 J774.IL4 cells (Figure 5b, lower left panel).
Activation of Mincle and TLR2 signaling is critical for the control of LVS replication by BCG pre-infection
BCG has an atypical cell wall that contains ligands for various PRRs. For example, lipoproteins, phosphatidylinositol mannans and lipomannan are all TLR2 ligands that are present on the surface of BCG.34 The adaptor molecule for TLR2 signaling is MyD88, which plays a central role in activation of the NF-kB pathway and the production of pro-inflammatory cytokines.35 BCG also contains an unique glycolipid, trehalose 6,6'-dimycolate (TDM), which binds to C-type lectin receptors such as Mincle that signal through the adaptor protein CARD9 to stimulate inflammatory responses.36 In order to determine if these PRR signaling components are essential for BCG-mediated NO-dependent LVS inhibition, we generated BMMØs from C57BL/6 Mincle, CARD9, TLR2 and MyD88 knockout mice. We also used C57BL/6 WT and TLR4−/− BMMØs for comparison. In contrast to TLR2, TLR4 has been shown to play a minimal role in macrophage activation by BCG.37 We infected BMMØs with BCG for 48 h, measured NO levels in culture media, and observed that deficiency of Mincle, CARD9, TLR2 and MyD88, but not TLR4, resulted in a significant reduction of NO production (Figure 6a), suggesting that activation of Mincle-CARD9 and TLR2-MyD88 signaling was critical for BCG-induced NO production in macrophages. Similar reductions of NO levels were observed in BCG + LVS superinfected Mincle-, CARD9-, TLR2- and MyD88-deficient BMMØs (Figure 6b), and these NO reductions correlated with loss of BCG-mediated LVS inhibition in these four cell types (Figure 6c). In contrast, TLR4 was not essential in BCG-mediated LVS inhibition.
Figure 6.

Innate signaling is required for BCG-mediated LVS inhibition during BCG–LVS superinfection. BMMØs prepared from WT C57BL6 and various gene-deficient mice, including Mincle, CARD9, TLR2, TLR4 and MyD88, were infected with BCG for 48 h and NO production was measured (a). Similarly prepared BMMØs were infected with LVS for 24 h with (BCG + LVS) or without (mock + LVS) prior BCG (48 h) infection, and the NO levels in culture medium (b) and viable LVS within macrophages (c) were analyzed. *P < 0.05 between indicated groups. ǂP < 0.05 between the indicated gene-deficiency and WT groups.
BCG: Bacillus Calmette Guẻrin; BMMØ: bone marrow-derived macrophages; CARD9: caspase recruitment domain family member 9; LVS: live vaccine strain; WT, wild type.
Discussion
In this study, we investigated the influence of pre-BCG exposure in M0 or M2 polarized macrophages on subsequent LVS infection. Based upon our published observations of IL-4-mediated LVS inhibition10,11 and data obtained from this study, we propose a model (Figure 7) exhibiting the two distinct defense mechanisms by which macrophages control LVS infection. When the M0 J774 and BMMØ cells are infected with BCG, their surface PPRs, i.e. TLR2 and Mincle, may recognize respective ligands (e.g. liproproteins and TDM) from BCG and subsequently activate NF-κB through the MyD88 and CARD9 adaptors. Activation of NF-κB may then up-regulate inducible NO synthase (iNOS) gene transcription and protein expression, resulting in increased NO production. Subsequent LVS infection of BCG-activated macrophages allows most LVS to escape from phagosomes followed by elimination via NO-mediated killing. In contrast, in M2 J774.IL4 macrophages, IL-4 may upregulate arginase (ARG1) gene transcription and protein expression via IL-4 R binding and STAT6 phosphorylation.38 The induced arginase converts arginine to ornithine, a precursor of polyamines and hydroxyproline that induces cell proliferation and collagen production.39 Because of the high ARG1 activity in M2-polarized J774.IL4 cells, minimal arginine, the common substrate for iNOS and ARG1, is available for conversion to L-hydroxy-arginine by BCG induced iNOS, with resulting minimal NO production following BCG infection. During subsequent LVS infection in M2 macrophages with prior BCG exposure, fewer LVS escape from the phagosome into the cytosol, and the macrophages control LVS growth by enhancing ATP production and phagosomal acidification as previously reported.11 In contrast, reduction of LVS replication in M2 J774.IL4 is independent of BCG activation during BCG–LVS superinfection.
Figure 7.
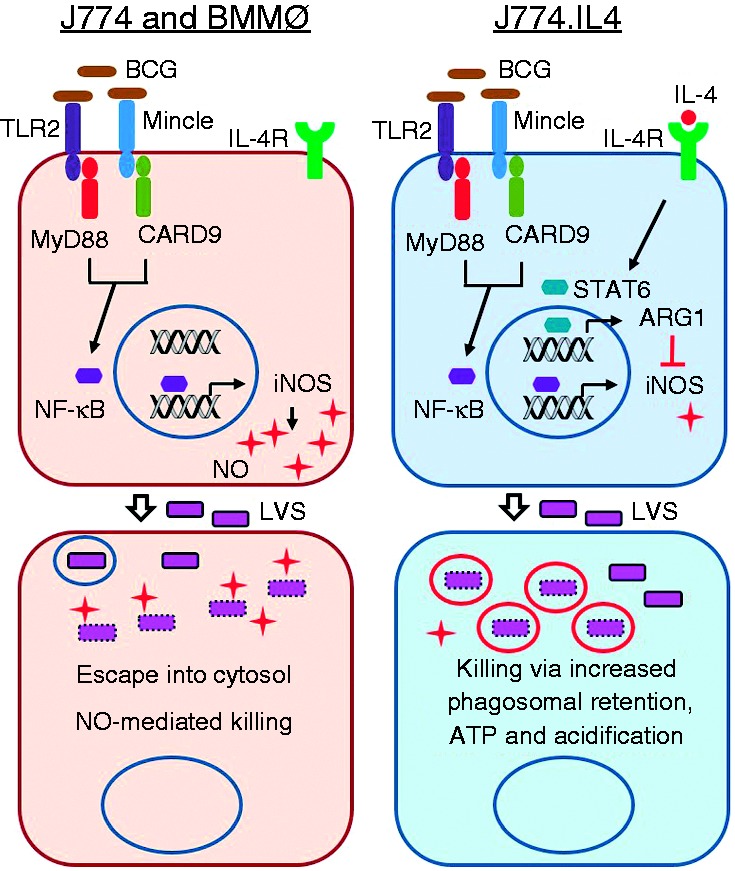
Working model for BCG-mediated LVS inhibition during BCG–LVS superinfection. Shown in the proposed model are distinct LVS inhibition mechanisms following BCG–LVS superinfection in M0 J774/BMMØs and M2 J774.IL4 cells. In the M0 macrophage, BCG infection activates TLR2-MyD88 and Mincle-CARD9 signal pathways leading to NO production for subsequent LVS killing. In contrast, BCG has minimal effect on IL-4 mediated LVS killing in J774.IL4 M2 macrophages.
BCG: Bacillus Calmette Guẻrin; BMMØ: bone marrow-derived macrophages; iNOS: inducible NO synthase; CARD9: caspase recruitment domain family member 9; LVS: live vaccine strain; M0: non-activated.
Both BCG and LVS can evade the endosomal–lysosomal degradation pathway, but by different mechanisms. Following phagocytosis, typical pathogen-containing phagosomes acquire markers such as Rab5 (a small GTPase) and EEA1 (early endosomal antigen 1), which directs the fusion of phagosomes with early endosomal vesicles.40 Endosomes continue to mature into late endosomes by replacing Rab5 with Rab7, and become acidified after the acquisition of vacuolar proton-ATPase molecules, eventually fusing with lysosomes for ultimate pathogen degradation. In the case of F. tularensis uptake by macrophages, the bacterium-containing phagosome matures into early and late endosomes, but fails to become acidified. Non-acidified late endosome-like phagosomes containing F. tularensis do not fuse with lysosomes and allow the bacteria to escape into the cytoplasm following gradual disruption of the vesicle.8 However, in J774.IL4 cells, IL-4 activation most likely enhances endosomal acidification, as suggested by our previous studies,11 improving LVS killing. Conversely, BCG phagosomes acquire some endosomal markers such as Rab 5, but do not mature into late endosomes by preventing Rab5/Rab7 conversion, and BCG replicates in these arrested vesicles.17,41 However, pre-infection with BCG appears to have little effect on LVS escape from the endosomes in both J774 and BMMØs.
It should be noted that the results presented here were obtained from a population of macrophages pre-infected with BCG and then infected with LVS. We have not specifically investigated the macrophages during simultaneous co-infection with both BCG and LVS. However, > 90% of LVS-infected cells were pre-infected with BCG (Figure 3). Thus, the observed LVS replication was a collective outcome from BCG pre-infected macrophages and single LVS-infected macrophages activated by BCG-induced secretory factors. A mixture of singly and multi-infected macrophages may exist in naturally occurring co-infection. Furthermore, our results imply a complexity of disease outcome via pre-infection due to the plasticity and dynamics of macrophage phenotypes in the infection compartment. It also needs to be noted that experiments conducted in this study also used BMMØ but not the more disease relevant alveolar macrophages (ALVM); however, we have compared rat BMMØ and ALVM and demonstrated comparable Francisella replication in these two types of macrophages.42 Additional studies using mouse ALVM are required to confirm the similar mechanisms of BCG-mediated inhibition of LVS replication that were observed in BMMØ and J774 cells.
In summary, we have provided evidence of BCG-mediated suppression of LVS replication using an in vitro macrophage infection model and have further characterized the mechanisms of LVS killing by M0, M1 (equivalent to BCG infection alone) and M2 macrophage phenotypes. Additionally, we have answered the question of whether an M1 polarizing pathogen influences the outcome of secondary infection in already M2 polarized macrophages by altering the bacterial killing mechanism(s). However, the effect of BCG on pneumonic tularemia in animals following superinfection remains to be elucidated. In this regard, mycobacterium-mediated protection against lethal malaria infection in a co-infection mouse model has been demonstrated.43
Acknowledgements
We thank Mr Srikanth Manam and Dr Jilani Chaudry for their technical support for the construction of the J774.vec and J774.IL4 cell lines. We also thank Dr Chinnaswamy Jagannath at the University of Texas Health Science Center at Houston who kindly provided the GFP-expressing BCG under support from his National Institutes of Health (NIH) grant (AI-78420). Image flow analyses were conducted in the University of Texas at San Antonio Immune Defense Core (supported by Research Centers in Minority Institutions, NIH grant G12MD007591).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: funding from the Army Research Office of the Department of Defense under contract no. W911NF-11-1-0136, and by the Jane and Roland Blumberg Professorship in Biology, to BPA.
References
- 1.Lijek RS, Weiser JN. Co-infection subverts mucosal immunity in the upper respiratory tract. Curr Opin Immunol 2012; 24: 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakaletz LO. Developing animal models for polymicrobial diseases. Nat Rev Microbiol 2004; 2: 552–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrevillaga G, Gaona J, Sanchez C, et al. Respiratory syncytial virus persistence in macrophages downregulates intercellular adhesion molecule-1 expression and reduces adhesion of non-typeable Haemophilus influenzae. Intervirology 2012; 55: 442–450. [DOI] [PubMed] [Google Scholar]
- 4.Gomes MS, Paul S, Moreira AL, et al. Survival of Mycobacterium avium and Mycobacterium tuberculosis in acidified vacuoles of murine macrophages. Infect Immun 1999; 67: 3199–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Draijer C, Robbe P, Boorsma CE, et al. Characterization of macrophage phenotypes in three murine models of house-dust-mite-induced asthma. Mediators Inflamm 2013; 2013: 632049–632049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melgert BN, ten Hacken NH, Rutgers B, et al. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immunol 2011; 127: 831–833. [DOI] [PubMed] [Google Scholar]
- 7.Shaykhiev R, Krause A, Salit J, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 2009; 183: 2867–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santic M, Molmeret M, Klose KE, et al. Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol 2006; 14: 37–44. [DOI] [PubMed] [Google Scholar]
- 9.Karabay O, Kilic S, Gurcan S, et al. Cervical lymphadenitis: tuberculosis or tularaemia? Clin Microbiol Infect 2013; 19: E113–E117. [DOI] [PubMed] [Google Scholar]
- 10.Ketavarapu JM, Rodriguez AR, Yu JJ, et al. Mast cells inhibit intramacrophage Francisella tularensis replication via contact and secreted products including IL-4. Proc Natl Acad Sci USA 2008; 105: 9313–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez AR, Yu JJ, Murthy AK, et al. Mast cell/IL-4 control of Francisella tularensis replication and host cell death is associated with increased ATP production and phagosomal acidification. Mucosal Immunol 2011; 4: 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redente EF, Higgins DM, Dwyer-Nield LD, et al. Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. J Leukoc Biol 2010; 88: 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyston PC. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J Med Microbiol 2008; 57: 921–930. [DOI] [PubMed] [Google Scholar]
- 14.Pechous RD, McCarthy TR, Zahrt TC. Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol Mol Biol Rev 2009; 73: 684–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones CL, Napier BA, Sampson TR, et al. Subversion of host recognition and defense systems by Francisella spp. Microbiol Mol Biol Rev 2012; 76: 383–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong A, Wehrly TD, Nair V, et al. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun 2008; 76: 5488–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee BY, Jethwaney D, Schilling B, et al. The Mycobacterium bovis bacille Calmette-Guerin phagosome proteome. Mol Cell Proteomics 2010; 9: 32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vergne I, Chua J, Singh SB, et al. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol 2004; 20: 367–394. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999; 11: 443–451. [DOI] [PubMed] [Google Scholar]
- 20.Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162: 3749–3752. [PubMed] [Google Scholar]
- 21.Yu JJ, Goluguri T, Guentzel MN, et al. Francisella tularensis T-cell antigen identification using humanized HLA-DR4 transgenic mice. Clin Vaccine Immunol 2010; 17: 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells CA, Salvage-Jones JA, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol 2008; 180: 7404–7413. [DOI] [PubMed] [Google Scholar]
- 23.Hsu YM, Zhang Y, You Y, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 2007; 8: 198–205. [DOI] [PubMed] [Google Scholar]
- 24.Green LC, Wagner DA, Glogowski J, et al. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 1982; 126: 131–138. [DOI] [PubMed] [Google Scholar]
- 25.Marletta MA, Yoon PS, Iyengar R, et al. Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry 1988; 27: 8706–8711. [DOI] [PubMed] [Google Scholar]
- 26.Checroun C, Wehrly TD, Fischer ER, et al. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A 2006; 103: 14578–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009; 27: 451–483. [DOI] [PubMed] [Google Scholar]
- 28.Katti MK, Dai G, Armitige LY, et al. The Delta fbpA mutant derived from Mycobacterium tuberculosis H37Rv has an enhanced susceptibility to intracellular antimicrobial oxidative mechanisms, undergoes limited phagosome maturation and activates macrophages and dendritic cells. Cell Microbiol 2008; 10: 1286–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh CR, Bakhru P, Khan A, et al. Cutting edge: Nicastrin and related components of gamma-secretase generate a peptide epitope facilitating immune recognition of intracellular mycobacteria, through MHC class II-dependent priming of T cells. J Immunol 2011; 187: 5495–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray HJ, Cong Y, Murthy AK, et al. Oral live vaccine strain-induced protective immunity against pulmonary Francisella tularensis challenge is mediated by CD4+ T cells and Abs, including immunoglobulin A. Clin Vaccine Immunol 2009; 16: 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham AL, Guentzel MN, Yu JJ, et al. M-Cells contribute to the entry of an oral vaccine but are not essential for the subsequent induction of protective immunity against Francisella tularensis. PLoS ONE 2016; 11: e0153402–e0153402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaufmann SH, Flesch IE. Cytokines in antibacterial resistance: possible applications for immunomodulation. Lung 1990; 168: 1025–1032. [DOI] [PubMed] [Google Scholar]
- 33.Reljic R, Stylianou E, Balu S, et al. Cytokine interactions that determine the outcome of Mycobacterial infection of macrophages. Cytokine 2010; 51: 42–46. [DOI] [PubMed] [Google Scholar]
- 34.Begum NA, Kobayashi M, Moriwaki Y, et al. Mycobacterium bovis BCG cell wall and lipopolysaccharide induce a novel gene, BIGM103, encoding a 7-TM protein: identification of a new protein family having Zn-transporter and Zn-metalloprotease signatures. Genomics 2002; 80: 630–645. [DOI] [PubMed] [Google Scholar]
- 35.Akira S. TLR signaling. Curr Top Microbiol Immunol 2006; 311: 1–16. [DOI] [PubMed] [Google Scholar]
- 36.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 2009; 9: 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heldwein KA, Liang MD, Andresen TK, et al. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J Leukoc Biol 2003; 74: 277–286. [DOI] [PubMed] [Google Scholar]
- 38.Weisser SB, McLarren KW, Voglmaier N, et al. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immunol 2011; 41: 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hesse M, Modolell M, La Flamme AC, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol 2001; 167: 6533–6544. [DOI] [PubMed] [Google Scholar]
- 40.Huotari J, Helenius A. Endosome maturation. EMBO J 2011; 30: 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun J, Deghmane AE, Soualhine H, et al. Mycobacterium bovis BCG disrupts the interaction of Rab7 with RILP contributing to inhibition of phagosome maturation. J Leukoc Biol 2007; 82: 1437–1445. [DOI] [PubMed] [Google Scholar]
- 42.Signarovitz AL, Ray HJ, Yu JJ, et al. Mucosal immunization with live attenuated Francisella novicida U112 DiglB protects against pulmonary F. tularensis SCHU S4 in the Fischer 344 rat model. PLoS ONE 2012; 7: e47639–e47639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Page KR, Jedlicka AE, Fakheri B, et al. Mycobacterium-induced potentiation of type 1 immune responses and protection against malaria are host specific. Infect Immun 2005; 73: 8369–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]