

Summary
The recently discovered comammox process encompasses both nitrification steps, the aerobic oxidation of ammonia and nitrite, in a single organism. All known comammox bacteria are affiliated with Nitrospira sublineage II and can be grouped into two distinct clades, referred to as A and B, based on ammonia monooxygenase phylogeny. In this study, we report high‐quality draft genomes of two novel comammox Nitrospira from the terrestrial subsurface, representing one clade A and one clade B comammox organism. The two metagenome‐assembled genomes were compared with other representatives of Nitrospira sublineage II, including both canonical and comammox Nitrospira. Phylogenomic analyses confirmed the affiliation of the two novel Nitrospira with comammox clades A and B respectively. Based on phylogenetic distance and pairwise average nucleotide identity values, both comammox Nitrospira were classified as novel species. Genomic comparison revealed high conservation of key metabolic features in sublineage II Nitrospira, including respiratory complexes I–V and the machineries for nitrite oxidation and carbon fixation via the reductive tricarboxylic acid cycle. In addition, the presence of the enzymatic repertoire for formate and hydrogen oxidation in the Rifle clades A and B comammox genomes, respectively, suggest a broader distribution of these metabolic features than previously anticipated.
Introduction
Nitrogen (N) is a key nutritional element for life on Earth and is essential for the biosynthesis of nucleic acids and proteins. In many environments, including unperturbed terrestrial ecosystems, N represents a growth‐limiting factor. Thus, artificial N fertilizers are intensively used in agriculture to enhance crop production, resulting in a doubling of the N flux into terrestrial environments and a severe perturbation of the global N cycle (Galloway et al., 2008). The biogeochemical N cycle comprises a series of aerobic and anaerobic processes mainly performed by microorganisms. Among these, nitrifying microorganisms play an essential role by performing the stepwise aerobic oxidation of ammonia to nitrate. Nitrification is mediated by functionally distinct groups of chemolithoautotrophic microorganisms: the ammonia‐oxidizing bacteria (AOB) or archaea (AOA), which operate in a tight interplay with nitrite‐oxidizing bacteria (NOB). However, nitrification can also be catalysed in a single organism by the recently discovered complete ammonia‐oxidizing (comammox) Nitrospira (Daims et al., 2015; van Kessel et al., 2015). The genus Nitrospira, which prior to the discovery of comammox had been regarded to comprise specialized nitrite oxidizers only, represents the most diverse NOB clade, harbouring at least six phylogenetic sublineages observed in a wide range of natural aquatic and terrestrial habitats, and engineered environments like drinking and wastewater treatment plants (Daims et al., 2001; Lebedeva et al., 2011; Daebeler et al., 2014; Daims et al., 2016; Gülay et al., 2016). All complete nitrifiers known to date are affiliated with Nitrospira sublineage II (Daims et al., 2015; van Kessel et al., 2015; Pinto et al., 2016; Palomo et al., 2018). Furthermore, comammox Nitrospira form two divergent clades, referred to as comammox clades A and B, based on phylogenetic analysis of the ammonia monooxygenase (AMO), the enzyme catalysing ammonia oxidation (Daims et al., 2015).
So far, most comammox Nitrospira genomes were obtained from engineered systems (Daims et al., 2015; van Kessel et al., 2015; Pinto et al., 2016; Wang et al., 2017; Palomo et al., 2018), some of which are characterized by low concentrations of ammonium. Especially in these, comammox Nitrospira appeared to dominate the nitrifying community (Bartelme et al., 2017; Pjevac et al., 2017; Koch et al., 2018), which is in line with the hypothesis that the comammox process is beneficial under substrate‐limited conditions selecting for high growth yields (Costa et al., 2006). Recent kinetic characterization of a complete nitrifier confirmed that comammox Nitrospira are indeed adapted to highly oligotrophic conditions and due to a higher affinity for ammonia might out‐compete canonical ammonia‐oxidizing microorganisms (Kits et al., 2017).
By now, comammox Nitrospira were also detected in several natural environments, including lake sediment and forest and agricultural soils (Orellana et al., 2017; Parks et al., 2017; Pjevac et al., 2017; Xia et al., 2018). Notably, in fertilized soils (Orellana et al., 2017) and acidic subtropical forest soils (Shi et al., 2018), the increased abundances of comammox Nitrospira in response to human‐induced N loadings indicate that they can drive nitrification also under less oligotrophic conditions. The observed diversity together with the identification of comammox Nitrospira as the most abundant nitrifiers in acidic forest soil (Hu and He, 2017) as well as in estuary and coastal environments (Xia et al., 2018) indicates their vital contribution to nitrification also in natural systems. However, comammox Nitrospira genomes from natural environments were rarely recovered and analysed so far, and thus the metabolic capabilities of complete nitrifiers in these ecosystems are poorly understood.
In this study, we recovered two high‐quality draft genome sequences of novel comammox Nitrospira from the Rifle sampling site, an aquifer adjacent to the Colorado River. Microbial community composition and inter‐species interactions in this subsurface environment have been extensively studied (Castelle et al., 2013; Brown et al., 2015; Hug et al., 2015; Anantharaman et al., 2016). A recent metagenomic characterization of this aquifer revealed that the nitrifying community comprises mainly Nitrospira‐like bacteria and canonical ammonia oxidizers appeared to be absent (Anantharaman et al., 2016). Here, a comparative genomic approach was used to analyse the two novel comammox genomes from the Rifle terrestrial subsurface in comparison to other sublineage II Nitrospira species, including canonical nitrite oxidizing and comammox organisms. To the best of our knowledge, this is the first genomic characterization of clades A and B comammox Nitrospira derived from the terrestrial subsurface, which is a valuable step towards understanding their environmental significance and distribution, and will help to identify metabolic drivers of niche differentiation between the comammox clades.
Result and discussion
General genomic information
This study reports two novel comammox Nitrospira metagenome‐assembled genomes (abbreviated as RCA and RCB, designating the comammox clade A and B genomes respectively) retrieved from the terrestrial subsurface of the Rifle sampling site, an aquifer adjacent to the Colorado River (CO). The high‐quality draft genomes are estimated to be 94% (RCA) and 91% (RCB) complete and comprise 88 and 296 contigs respectively (Table 1). The pairwise comparison of the Rifle comammox genomes with 32 other Nitrospira genomes showed that the maximum average nucleotide identity (ANI) values for these genomes were below the defined species cut off of 95% (Richter and Rossello‐Mora, 2009). This classifies them as novel species, which was also confirmed by the phylogenetic distances to their closest relatives in phylogenomic analyses (see below). The observed GC content, genome sizes and the number of predicted protein‐coding sequences (CDS) are in the range of previously published Nitrospira sublineage II genomes (Supporting Information Table S1). The pangenome of 24 sublineage II Nitrospira species was analysed by using reciprocal BLAST hit (RBH) analysis to identify shared and unique proteins (Supporting Information Tables S2 and S3). Only 12% of RCA and 11% of RCB CDS are conserved in all analysed sublineage II Nitrospira genomes and more than half of all CDS (~54%) in both Rifle genomes are predicted proteins of unknown function.
Table 1.
General characteristics of Rifle comammox Nitrospira genomes.
| Nitrospira sp. RCA | Nitrospira sp. RCB | |
|---|---|---|
| Completenessa | 94% | 91% |
| Redundancya | 2.7% | 2.7% |
| Genome size (Mb) | 3.29 | 3.55 |
| GC content | 56.8% | 57.1% |
| Number of contigs | 88 | 296 |
| N50 of contigs | 92 268 | 18 857 |
| Number of CDSsb | 3456 | 3711 |
| Coding density | 86% | 83.9% |
| rRNAs | 1 | 0 |
| tRNAs | 41 | 42 |
| CDSc in core genome | 407 (12%) | 400 (11%) |
| Species‐specific CDSc | 853 (25%) | 966 (26%) |
Based on lineage‐specific marker sets determined with CheckM (Parks et al., 2015).
Inferred with Prodigal (Hyatt, 2010).
CDS with RBH hits with an amino acid identity ≥45% and a minimum alignment length ≥70% were defined as homologues proteins. CDS with no RBH hit were considered species specific.
Phylogenetic affiliation of the novel comammox Nitrospira
To infer the phylogenetic affiliation of the Rifle comammox Nitrospira, we reconstructed a maximum likelihood (ML) phylogenomic tree based on a concatenated alignment of 91 single copy core genes (Fig. 1). This clearly affiliated the two species with comammox clades A and B respectively. Furthermore, according to this analysis, complete nitrifiers form two monophyletic groups within Nitrospira sublineage II. This separation of comammox into clades A and B is consistent with the amoA‐based phylogeny (Fig. 2) but in stark contrast to 16S rRNA gene‐based analyses where this monophyletic structure of the comammox clades is not observed (Pinto et al., 2016). Here, the clade A comammox Nitrospira inopinata clusters with the strict nitrite‐oxidizing Nitrospira moscoviensis, making it impossible to reliably distinguish canonical and comammox species based on their 16S rRNA gene. However, in the phylogenomic tree, the two distinct comammox clades are closely affiliated with canonical Nitrospira (Fig. 1), which can hamper the identification of novel comammox organisms when they cluster close to but not within the known comammox clades. An additional group containing canonical Nitrospira clustering in between the comammox clades further suggests a complex evolutionary history of the ammonia oxidation pathway within Nitrospira, as already discussed in the former studies (Palomo et al., 2018).
Figure 1.
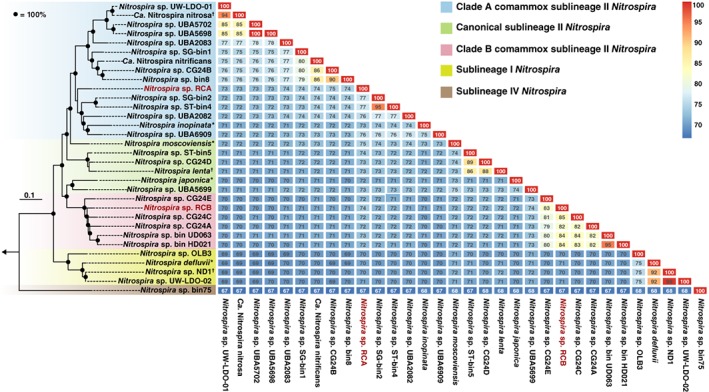
Phylogenomic analysis of the genus Nitrospira. The maximum likelihood tree was constructed using a concatenated alignment of 91 single copy core genes. Nitrospira sublineages and comammox clades are indicated by coloured boxes. Asterisks behind species names indicate closed genomes, daggers high‐quality assemblies with ≤22 contigs. Bootstrap support values = 100% are indicated by black circles. The arrow indicates the position of the outgroup, which consisted of two Leptospirillum species. The scale bar corresponds to 10% estimated sequence divergence. The genome similarity heatmap on the right gives the pairwise ANI values between all 32 Nitrospira genomes included in the analysis.
Figure 2.
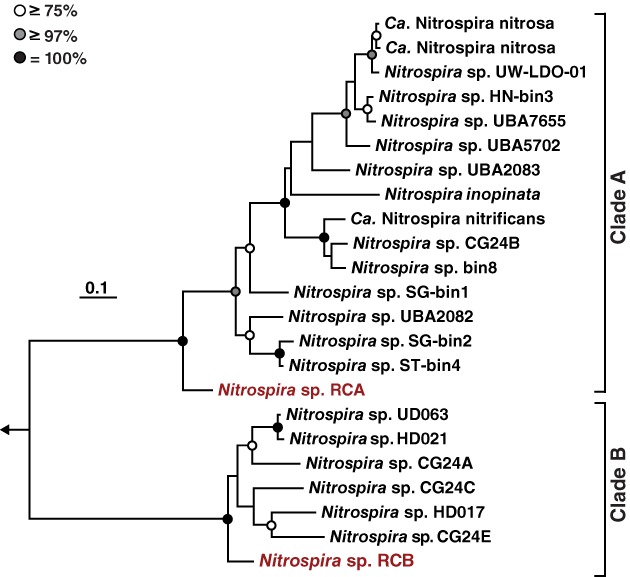
Maximum likelihood phylogenetic tree of 22 comammox Nitrospira amoA sequences. The arrow indicates the position of the outgroup, which consisted of two Nitrosomonas sequences. The Rifle comammox Nitrospira sequences are shown in red. Bootstrap support values ≥75%, ≥97% and =100% are indicated by white, grey and black circles, respectively. The scale bar corresponds to 10% estimated sequence divergence.
Respiratory chain and carbon metabolism
Genes for respiratory complexes I–V are highly conserved in all Nitrospira (Lücker et al., 2010; Koch et al., 2015; Palomo et al., 2018), including in the RCA and RCB genomes. Furthermore, the core genome contained all genes for glycolysis/gluconeogenesis, the non‐oxidative pentose phosphate pathway and the tricarboxylic acid cycle (Supporting Information Tables S2 and S3). The identification of key enzymes for the reductive tricarboxylic acid cycle (rTCA), including ATP‐citrate lyase, 2‐oxoglutarate:ferredoxin oxidoreductase and pyruvate:ferredoxin oxidoreductase in the RCA and RCB genomes suggests that, like all Nitrospira, these comammox species employ the rTCA for CO2 fixation. All analysed genomes furthermore contained pyruvate carboxylase subunits A and B, required for the carboxylation of pyruvate to form oxaloacetate in order to replenish TCA cycle intermediates withdrawn for biosynthesis reactions. Nitrospira furthermore has the genomic potential to utilize simple organic substrates such as pyruvate (Lücker et al., 2010; Koch et al., 2015), but their role to support mixotrophic growth is not fully understood yet. The uptake of pyruvate was shown for some uncultured Nitrospira in activated sludge (Daims et al., 2001), while no assimilation by sublineage I Nitrospira was observed in a later study (Gruber‐Dorninger et al., 2015). Moreover, N. moscoviensis did not use pyruvate as an electron donor under anoxic conditions (Koch et al., 2015).
Nitrogen metabolism
Complete nitrifiers grow chemolithoautotrophically by aerobic oxidation of ammonia to nitrate (van Kessel et al. 2015, Daims et al. 2015). Both Rifle comammox genomes contained the full gene set for AMO and hydroxylamine dehydrogenase (HAO) necessary for ammonia oxidation to nitrite and all subunits of the nitrite oxidoreductase (NXR) for nitrite oxidation. The AMO structural genes amoCAB of RCA were clustered together with the putative AMO subunits amoEDD2, haoAB and cycAB encoding HAO and the associated quinone‐interaction module, and a copper transporter (copCD). In RCB amoCAB were localized on a small contig along with few hypothetical proteins and a transposase family protein (Fig. 3). Similar to RCB, transposase genes were identified directly upstream of amoCAB also in the genome of N. inopinata, and the entire operon had a divergent tetranucleotide signature (Daims et al., 2015). These features might indicate that comammox Nitrospira acquired the ammonia‐oxidizing capability through lateral gene transfer (Daims et al., 2015; Palomo et al., 2018). Like betaproteobacterial AOB, all comammox Nitrospira contain genes for the cytochrome c maturation system I, which is absent in most canonical Nitrospira. Intriguingly, also few strict nitrite‐oxidizing sublineage II Nitrospira possess this gene cluster (Supporting Information Fig. S1), indicating either a loss of the ammonia‐oxidizing potential in these species or an alternative function of the cytochrome c proteins synthesized by this system.
Figure 3.
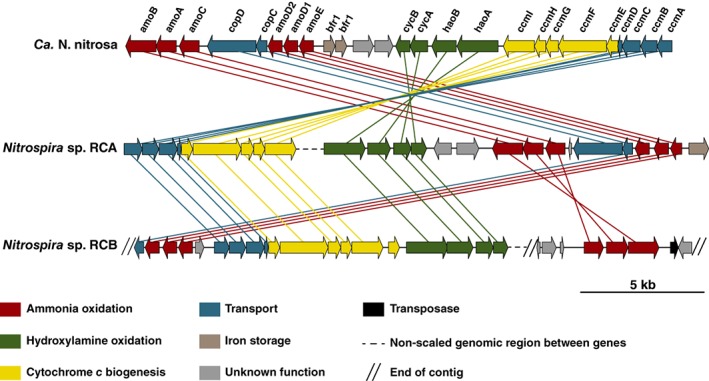
Schematic representation of the AMO genomic region in the Rifle clades A and B comammox Nitrospira in comparison to Ca. N. nitrosa. Arrows represent genes and indicate the transcriptional direction. Homologues genes are connected by lines. amo, ammonia monooxygenase; cop, copper transport; bfr, bacterioferritin; cyc, cytochrome c; hao, hydroxylamine dehydrogenase; ccm, cytochrome c biogenesis. Genes are drawn to scale.
Recently, it has been suggested for betaproteobacterial AOB, which possess an ammonia‐oxidizing machinery similar to comammox Nitrospira (Daims et al., 2015; van Kessel et al., 2015), that ammonia oxidation includes not only hydroxylamine but also nitric oxide (NO) as obligate intermediates (Caranto and Lancaster, 2017). In this model, NO is the product of hydroxylamine oxidation by HAO, which subsequently is converted to nitrite either abiotically or enzymatically, potentially by a bidirectional copper‐dependent dissimilatory nitrite reductase (NirK) (Caranto and Lancaster, 2017). All analysed Nitrospira genomes except RCA possess NirK (Fig. 4, Supporting Information Table S4), but its role in Nitrospira remains to be determined. However, it should be noted that the lack of NirK in RCA potentially is due to genome incompleteness. Like all other comammox Nitrospira (Palomo et al., 2018), both RCA and RCB lack the genetic potential for assimilatory nitrite reduction (Fig. 4, Supporting Information Table S4). Still, RCB encodes a MFS‐type nitrite/nitrate transporter (NarK), which is found in some clades A and B comammox and in all canonical Nitrospira genome (Fig. 4, Supporting Information Table S4).
Figure 4.
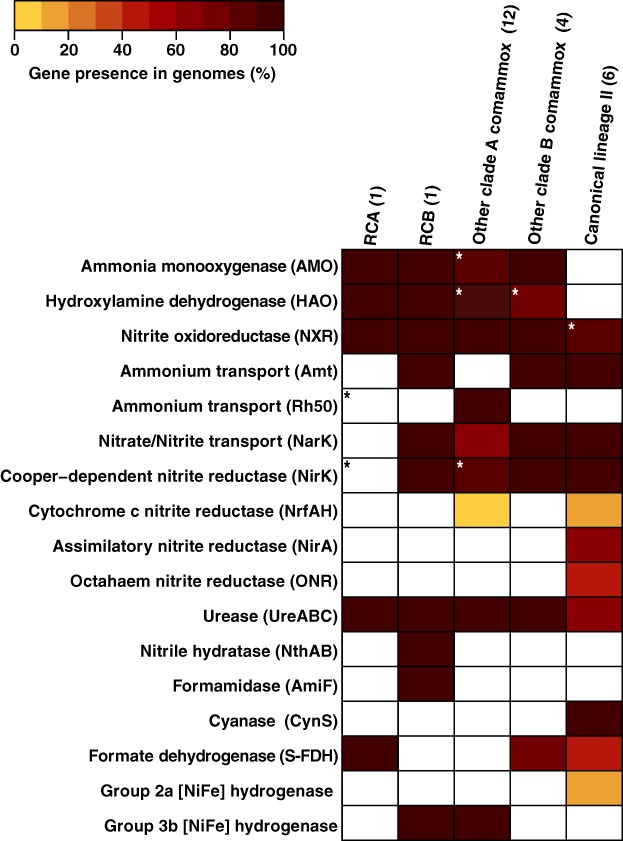
Distribution pattern of key metabolic features involved in nitrogen and alternative energy metabolisms. In total, 24 Nitrospira genomes were included in the analysis (Supporting Information Table S4). Numbers in parentheses indicate the number of genomes analysed in the respective group. Features shown in white were not detected. Asterisks indicate missing genes that potentially result from low‐quality assemblies, based on their universal presence in other Nitrospira genomes.
For ammonium uptake, clade A comammox (Daims et al., 2015; van Kessel et al., 2015; Palomo et al., 2018) and most betaproteobacterial AOB (Lupo et al., 2007) employ low‐affinity Rh‐type transporters. In contrast, clade B comammox, like canonical Nitrospira and ammonia‐oxidizing archaea (AOA; Offre et al., 2014), possess high‐affinity AmtB‐type transporters (Palomo et al., 2018). No ammonium transporter could be identified in RCA, which, however, is most likely due to incomplete recovery of the genome. The RCB genome encoded three copies of AmtB‐type transporters (Fig. 4) that had amino acid similarities ranging from 50% to 65%. Notably, one of these ammonium transporters shows the highest similarity to the Amt1 transporter of AOA based on BLAST analysis. In AOA, distinct copies of AmtB‐type transporters were differentially expressed when subjected to ammonium limitation, suggesting functional differentiation (Qin et al., 2018). In addition to external ammonium sources, ammonium can also originate from the intracellular hydrolysis of urea, and both Rifle genomes encoded ureases and the corresponding ABC transport systems. The presence and activity of urease in both canonical and comammox Nitrospira indicate that hydrolysis of urea is a common metabolic feature within this genus (Koch et al., 2015; van Kessel et al., 2015; Ushiki et al., 2018). Intriguingly, while canonical Nitrospira employ a cyanase to utilize cyanate as an additional metabolic source of ammonium (Palatinszky et al., 2015; Ushiki et al., 2018) this function is absent in complete nitrifiers (Palomo et al., 2018).
One of the unique genomic regions of RCB encoded the two subunits of a cobalt‐containing nitrile hydratase and an accessory protein partly conserved also in Nitrospira sp. UD063 (Supporting Information Table S3). This class of enzymes catalyses the hydration of nitriles, of which cyanide (HCN) is the simplest, to amides that can be subsequently hydrolyzed by amidases to produce monocarboxylate and ammonium (Kobayashi and Shimizu, 1998). Besides, the RCB genome contained a putative formamidase, an aliphatic amidase that converts formamide to ammonium and formate (Tauber et al., 2000; Skouloubris et al., 2001). These results indicate that Rifle clade B comammox Nitrospira use a nitrile hydratase/formamidase system to detoxify cyanide or other nitriles and produce ammonium, which might be used as an energy source and for assimilation (Fig. 5).
Figure 5.
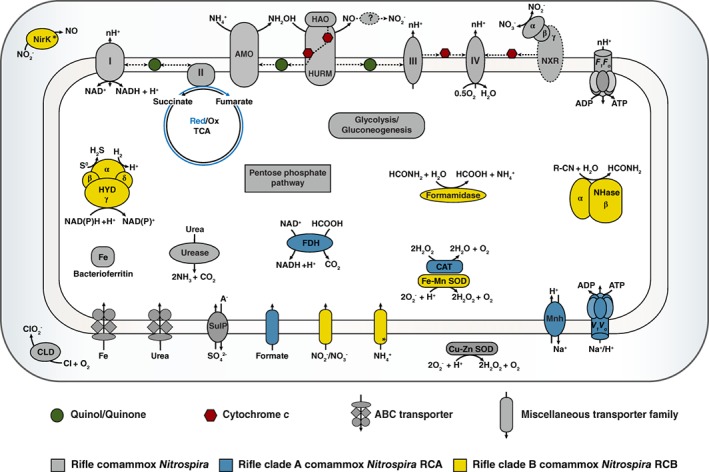
Comparison of central metabolic features of the Rifle comammox Nitrospira. AMO, ammonia monooxygenase; CAT, catalase; CLD, chlorite dismutase; FDH, formate dehydrogenase; HAO, hydroxylamine dehydrogenase; HURM, hydroxylamine‐ubiquinone reaction module; HYD, group 3b bifunctional NAD(P) hydrogenase; Mnh, multisubunit Na+/H+ antiporter; NHase, nitrile hydratase; NirK, Cu‐dependent nitrite reductase; SulP, sulfate permease. Asterisks indicate missing genes that potentially result from low‐quality assemblies. The question mark indicates that the exact pathway of nitrite formation from NO is uncertain.
Alternative energy metabolisms
The variable genome of the analysed sublineage II Nitrospira species includes genes for hydrogen and formate oxidation as alternative energy sources. Physiological analyses of N. moscoviensis revealed that hydrogen and formate sustained growth in the absence of nitrite (Koch et al., 2014; Koch et al., 2015). Interestingly, in contrast to N. moscoviensis that has a group 2a [NiFe] hydrogenase, clade A comammox employ a group 3b bidirectional [NiFe] hydrogenase (sulfhydrogenase), whereas clade B apparently lacks this enzyme (Palomo et al., 2018). Here, the complete operon encoding the group 3b hydrogenase was identified in RCB but was absent from the RCA genome. However, the metabolic role of this hydrogenase in complete nitrifiers remains to be determined. Potential functions of group 3b hydrogenases include NAD(P)‐dependent H2 oxidation (Yoon et al., 1996), H2 evolution (Berney et al., 2014) and reduction of elemental sulphur (S0) to H2S (Ma et al., 2000).
Similarly, the capability to oxidize formate seems to be more broadly distributed within Nitrospira than previously assumed. Some members of the genus Nitrospira can oxidize formate as an alternative energy source, using either oxygen or nitrate as a terminal electron acceptor (Koch et al., 2015). Fascinatingly, some uncultured Nitrospira from activated sludge only assimilate formate‐derived carbon in the presence of nitrite (Gruber‐Dorninger et al., 2015), and N. moscoviensis was shown to perform simultaneous formate and nitrite oxidation (Koch et al., 2015). So far, canonical and comammox clade B Nitrospira were described to possess a NAD‐dependent formate dehydrogenase and a formate transporter (Lücker et al., 2010; Koch et al., 2015; Palomo et al., 2018; Ushiki et al., 2018), which were absent in RCB. Contrastingly, RCA possessed all genes necessary for formate uptake and oxidation (Fig. 5, Supporting Information Table S2), making this the first clade A comammox organism with the genomic potential for formate oxidation. In natural environments, a mixotrophic lifestyle could be beneficial in oxic‐anoxic transition zones, where hydrogen and formate are supplied by fermentative microorganisms.
In addition, both Rifle comammox genomes encode for high‐affinity SulP/SLC26‐type transporters that may function as inorganic anion uptake transporters or anion:anion exchangers that could transport a broad range of substrates, including sulphate, bicarbonate, chloride, oxalate, iodide and formate (Alper and Sharma, 2013). This, in combination with the identification of genes for formate and hydrogen oxidation in RCA and RCB, respectively, indicates an enhanced genomic and metabolic plasticity of both comammox clades.
Environmental adaptation and defence
The RCA genome contained a complete V1Vo ATPase complex in addition to the F‐type ATPase (respiratory complex V) present in all Nitrospira (Fig. 5, Supporting Information Table S2). V‐type ATPases can couple both ATP synthesis and hydrolysis to the translocation of H+ or Na+ ions across the membrane. In Thermus thermophilus, the V‐type ATPase operates predominantly as H+‐driven ATP synthase (Nakano et al., 2008), while in Gram‐positive bacteria, this complex functions as a Na+ pump (Boekema et al., 1999). It has been shown that under slightly alkaline conditions, expression and activity of the F1Fo ATPase are reduced, while the V1Vo ATPase is induced to generate a Na+/H+ motive force (Ikegami et al., 1999), which may be necessary for pH homeostasis (Krulwich et al., 2011). The exact function of V‐type ATPase in RCA, however, remains uncertain; it may be involved either in energy conservation or ATP‐dependent sodium extrusion. Additionally, a putative Mnh‐type secondary Na+/H+ antiporter is encoded within the RCA genome (Fig. 5). Multisubunit Na+/H+ antiporters are also found in some marine nitrite oxidizers (Lücker et al., 2013; Ngugi et al., 2016) and are potentially involved in salt tolerance. Interestingly, BLAST surveys indicated that these monovalent Na+/H+ antiporters and the V1Vo ATPase were present in several genomes obtained from the Rifle site in the previous studies (Anantharaman et al., 2016), which hints at their importance in this environment.
For response to environmental stress, the RCA and RCB genomes contained genes for reactive oxygen stress defence and heavy metal resistance, including superoxide dismutase (SOD), catalase, peroxiredoxins and arsenic detoxification mechanisms. Like some sublineage II Nitrospira (Supporting Information Table S2 and S3), both Rifle comammox genomes contained genes encoding Cu‐Zn family SODs, which are periplasmic metalloenzymes potentially protecting periplasmic proteins against reactive oxygen during the stationary phase (John and Steinman, 1996). Similar to Nitrospira lenta, RCB additionally encoded a cytoplasmic Fe‐Mn family SOD, which was predicted to be a Fe‐tetramer SOD by SODa (Kwasigroch et al., 2008). Furthermore, both Rifle comammox Nitrospira along with several sublineage II Nitrospira, encoded an arsenate reductase in their genomes and could further detoxify arsenite [As(III)] through methylation (Supporting Information Table S2 and S3). Microbially mediated methylation of As has been proposed as one of the main detoxification mechanisms in terrestrial and aquatic environments (Bhattacharjee and Rosen, 2007). Interestingly, the Rifle site is a former milling facility that is rich in uranium and other redox‐sensitive metals such as vanadium, selenium and arsenic, and resistance mechanisms against these heavy metals might thus confer a selective advantage.
Conclusions
Our understanding of the evolution and metabolic flexibility of comammox Nitrospira is mainly based on genomes obtained from engineered systems, because only few draft genome sequences from natural environments have been obtained so far (Orellana et al., 2017; Parks et al., 2017). In this study, we analysed two novel comammox Nitrospira genomes acquired from the terrestrial subsurface. They were obtained from sites with extremely low ammonium concentrations (Hug et al., 2015), fitting to the high substrate affinity of the complete nitrifier N. inopinata (Kits et al., 2017). Comparative analysis of the two novel comammox genomes with other sublineage II Nitrospira species, including canonical nitrite oxidizers and complete nitrifiers, revealed strong conservation of metabolic key features, but also revealed a large genomic flexibility and adaptability of these enigmatic organisms. Metabolic features were identified in the novel comammox genomes that were assumed to be specific for certain functional clades within Nitrospira sublineage II and the observed broader distribution of formate and hydrogen oxidation machineries indicates an expanded ecophysiological role of these substrates within the energy metabolism of comammox Nitrospira. Previous studies performed at this aquifer system failed to identify known ammonia‐oxidizing microorganisms but found members of the genus Nitrospira as the main nitrifiers (Anantharaman et al., 2016). Our identification of comammox Nitrospira at this site indicates that complete nitrifiers can apparently be the main drivers of ammonia oxidation in the terrestrial subsurface. This warrants future studies to investigate in more depth the distribution, abundance and activity of comammox Nitrospira in a range of natural ecosystems to further elucidate their role in the biogeochemical nitrogen cycle.
Experimental procedures
Genome sequencing and assembly
Sampling and sequencing of metagenomes from two sediment cores taken at the Rifle research site, adjacent to the Colorado River are described elsewhere (Hug et al., 2015). Raw reads were trimmed with Sickle (Joshi and Fass, 2011) using default parameters, and assembled with IDBA‐UD v1.1.1 (Peng et al., 2012) using a minimal kmer size of 40, a maximum of 140 and steps of 20. Open reading frames were predicted for scaffolds longer than 1 Kbp with Prodigal (Hyatt, 2010) and functional predictions determined through similarity searches against the UniRef90 (Suzek et al., 2007), KEGG (Ogata et al., 1999) and UniProt (Bateman et al., 2017) databases. Reads were mapped to scaffolds with bowtie2 (Langmead and Salzberg, 2012) to determine their relative abundance. Automated binning was conducted with Metabat (Kang et al., 2015) and Concoct (Alneberg et al., 2014) using differential coverage information and the best genomic bins were selected with DAStool (Sieber et al., 2018). Bins were imported into ggKbase (https://ggkbase.berkeley.edu) for further manual refinement based on their GC, coverage, taxonomy of scaffolds, as well as completion assessed according to the number of bacterial single copy genes present in each bin. Scaffolding errors in two bins that were found to contain amoA genes were fixed using a published script as described elsewhere (Brown et al., 2015). To determine completeness and contamination (referred to as redundancy in this study) of the assembled genomes CheckM 1.0.7 was used (Parks et al., 2015).
Annotation
Genome annotation was performed using Prokka (version 1.12‐beta; Seemann, 2014). For annotation, a modified version of Prokka was used which employs BLASTP to search all predicted CDSs against the NCBI RefSeq non‐redundant protein database (O'Leary et al., 2016). Automatic annotations of genes of interest were confirmed by BLAST against the TrEMBL, Swiss‐Prot and NCBI nr databases. The presence of signal peptides was checked using SignalP (Petersen et al., 2011) and Phobius (Kall et al., 2004).
Phylogenomic analysis
For genome‐based phylogenetic analyses, we used the up‐to‐date bacterial core gene set and pipeline for phylogenomic tree reconstruction (UBCG; Na et al., 2018) to identify and extract 92 universal bacterial core genes in 32 Nitrospira and two Leptospirillum genomes. As all included genomes were lacking a gene for the phenylalanine‐tRNA ligase, beta subunit (pheT), all downstream analyses were performed using the 91 remaining core genes identified by Na and colleagues. These genes then were aligned and concatenated within UBCG using default parameters. Using the concatenated nucleotide alignment, a tree was calculated using RAxML version 8.2.10 (Stamatakis, 2014) on the CIPRES science gateway (Miller et al., 2010) with the GTR substitution and GAMMA rate heterogeneity models and 100 bootstrap iterations. The two Leptospirillum species were used as outgroup to root the tree. Alignments of the AMO subunit A nucleotide sequences (amoA) were obtained using ClustalW as implemented in MEGA7 (Kumar et al., 2016) and the phylogenetic tree was inferred by RAxML on CIPRES with the GTR‐GAMMA model and 1000 bootstrap replications.
Genome comparisons
Average nucleotide identities between the 32 Nitrospira genomes were calculated using the OrthoANIu algorithm (Yoon et al., 2017). Orthologues and strain‐specific proteins were identified by reciprocal best BLAST (Altschul et al., 1990) using a custom in‐house script. BLAST hits with an E‐value of 1e‐6, amino acid identities ≥45% and a minimum alignment length ≥ 70% were considered as orthologues. The AMO gene clusters in Nitrospira genomes were visualized using the genoPlotR package (Guy et al., 2010).
Supporting information
Table S1 General and genomic characteristics of all analysed Nitrospira.
Table S2 Reciprocal best BLAST hits between the RCA and selected sublineage II Nitrospira genomes. Highlighted cells indicate manually curated annotations.
Table S3 Reciprocal best BLAST hits between the RCB and selected sublineage II Nitrospira genomes. Highlighted cells indicate manually curated annotations.
Table S4 Summary of key metabolic features involved in nitrogen and alternative energy metabolism in selected sublineage II Nitrospira genomes.
Figure S1 Schematic representation of the AMO genomic region in sublineage II Nitrospira genomes. Homologues genes are connected by lines. amo, ammonia monooxygenase; cop, copper transport; bfr, bacterioferritin; cyc, cytochrome c; hao, hydroxylamine dehydrogenase; ccm, cytochrome c biogenesis. Genes are drawn to scale.
Acknowledgements
We are grateful to Christopher Lawson for helpful discussions. Financial support was provided by the Netherlands Organization for Scientific Research (grants 863.14.019, 016.Veni.192.062 and 016.Vidi.189.050, and Gravitation grants 024.002.001 [NESSC] and 024.002.002 [SIAM]) and the Radboud Excellence Initiative. This work was further supported by Lawrence Berkeley National Laboratory's Sustainable Systems Scientific Focus Area funded by the US Department of Energy, Office of Science, Office of Biological and Environmental Research under contract DE‐AC02‐05CH11231. DNA sequencing was conducted at the DOE Joint Genome Institute, a DOE Office of Science User Facility, via the Community Science Program.
Data Availability: The genome sequences of the two comammox Nitrospira genomes recovered in this study have been deposited in GenBank under accession numbers SPAW00000000 and SPAX00000000 (BioProject PRJNA513947), the raw sequencing data are available under BioProject number SRX1990948.
Data availability
The genome sequences of the two comammox Nitrospira genomes recovered in this study have been deposited in GenBank under accession numbers SPAW00000000 and SPAX00000000 (BioProject PRJNA513947), the raw sequencing data are available under BioProject number SRX1990948.
References
- Alneberg, J. , Bjarnason, B.S. , de Bruijn, I. , Schirmer, M. , Quick, J. , Ijaz, U.Z. , et al (2014) Binning metagenomic contigs by coverage and composition. Nat Methods 11: 1144–1146. [DOI] [PubMed] [Google Scholar]
- Alper, S.L. , and Sharma, A.K. (2013) The SLC26 gene family of anion transporters and channels. Mol Aspects Med 34: 494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. , and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Anantharaman, K. , Brown, C.T. , Hug, L.A. , Sharon, I. , Castelle, C.J. , Probst, A.J. , et al (2016) Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 7: 13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelme, R.P. , McLellan, S.L. , and Newton, R.J. (2017) Freshwater recirculating aquaculture system operations drive biofilter bacterial community shifts around a stable nitrifying consortium of ammonia‐oxidizing Archaea and Comammox Nitrospira . Front Microbiol 8: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A. , Martin, M.J. , O'Donovan, C. , Magrane, M. , Alpi, E. , Antunes, R. , et al (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45: D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney, M. , Greening, C. , Conrad, R. , Jacobs, W.R. , and Cook, G.M. (2014) An obligately aerobic soil bacterium activates fermentative hydrogen production to survive reductive stress during hypoxia. Proc Natl Acad Sci U S A 111: 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee, H. , and Rosen, B.P. (2007) Arsenic metabolism in prokaryotic and eukaryotic microbes In Molecular Microbiology of Heavy Metals, Nies D.H., and Silver S. (eds). Berlin, Heidelberg: Springer, pp. 371–406. [Google Scholar]
- Boekema, E.J. , van Breemen, J.F.L. , Brisson, A. , Ubbink‐Kok, T. , Konings, W.N. , and Lolkema, J.S. (1999) Biological motors ‐ connecting stalks in V‐type ATPase. Nature 401: 37–38. [DOI] [PubMed] [Google Scholar]
- Brown, C.T. , Hug, L.A. , Thomas, B.C. , Sharon, I. , Castelle, C.J. , Singh, A. , et al (2015) Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523: 208–211. [DOI] [PubMed] [Google Scholar]
- Caranto, J.D. , and Lancaster, K.M. (2017) Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc Natl Acad Sci U S A 114: 8217–8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle, C.J. , Hug, L.A. , Wrighton, K.C. , Thomas, B.C. , Williams, K.H. , Wu, D.Y. , et al (2013) Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, E. , Pérez, J. , and Kreft, J.U. (2006) Why is metabolic labour divided in nitrification? Trends Microbiol 14: 213–219. [DOI] [PubMed] [Google Scholar]
- Daebeler, A. , Bodelier, P.L.E. , Yan, Z. , Hefting, M.M. , Jia, Z.J. , and Laanbroek, H.J. (2014) Interactions between Thaumarchaea, Nitrospira and methanotrophs modulate autotrophic nitrification in volcanic grassland soil. ISME J 8: 2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daims, H. , Lücker, S. , and Wagner, M. (2016) A new perspective on microbes formerly known as nitrite‐oxidizing bacteria. Trends Microbiol 24: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daims, H. , Nielsen, J.L. , Nielsen, P.H. , Schleifer, K.H. , and Wagner, M. (2001) In situ characterization of Nitrospira‐like nitrite oxidizing bacteria active in wastewater treatment plants. Appl Environ Microbiol 67: 5273–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daims, H. , Lebedeva, E.V. , Pjevac, P. , Han, P. , Herbold, C. , Albertsen, M. , et al (2015) Complete nitrification by Nitrospira bacteria. Nature 528: 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway, J.N. , Townsend, A.R. , Erisman, J.W. , Bekunda, M. , Cai, Z.C. , Freney, J.R. , et al (2008) Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320: 889–892. [DOI] [PubMed] [Google Scholar]
- Gruber‐Dorninger, C. , Pester, M. , Kitzinger, K. , Savio, D.F. , Loy, A. , Rattei, T. , et al (2015) Functionally relevant diversity of closely related Nitrospira in activated sludge. ISME J 9: 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy, L. , Roat Kultima, J. , and Andersson, S.G.E. (2010) genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26: 2334–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gülay, A.M. , Albrechtsen, H.J. , Abu Al‐Soud, W. , Sorensen, S.J. , and Smets, B.F. (2016) Ecological patterns, diversity and core taxa of microbial communities in groundwater‐fed rapid gravity filters. ISME J 10: 2209–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H.W. , and He, J.Z. (2017) Comammox‐a newly discovered nitrification process in the terrestrial nitrogen cycle. J Soil Sediment 17: 2709–2717. [Google Scholar]
- Hug, L.A. , Thomas, B.C. , Brown, C.T. , Frischkorn, K.R. , Williams, K.H. , Tringe, S.G. , and Banfield, J.F. (2015) Aquifer environment selects for microbial species cohorts in sediment and groundwater. ISME J 9: 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami, M. , Kawano, M. , Takase, K. , Yamato, I. , Igarashi, K. , and Kakinuma, Y. (1999) Enterococcus hirae vacuolar ATPase is expressed in response to pH as well as sodium. FEBS Lett 454: 67–70. [DOI] [PubMed] [Google Scholar]
- John, S.G. , and Steinman, H.M. (1996) Periplasmic copper‐zinc superoxide dismutase of Legionella pneumophila: role in stationary‐phase survival. J Bacteriol 178: 1578–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, N.A. , and Fass, J.N. (2011) Sickle: A sliding‐window, adaptive, quality‐based trimming tool for FastQ files. URL http://github.com/najoshi/sickle.
- Kall, L. , Krogh, A. , and Sonnhammer, E.L.L. (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338: 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kang, D.W.D. , Froula, J. , Egan, R. , and Wang, Z. (2015) MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer J 3: e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kits, K.D. , Sedlacek, C.J. , Lebedeva, E.V. , Han, P. , Bulaev, A. , Pjevac, P. , et al (2017) Kinetic analysis of a complete nitrifier reveals an oligotrophic lifestyle. Nature 549: 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, M. , and Shimizu, S. (1998) Metalloenzyme nitrile hydratase: structure, regulation, and application to biotechnology. Nat Biotech 16: 733–736. [DOI] [PubMed] [Google Scholar]
- Koch, H. , van Kessel, M.A.H.J. , and Lücker, S. (2018) Complete nitrification: insights into the ecophysiology of comammox Nitrospira . Appl Microbiol Biotechnol 103: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, H. , Lücker, S. , Albertsen, M. , Kitzinger, K. , Herbold, C. , Spieck, E. , et al (2015) Expanded metabolic versatility of ubiquitous nitrite‐oxidizing bacteria from the genus Nitrospira . Proc Natl Acad Sci U S A 112: 11371–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, H. , Galushko, A. , Albertsen, M. , Schintlmeister, A. , Gruber‐Dorninger, C. , Lücker, S. , et al (2014) Growth of nitrite‐oxidizing bacteria by aerobic hydrogen oxidation. Science 345: 1052–1054. [DOI] [PubMed] [Google Scholar]
- Krulwich, T.A. , Sachs, G. , and Padan, E. (2011) Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol 9: 330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasigroch, J.M. , Wintjens, R. , Gilis, D. , and Rooman, M. (2008) SODa: an Mn/Fe superoxide dismutase prediction and design server. BMC Bioinformatics 9: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva, E.V. , Off, S. , Zumbraegel, S. , Kruse, M. , Shagzhina, A. , Lücker, S. , et al (2011) Isolation and characterization of a moderately thermophilic nitrite‐oxidizing bacterium from a geothermal spring. FEMS Microbiol Ecol 75: 195–204. [DOI] [PubMed] [Google Scholar]
- Lupo, D. , Li, X.D. , Durand, A. , Tomizaki, T. , Cherif‐Zahar, B. , Matassi, G. , et al (2007) The 1.3‐a resolution structure of Nitrosomonas europaea Rh50 and mechanistic implications for NH3 transport by rhesus family proteins. Proc Natl Acad Sci U S A 104: 19303–19308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücker, S. , Nowka, B. , Rattei, T. , Spieck, E. , and Daims, H. (2013) The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front Microbiol 4: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücker, S. , Wagner, M. , Maixner, F. , Pelletier, E. , Koch, H. , Vacherie, B. , et al (2010) A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite‐oxidizing bacteria. Proc Natl Acad Sci U S A 107: 13479–13484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, K.S. , Weiss, R. , and Adams, M.W.W. (2000) Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J Bacteriol 182: 1864–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.A. , Pfeiffer, W. , and Schwartz, T. (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees In 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA: IEEE, pp. 1–8. 10.1109/GCE.2010.5676129. [DOI] [Google Scholar]
- Na, S.I. , Kim, Y.O. , Yoon, S.H. , Ha, S.M. , Baek, I. , and Chun, J. (2018) UBCG: up‐to‐date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J Microbiol 56: 280–285. [DOI] [PubMed] [Google Scholar]
- Nakano, M. , Imamura, H. , Toei, M. , Tamakoshi, M. , Yoshida, M. , and Yokoyama, K. (2008) ATP hydrolysis and synthesis of a rotary motor V‐ATPase from Thermus thermophilus . J Biol Chem 283: 20789–20796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngugi, D.K. , Blom, J. , Stepanauskas, R. , and Stingl, U. (2016) Diversification and niche adaptations of Nitrospina‐like bacteria in the polyextreme interfaces of Red Sea brines. ISME J 10: 1383–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary, N.A. , Wright, M.W. , Brister, J.R. , Ciufo, S. , Haddad, D. , McVeigh, R. , et al (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offre, P. , Kerou, M. , Spang, A. , and Schleper, C. (2014) Variability of the transporter gene complement in ammonia‐oxidizing archaea. Trends Microbiol 22: 665–675. [DOI] [PubMed] [Google Scholar]
- Ogata, H. , Goto, S. , Sato, K. , Fujibuchi, W. , Bono, H. , and Kanehisa, M. (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana, L.H. , Chee‐Sanford, J.C. , Sanford, R.A. , Loffler, F.E. , and Konstantinidis, K.T. (2017) Year‐round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl Environ Microbiol 84: UNSP e01646–UNSP e01617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatinszky, M. , Herbold, C. , Jehmlich, N. , Pogoda, M. , Han, P. , von Bergen, M. , et al (2015) Cyanate as an energy source for nitrifiers. Nature 524: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo, A. , Pedersen, A.G. , Fowler, S.J. , Dechesne, A. , Sicheritz‐Ponten, T. , and Smets, B.F. (2018) Comparative genomics sheds light on niche differentiation and the evolutionary history of comammox Nitrospira . ISME J 12: 1779–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D.H. , Imelfort, M. , Skennerton, C.T. , Hugenholtz, P. , and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D.H. , Rinke, C. , Chuvochina, M. , Chaumeil, P.A. , Woodcroft, B. , Evans, P.N. , et al (2017) Recovery of nearly 8,000 metagenome‐assembled genomes substantially expands the tree of life. Nat Microbiol 3: 253–253. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Leung, H.C.M. , Yiu, S.M. , and Chin, F.Y.L. (2012) IDBA‐UD: a de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Petersen, T.N. , Brunak, S. , von Heijne, G. , and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- Pinto, A.J. , Marcus, D.N. , Ijaz, U.Z. , Santos, Q. , Dick, G.J. , and Raskin, L. (2016) Metagenomic evidence for the presence of Comammox Nitrospira‐like bacteria in a drinking water system. Msphere 1: e00054–e00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pjevac, P. , Schauberger, C. , Poghosyan, L. , Herbold, C.W. , van Kessel, M.A.H.J. , Daebeler, A. , et al (2017) AmoA‐targeted polymerase chain reaction primers for the specific detection and quantification of Comammox Nitrospira in the environment. Front Microbiol 8: 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, W. , Amin, S.A. , Lundeen, R.A. , Heal, K.R. , Martens‐Habbena, W. , Turkarslan, S. , et al (2018) Stress response of a marine ammonia‐oxidizing archaeon informs physiological status of environmental populations. ISME J 12: 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, M. , and Rossello‐Mora, R. (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106: 19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069.24642063 [Google Scholar]
- Shi, X. , Hu, H.‐W. , Wang, J. , He, J.‐Z. , Zheng, C. , Wan, X. , and Huang, Z. (2018) Niche separation of comammox Nitrospira and canonical ammonia oxidizers in an acidic subtropical forest soil under long‐term nitrogen deposition. Soil Biol Biochem 126: 114–122. [Google Scholar]
- Sieber, C.M.K. , Probst, A.J. , Sharrar, A. , Thomas, B.C. , Hess, M. , Tringe, S.G. , and Banfield, J.F. (2018) Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 3: 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouloubris, S. , Labigne, A. , and De Reuse, H. (2001) The AmiE aliphatic amidase and AmiF formamidase of Helicobacter pylori: natural evolution of two enzyme paralogues. Mol Microbiol 40: 596–609. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek, B.E. , Huang, H.Z. , McGarvey, P. , Mazumder, R. , and Wu, C.H. (2007) UniRef: comprehensive and non‐redundant UniProt reference clusters. Bioinformatics 23: 1282–1288. [DOI] [PubMed] [Google Scholar]
- Tauber, M.M. , Cavaco‐Paulo, A. , Robra, K.H. , and Gubitz, G.M. (2000) Nitrile hydratase and amidase from Rhodococcus rhodochrous hydrolyze acrylic fibers and granular polyacrylonitriles. Appl Environ Microbiol 66: 1634–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushiki, N. , Fujitani, H. , Shimada, Y. , Morohoshi, T. , Sekiguchi, Y. , and Tsuneda, S. (2018) Genomic analysis of two phylogenetically distinct Nitrospira species reveals their genomic plasticity and functional diversity. Front Microbiol 8: 2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel, M. , Speth, D.R. , Albertsen, M. , Nielsen, P.H. , Op den Camp, H.J.M. , Kartal, B. , et al (2015) Complete nitrification by a single microorganism. Nature 528: 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Ma, L. , Mao, Y. , Jiang, X. , Xia, Y. , Yu, K. , et al (2017) Comammox in drinking water systems. Water Res 116: 332–341. [DOI] [PubMed] [Google Scholar]
- Xia, F. , Wang, J.‐G. , Zhu, T. , Zou, B. , Rhee, S.‐K. , and Quan, Z.‐X. (2018) Ubiquity and diversity of complete ammonia oxidizers (comammox). Appl Environ Microbiol 84: e01390–e01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, K.S. , Ueda, Y. , Ishii, M. , Igarashi, Y. , and Kodama, T. (1996) NADH:ferredoxin reductase and NAD‐reducing hydrogenase activities in Hydrogenobacter thermophilus strain TK‐6. FEMS Microbiol Lett 139: 139–142. [Google Scholar]
- Yoon, S.H. , Ha, S.M. , Lim, J. , Kwon, S. , and Chun, J. (2017) A large‐scale evaluation of algorithms to calculate average nucleotide identity. Antonie Leeuwenhoek 110: 1281–1286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 General and genomic characteristics of all analysed Nitrospira.
Table S2 Reciprocal best BLAST hits between the RCA and selected sublineage II Nitrospira genomes. Highlighted cells indicate manually curated annotations.
Table S3 Reciprocal best BLAST hits between the RCB and selected sublineage II Nitrospira genomes. Highlighted cells indicate manually curated annotations.
Table S4 Summary of key metabolic features involved in nitrogen and alternative energy metabolism in selected sublineage II Nitrospira genomes.
Figure S1 Schematic representation of the AMO genomic region in sublineage II Nitrospira genomes. Homologues genes are connected by lines. amo, ammonia monooxygenase; cop, copper transport; bfr, bacterioferritin; cyc, cytochrome c; hao, hydroxylamine dehydrogenase; ccm, cytochrome c biogenesis. Genes are drawn to scale.
Data Availability Statement
The genome sequences of the two comammox Nitrospira genomes recovered in this study have been deposited in GenBank under accession numbers SPAW00000000 and SPAX00000000 (BioProject PRJNA513947), the raw sequencing data are available under BioProject number SRX1990948.