

Abstract
Periventricular nodular heterotopia (PNH) is a common structural malformation of cortical development. Mutations in the filamin A gene are frequent in familial cases with X‐linked PNH. However, many cases with sporadic PNH remain genetically unexplained. Although medically refractory epilepsy often brings attention to the underlying PNH, patients are often not candidates for surgical resection. This limits access to neuronal tissue harboring causal mutations. We evaluated a patient with PNH and medically refractory focal epilepsy who underwent a presurgical evaluation with stereotactically placed electroencephalographic (SEEG) depth electrodes. Following SEEG explantation, we collected trace tissue adherent to the electrodes and extracted the DNA. Whole‐exome sequencing performed in a Clinical Laboratory Improvement Amendments–approved genetic diagnostic laboratory uncovered a de novo heterozygous pathogenic variant in novel candidate PNH gene MEN1 (multiple endocrine neoplasia type 1; c.1546dupC, p.R516PfsX15). The variant was absent in an earlier exome profiling of the venous blood–derived DNA. The MEN1 gene encodes the ubiquitously expressed, nuclear scaffold protein menin, a known tumor suppressor gene with an established role in the regulation of transcription, proliferation, differentiation, and genomic integrity. Our study contributes a novel candidate gene in PNH generation and a novel practical approach that integrates electrophysiological and genetic explorations of epilepsy.
Keywords: epilepsy, MEN1 gene, periventricular nodular heterotopia, somatic mosaicism, stereotactically placed EEG
1. INTRODUCTION
Periventricular nodular heterotopia (PNH) is a frequent structural malformation of cortical development with a variable radiological and clinical presentation and a frequent association with epilepsy.1 Mutations in the filamin A gene (FLNA) are common in familial cases with X‐linked PNH.1 However, they provide a causal explanation only in about 8%‐26% of sporadic cases and most are women.1, 2 Sporadic PNH is genetically heterogeneous, and it has been linked to genes involved in vesicle trafficking and molecular transport (ARFGEF2), in regulation of gene expression (INTS8, MCPH1), in neuronal proliferation and differentiation (DCHS1, FAT4, NEEDL4), and in synapse development (MAP1B, TMTC3), and to genes with unclear function (ERMARD) (C6orf70).1, 3, 4 PNH has been also described in association with chromosomal abnormalities and with an enrichment in large and cryptic copy number variants.4 The challenge of molecular diagnosis of sporadic PNH is compounded by the lack of access to the dysplastic brain tissue, as many patients with PNH and epilepsy are not suitable candidates for a resective surgery. It has been shown that even deep high‐coverage sequencing of peripheral blood cells of patients with malformations of cortical development limits mutation detection to about 17%.5 We evaluated a patient with PNH and medically refractory, surgically nonremediable focal epilepsy and developed a method for sampling dysplastic tissue from cells adherent to diagnostic stereotactically placed electroencephalographic (SEEG) depth electrodes. We identified a somatic pathogenic variant in a novel candidate PNH gene, MEN1.
2. MATERIALS AND METHODS
The Baylor Institutional Review Board (IRB H‐13798) approved the study. Epilepsy was classified according to the established guidelines of the International League Against Epilepsy.6 Intracranial EEG was performed with commercial 16‐contact, 0.9‐mm‐diameter depth electrodes (Ad‐Tech) using the Nihon Kohden Neurofax EEG‐1200 Platform and a 2000‐Hz sampling rate. Brain magnetic resonance imaging (MRI) was performed on a Philips Healthcare Ingenia using a 1.5‐T magnet and an established clinical protocol for epilepsy with 1‐mm slices through the hippocampus. The SEEG electrode design was guided by a clinical expert panel of the Baylor Comprehensive Epilepsy Program, and placement was done with the assistance of the ROSA Brain (http://www.medtech.fr/en/rosa-brain) device, targeting the PNH and hippocampal body following a published technique.7 SEEG electrodes were removed under general anesthesia. Anchor bolts were removed, electrodes were carefully pulled out, electrode tips were cut off, and each electrode tip was placed in a separate conical tube filled with phosphate‐buffered saline solution (PBS) in the operating room. The tubes were immediately transferred to 4°C and soaked in PBS for 4 hours. Wires were then discarded and cells were pelleted by centrifugation at 3500 rpm for 10 minutes. The supernatant was removed, cells were resuspended in 50 µL of the remaining saline buffer, and 4 µL of the cellular suspension was used to amplify the genomic DNA using the Qiagen REPLI‐g Single Cell kit (cat #150373) in a single isothermal reaction at 30°C over 16 hours according to the manufacturer's instructions. gDNA amplification quantity and quality were evaluated on a TapeStation (https://www.agilent.com/). The final DNA yields from the cells attached to the SEEG electrodes inserted into the left and the right PNH were 33.15 μg (concentration = 663 ng/μL) and 18.95 μg (concentration 379 ng/μL), respectively. Venous blood samples of the patient and both parents were processed as published before.8 One microgram of each DNA sample derived from the blood and the brain underwent a next‐generation whole‐exome sequencing in a Clinical Laboratory Improvement Amendments–approved laboratory (https://www.genedx.com/) according to the published methods.9 The mean depth coverage was 155× and the quality threshold was 97.6%. For the MEN1 gene, 100% of the coding region was covered at a minimum of 10×. The clinical analysis adhered to the established guidelines.10 MEN1 gene variant validation by Sanger sequencing was performed in the clinical genetic laboratory and subsequently in the research laboratory directed by A.M.G. on two samples independently amplified respectively from the right and the left PNH lesions. We used forward (CTTGCTCTCACCTTGCTCT) and reverse (TAGGGGTGGACACTTTCTGC) primers designed with Primer 3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) for an expected product size of 470 bp. We performed a PubMed literature review for MEN1 gene association with (1) PNH using the keywords “MEN1 gene and either malformations of cortical development, focal cortical dysplasia or periventricular nodular heterotopia”, yielding no results; and (2) epilepsy or seizures using the keywords “MEN1 gene and epilepsy” and “MEN1 gene and seizures”, yielding 14 search results summarized in Table S2.
3. RESULTS
The patient was a 37‐year‐old male with medically refractory focal seizures with altered awareness starting at the age of 30 years. The patient had no known epilepsy risk factors or a family history of epilepsy. He suffered from obesity (body mass index = 37) and an associated obstructive sleep apnea managed with a nightly use of a continuous positive airway pressure machine. Aside from morbid obesity, general and neurological examinations were within normal limits. Results of the diagnostic studies are summarized in Table S1. The brain MRI showed a bilateral PNH (Figure S1) most consistent with the bilateral posterior periventricular nodular heterotopia type described by Mandelstam et al.11 The SEEG evaluation targeting the PNH areas and bilateral hippocampi (Figure 1) captured habitual seizures emanating independently from bilateral hippocampal structures (Figure S2), and the decision was made to implant a responsive neurostimulator (Neuropace), using two depth electrodes in the bilateral hippocampi. Whole‐exome sequencing and a chromosomal microarray analysis of blood‐derived DNA were unrevealing as to the genetic etiology of the patient's PNH and epilepsy. We then analyzed the tissue stripped from the SEEG electrodes. We did not identify pathogenic variants in the coding regions in any of the genes previously reported in association with either familial or sporadic PNH.4 However, we uncovered a mosaic de novo heterozygous pathogenic variant in the gene MEN1 (multiple endocrine neoplasia type 1; NM_130799.2, c.1546dupC, p.R516PfsX15) in 16.7% of the sequenced alleles. Targeted Sanger sequencing confirmed the absence of the variant in blood‐derived DNA of the proband and his parents (Figure 2). The variant identified in the brain sample of our patient was a known frameshift mutation that caused a codon change at a conserved arginine 516 (Figure 2A), thus creating a premature stop codon at position 15 of the new reading frame and leading to a loss of 95 amino acids of the protein. The c.1546dupC variant has been previously reported at a high frequency in patients with MEN1 syndrome and in a multigenerational Finnish family.12 This truncating mutation affects one of the two nuclear localization signals (NLSs), and a published in vitro functional study has shown that this variant impairs nuclear localization of protein compared to wild type.13 Thus, the MEN1 variant was classified as pathogenic (class 5) using the American College of Medical Genetics and Genomics recommendations (Table S3).10, 14 Prior research has shown that a variable fraction of cells of MEN1‐associated tumors frequently display loss of heterozygosity (LOH).15 However, we could not achieve a reliable exon‐level deletion/duplication analysis using the available exome sequence data.
Figure 1.
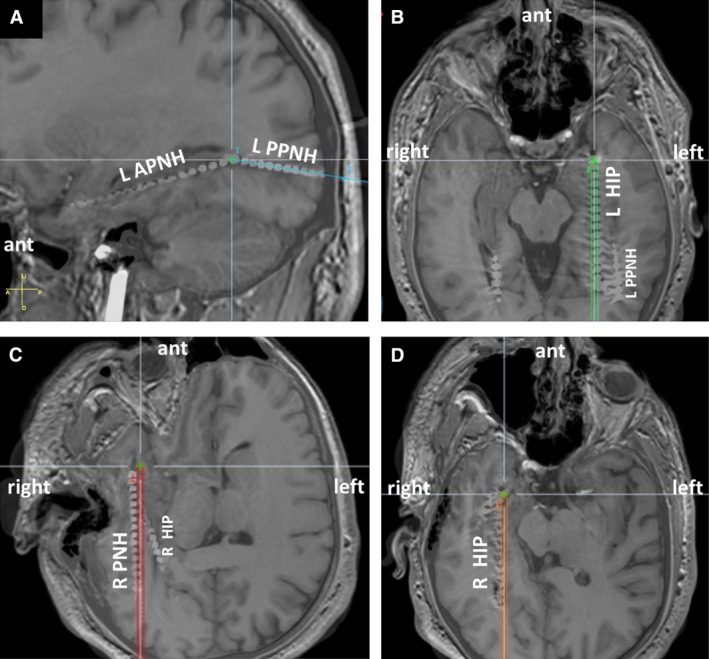
Stereotactically placed electroencephalographic (SEEG) electrode placement. SEEG electrodes were placed to sample bilateral periventricular nodular heterotopia (PNH) and bilateral hippocampi. Images are 1.5‐T noncontrast brain magnetic resonance imaging fused with head computed tomography and coregistered with the inserted SEEG electrodes. A, Sagittal view. L APNH, SEEG electrode sampling the anterior (ant) to midtemporal aspect of the left PNH; L PPNH, SEEG electrode sampling midtemporal to occipital aspect of the left PNH. Both the L APNH and L PPNH are lateral to the electrode sampling left hippocampus (L HIP). B, C, Modified axial views following the trajectories of the L HIP and R HIP orthogonally placed occipitotemporal SEEG electrodes sampling the left and the right hippocampus. D, Modified axial view along the trajectory of the SEEG electrode sampling the right PNH
Figure 2.
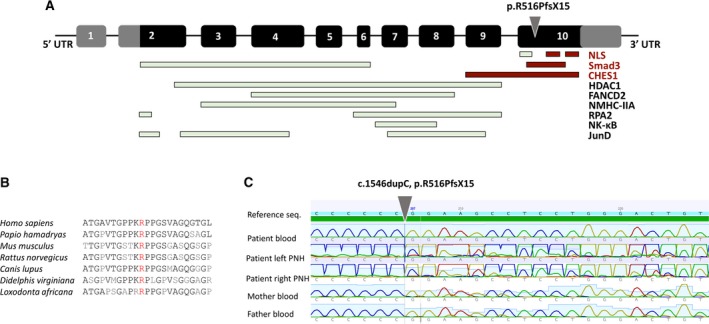
MEN1 gene schema and Sanger validation. A, MEN1 gene consists of 10 exons. Black panels represent transcribed and translated regions, whereas gray panels represent untranslated regions (UTRs). Exon 2 contains the start codon (ATG), and exon 10 the stop codon (TGA). Gray arrowhead indicates the location of the variant [c.1546dupC, p.R516PfsX15] found in the case presented in the article. NLS, nuclear localization signal. B, The affected variant is highly conserved, and the gene itself is highly conserved. Mouse Men1 demonstrates 97% identity/98% similarity to human MEN1 at the amino acid level.18 C, Sanger validation confirmed the absence of the variant in the peripheral blood DNA and the presence of the variant in a mosaic form in periventricular nodular heterotopia (PNH) on either side
4. DISCUSSION
PNH is one of the most frequent structural malformations of cortical development.4 A sporadic PNH is genetically heterogeneous,4 and a majority of cases will be negative for FLNA pathogenic variants.1, 2 They are likely to remain undiagnosed because the migration abnormalities often occur due to brain‐restricted somatic mosaicism, the genetic testing of peripheral tissues remains nondiagnostic, and a patient may not be a candidate for resective epilepsy surgery. This eliminates the possibility of direct examination of brain tissue. We successfully employed a novel, minimally invasive detection method taking advantage of small amounts of tissue adherent to the SEEG electrodes. Genetic analysis of the tissue sample stripped from the electrodes that probed the PNH uncovered tissue mosaicism akin to an examination of resected brain tissue.16 We identified a known pathogenic c.1546dupC variant in a novel candidate PNH gene, MEN1, in 16.7% of the sequenced alleles (Figure 2 and Figure S3). The gene is located on chromosome 11q13 and encodes a multifunctional nuclear scaffold protein menin with a known role in the regulation of transcription, proliferation, differentiation, and genomic integrity.17 It is highly conserved among species (Figure 2B) and shows a ubiquitous expression beyond tissues typically affected by MEN1 syndrome, including cortex, hippocampus, brainstem, and cerebellum.18, 19 Immunohistochemistry and antibody staining in the murine brain show the strongest labeling in the pyramidal cells of the hippocampus (http://mouse.brain‐map.org).18 It has been most extensively studied in connection to the MEN1 syndrome as a tumor suppressor since germline mutations in the MEN1 gene have been detected in about 85% of familial MEN1 cases.20 We performed a detailed literature search for MEN1 gene connection to epilepsy, seizures, or neuronal migrational defects and found that the reported seizures were typically associated with hypoglycemic episodes due to pancreatic insulinomas (Table S2). Brain MRI was reported in six of 32 (18%) patients, of whom one case harbored a dual lesion of pituitary adenoma and optic chiasm glioma. Because genetic analysis was not performed, the role of menin in the development of this brain lesion is uncertain (Table S2). Thus, the connection of MEN1 to neuronal migrational defects or to epilepsy outside of endocrinological disturbances of MEN1 syndrome has not been previously described. However, there is evidence that menin plays an important role in neurogenesis and synaptic function.17, 21 Homozygous deletions in mouse models are embryonically lethal and lead to developmental abnormalities, including neural tube defects.19 Menin has several key interactions with transcriptional activators and repressors (Figure 2A). For example, menin enhances transcription of a neuronal modulator p35, which is a dominant activator of neuronal cyclin‐dependent kinase 5 (Cdk5),21 an important regulator of neuronal migration, neurite outgrowth, axon guidance, and synapse formation.22 Moreover, Cdk5 deficiency in Cdk5 and p35 knockout mouse models triggers aberrant neuronal migration and limbic epilepsy.23 Importantly, the truncating mutation of our patient (p.R516PfsX15) affects one of the two NLS domains, and it was shown that this variant impairs nuclear localization of a protein, thus directly affecting the ability of menin to exert its key function in transcriptional regulation.13 The mutation in our patient also affects the domain interacting with Smad3, and menin supports Smad3/TGFβ transcriptional control of cell growth.24 Also, the Smad3/TGFβ signaling cascade is implicated in enhancing neuronal excitability and was shown to be altered in patients with temporal lobe epilepsy and hippocampal sclerosis.25, 26 Equally intriguing is menin involvement in DNA repair through the checkpoint kinase 1 (CHES1), because a loss of function may lead to additional mutations and possible cell proliferation through impaired DNA repair.27 Lastly, menin undergoes proteolytic cleavage, and the C‐terminal fragment colocalizes in the glutamatergic presynaptic terminals with the nicotinic acetylcholine receptor α7 subunits.17 The absence of C‐terminal fragment in mouse hippocampal neurons impacts synapse formation, plasticity, survival, maturation, and synaptic integration of adult‐born neurons.17 It is, thus, intriguing to speculate that in our patient, the predicted MEN1 gene haploinsufficiency not only triggered problems with neuronal proliferation, differentiation, and positioning through a downstream effect on the p35/Cdk5, Smad3/TGFβ, and CHES1 systems, but also contributed to the epileptogenic circuitry through the cholinergic dysregulation at the hippocampal synapses.
Our pilot investigation has limitations to be reconciled in future studies. (1) Our discovery was made in a single patient and needs to be validated in additional cases. We have submitted the MEN1 gene in connection to nodular heterotopia and malformations of cortical development to GeneMatcher (https://genematcher.org, submission ID = 37289) but have not yet identified additional patients. (2) We identified a somatic pathogenic variant of MEN1 in the brain‐derived DNA and showed its absence in the peripheral blood‐derived sample. However, we did not have access to other tissue types or to tissue from other brain regions. Thus, we could not determine the true extent of mosaicism. (3) Prior research has shown that tumor cells of the MEN1 syndrome often display LOH.15, 19 We attempted a copy number variant analysis but could not achieve reliable exon‐level deletion/duplication results using the available exome sequence data. A reliable determination of copy number change in a limited amount of the DNA source derived from a mixed cellular population will require future, more extensive experimentation with quantitative molecular assays. However, despite these limitations, our study contributes a novel candidate gene for one of the most common forms of structural malformations of cortical development, and this discovery complements and advances our understanding of the complex regulatory processes involved in neuronal development and in epileptogenic and cognitive networks. Additionally, our study contributes a practical, minimally invasive molecular diagnostic approach using trace tissues adherent to diagnostic SEEG electrodes. This allows integration of the electrophysiological and genetic explorations of epilepsy, which may be especially valuable for patients not eligible for resective epilepsy surgery in whom genetic testing of blood DNA remains unrevealing.
CONFLICT OF INTEREST
P.C.V. is a site PI for Cenobamate and received travel fees to attend their annual investigator's meeting. P.V. also received travel fees to attend the June 2019 JAMA Neurology editorial board meeting.
AUTHOR CONTRIBUTIONS
L.M.: data collections, analysis, manuscript preparation. Z.H., J.G., and P.C.V.: clinical care and evaluation of the patient, critical review of the manuscript. D.Y., R.N., and T.V.: clinical care and evaluation of the patient, SEEG electrode implantation and removal, critical review of the manuscript. J.D.: tissue sample processing, genetic data analysis, variant validation, critical review of the manuscript. A.M.G.: conceptual design of the study, sample collection, clinical care and evaluation of the patient, data analysis, manuscript preparation, critical review of the manuscript.
Supporting information
ACKNOWLEDGMENTS
We appreciate the kind assistance of Eric Raap, research coordinator at the Translational Epilepsy Neurogenetics Laboratory and Baylor Adult Epilepsy Genetics Clinic, Martine Loeb, MS, Genetic Counselor, and her colleagues at the GeneDx Clinical Diagnostic Laboratory.
Montier L, Haneef Z, Gavvala J, et al. A somatic mutation in MEN1 gene detected in periventricular nodular heterotopia tissue obtained from depth electrodes. Epilepsia. 2019;60:e104–e109. 10.1111/epi.16328
Funding information
This study was supported by NIH/NINDS 1 U01 NS090406 (A.M.G., L.M.) and 1 U01 NS090362 (A.M.G., J.D.)
REFERENCES
- 1. Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with filamin A mutations. Brain. 2006;129:1892–906. [DOI] [PubMed] [Google Scholar]
- 2. Liu W, Yan B, An D, Xiao J, Hu F, Zhou D. Sporadic periventricular nodular heterotopia: classification, phenotype and correlation with filamin A mutations. Epilepsy Res. 2017;133:33–40. [DOI] [PubMed] [Google Scholar]
- 3. Heinzen EL, O'Neill AC, Zhu X, et al. De novo and inherited private variants in MAP1B in periventricular nodular heterotopia. PLoS Genet. 2018;14:e1007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cellini E, Vetro A, Conti V, et al. Multiple genomic copy number variants associated with periventricular nodular heterotopia indicate extreme genetic heterogeneity. Eur J Hum Genet. 2019;27:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jamuar SS, Walsh CA. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:2038. [DOI] [PubMed] [Google Scholar]
- 6. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGovern RA, Ratneswaren T, Smith EH, et al. Investigating the function of deep cortical and subcortical structures using stereotactic electroencephalography: lessons from the anterior cingulate cortex. J Vis Exp. 2015(98). 10.3791/52773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klassen TL, Bomben VC, Patel A, et al. High‐resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Retterer K, Juusola J, Cho MT, et al. Clinical application of whole‐exome sequencing across clinical indications. Genet Med. 2016;18:696–704. [DOI] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mandelstam SA, Leventer RJ, Sandow A, et al. Bilateral posterior periventricular nodular heterotopia: a recognizable cortical malformation with a spectrum of associated brain abnormalities. AJNR Am J Neuroradiol. 2013;34:432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agarwal SK, Kester MB, Debelenko LV, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997;6:1169–75. [DOI] [PubMed] [Google Scholar]
- 13. Ikeo Y, Sakurai A, Hashizume K. Characterization of the MEN1 gene product, menin, by site‐specific polyclonal antibodies. Jpn J Cancer Res. 1999;90:1088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2. 0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55. [DOI] [PubMed] [Google Scholar]
- 15. Loffler KA, Biondi CA, Gartside M, et al. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. Int J Cancer. 2007;120:259–67. [DOI] [PubMed] [Google Scholar]
- 16. Mirzaa GM, Campbell CD, Solovieff N, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 2016;73:836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Getz AM, Xu F, Visser F, Persson R, Syed NI. Tumor suppressor menin is required for subunit‐specific nAChR alpha5 transcription and nAChR‐dependent presynaptic facilitation in cultured mouse hippocampal neurons. Sci Rep. 2017;7:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart C, Parente F, Piehl F, et al. Characterization of the mouse Men1 gene and its expression during development. Oncogene. 1998;17:2485–93. [DOI] [PubMed] [Google Scholar]
- 19. Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17:1880–92. [DOI] [PubMed] [Google Scholar]
- 20. Mohr H, Pellegata NS. Animal models of MEN1. Endocr Relat Cancer. 2017;24:T161–77. [DOI] [PubMed] [Google Scholar]
- 21. Zhuang K, Huang C, Leng L, et al. Neuron‐specific menin deletion leads to synaptic dysfunction and cognitive impairment by modulating p35 expression. Cell Rep. 2018;24:701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shah K, Lahiri DK. A tale of the good and bad: remodeling of the microtubule network in the brain by Cdk5. Mol Neurobiol. 2017;54:2255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patel LS, Wenzel HJ, Schwartzkroin PA. Physiological and morphological characterization of dentate granule cells in the p35 knock‐out mouse hippocampus: evidence for an epileptic circuit. J Neurosci. 2004;24:9005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hendy GN, Kaji H, Sowa H, Lebrun JJ, Canaff L. Menin and TGF‐beta superfamily member signaling via the Smad pathway in pituitary, parathyroid and osteoblast. Horm Metab Res. 2005;37:375–9. [DOI] [PubMed] [Google Scholar]
- 25. Paul D, Dixit A, Srivastava A, et al. Altered transforming growth factor beta/SMAD3 signalling in patients with hippocampal sclerosis. Epilepsy Res. 2018;146:144–50. [DOI] [PubMed] [Google Scholar]
- 26. Zhang W, Du Y, Zou Y, Luo J, Lü Y, Yu W. Smad anchor for receptor activation and phospho‐Smad3 were upregulated in patients with temporal lobe epilepsy. J Mol Neurosci. 2019;68:91–8. [DOI] [PubMed] [Google Scholar]
- 27. Bartsch DK, Slater EP, Albers M, et al. Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab 2014;99:E2387–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials