

Abstract
Background
Fibrosing, non-idiopathic pulmonary fibrosis (non-IPF) interstitial lung diseases (fILDs) are a heterogeneous group of diseases characterized by a different amount of inflammation and fibrosis. Therapy is currently based on corticosteroids and/or immunomodulators. However, response to these therapies is highly variable, sometimes without meaningful improvement, especially in more fibrosing forms. Pirfenidone and nintedanib have recently demonstrated to reduce functional decline in patients with IPF. However, their antifibrotic mechanism makes these two drugs an interesting approach for treatment of fibrosing ILDs other than IPF.
Objectives
We here report our experience with antifibrotic drugs in fibrosing non-IPF ILDs patients having a progressive phenotype during immunosuppressive therapy.
Methods
Patients with a multidisciplinary team diagnosis of fibrosing non-IPF ILDs experiencing a progressive phenotype during treatment with corticosteroids and/or immunomodulators between October-2014 and January-2018 at our tertiary referral Center for ILDs were retrospectively analyzed. Antifibrotic therapy was administered after application with the respective health insurance company and after consent by the patient. Pulmonary-function-tests and follow-up visits were performed every 6 ± 1 months.
Results
Eleven patients were treated with antifibrotic drugs (8 males, mean age 62 ± 12.8 years, mean FVC% 62.8 ± 22.3, mean DLCO% 35.5 ± 10.7, median follow-up under antifibrotic treatment 11.1 months). Patients had a diagnosis of unclassifiable ILD in 6 cases, pleuroparenchymal fibroelastosis in 2 cases, idiopathic-NSIP in 1 case, asbestos-related ILD in 1 case and Hermansky-Pudlak syndrome in 1 case. Treatment before antifibrotics consisted of corticosteroids in all patients: 5 combined with Azathioprin, 1 with either methotrexate or cyclophosphamide (i.v.). Ten patients were treated with pirfenidone (2403 mg/die) and 1 with nintedanib (300 mg/die). Median FVC was 56, 56, 50%, at time points − 24, − 12, − 6 before initiation, 44% at time of initiation and 46.5% at 6 months after initiation of antifibrotic treatment. Antifibrotic treatment was generally well tolerated with a need of dose reduction in 2 cases (rash and nausea) and early termination in 3 cases.
Conclusions
Antifibrotic treatment may be a valuable treatment option in patients with progressive fibrosing non-IPF ILD if currently no other treatment options exist. However, prospective, randomized clinical trials are urgently needed to assess the real impact of antifibrotic therapy in these patients.
Keywords: Interstitial lung disease, Nintedanib, Pirfenidone, Progressive fibrosing interstitial lung diseases, Real-world experience, IPAF
Background
Interstitial lung diseases comprise a heterogeneous group of almost 200 entities characterized by a different amount of inflammation and/or fibrosis [1, 2]. Idiopathic Pulmonary Fibrosis (IPF) is the most frequent and aggressive form, representing the prototype of progressive fibrosing interstitial lung diseases [3, 4]. However, recent evidence has shown that also other non-IPF fibrosing interstitial lung diseases may, similarly to IPF, reveal a progressive phenotype (PF-ILDs) characterized by a rapid functional decline, worsening of symptoms and a detrimental prognosis [5–9]. According to a recent study by Olson et al., the prevalence of this subset of patients has been estimated at 0.22–2 per 10,000 persons in Europe and 2.8 per 10,000 persons in USA, thus representing a considerable number [10].
Therapy of PF-ILDs is currently based on corticosteroids and/or immunomodulators. However, response to these therapies is highly variable, sometimes without meaningful improvement [11]. In recent years two antifibrotic drugs, pirfenidone and nintedanib, have been developed and approved for the treatment of IPF [12–17]. Their impact on the course of other fibrosing ILDs is unknown. However, given some pathobiological and clinical similarities between PF-ILDs and IPF, both pirfenidone and nintedanib, may represent an interesting and reasonable approach also for PF-ILDs [18–21].
We here report our experience with antifibrotic drugs in fibrosing non-IPF ILDs patients with a progressive phenotype despite immunosuppressive therapy.
Methods
Study population
A retrospective analysis of the database of our tertiary referral Center identified all patients with a multidisciplinary team (MDT) diagnosis of fibrosing non-IPF ILDs that experienced a progressive decline in lung function during treatment with corticosteroids and/or immunomodulators between October-2014 and January-2018. At the time of diagnosis, a complete evaluation of medical history, serological data including autoantibodies, all comorbidities and related treatments were assessed. A rheumatologic evaluation was also required to interpret any rheumatologic sign and serological data [1, 22–24]. Each patient underwent high resolution computed tomography (HRCT) exams at baseline time and on follow-up every 6–12 months. In-house software YACTA was used to automatically quantify lung density histogram. Longitudinal changes in the 40th and 80th percentiles of attenuation histogram were assessed [25]. If possible, Bronchoalveolar lavage (BAL) and transbronchial cryobiopsy were also performed. Age, physiology and distinct comorbidities were used to calculate the TORVAN index, an index predictive of mortality recently validated for IPF [26]. Patients were considered to have a progressive phenotype if there was evidence of any of the following criteria: a relative decline of ⩾10% in forced vital capacity (FVC); a relative decline of ⩾15% in diffusing capacity of the lung for carbon monoxide (DLCO); or worsening symptoms or a worsening radiological appearance accompanied by a ⩾5–< 10% relative decrease in FVC within a 24-month period prior to antifibrotic therapy initiation [5, 6]. Patients underwent routinely follow-up visits and pulmonary function tests (FVC and DLCO) every 6 ± 1 months. Antifibrotic therapy, either pirfenidone or nintedanib, was introduced after a proven progression of the disease and lack of response to corticosteroids and/or immunomodulators, after approval of the respective health insurance company and according to German laws for off-label use and after consent by the patient. As in IPF patients, pirfenidone was given as continuous oral treatment at a dose of 2403 mg·day− 1 (3 capsules three times·day− 1) and nintedanib 300 mg·day− 1 (1 capsule twice·day− 1) [12–17]. Patients under antifibrotic therapy were reevaluated every 6–12 weeks after initiation according to the standard of care in our department. Liver function monitoring was conducted on a monthly basis. All adverse events (related and not related to treatment), treatment compliance and interruptions for any reason were also recorded at each treatment visit. To assess difference in one-year mortality between PF-ILD and IPF, a comparison with a cohort of 257 IPF patients collected in our center was also performed. All clinical information was obtained from medical records. Pulmonary function tests (FVC and DLCO) were performed according to the ATS/ERS guidelines.
Statistical analysis
Characteristics of the study population were expressed as median (interquartile range) or as percentage of the relative frequency as appropriated. Wilcoxon test for paired data was used to assess differences in median FVC between each follow-up time. Kaplan-Meier survival analysis was used to assess overall survival. All the statistical analyses were performed using STATA/IC 14.2 version. A p value less than 0.05 was considered significant.
Results
Eleven patients were included in the analysis. Eight were males (72.72%) and 3 were females (27.27%). There were 5 former smokers (45.45%) and 6 never-smokers (54.54%). The mean age was 62.09 ± 12.80 and 63.72 ± 12.72 years at diagnosis and at antifibrotic initiation respectively. The mean FVC % predicted was 62.82 ± 22.30 while the mean DLCO % predicted was 35.55 ± 10.74 before initiation of the antifibrotic therapy. The mean TORVAN index was 17.18 ± 5.13. Patients were followed for a median follow-up time of 16.6 months before and 11.1 months under antifibrotic treatment. Visual assessment of HRCT findings at baseline is showed in Table 1 while Fig. 1 reports automatic histogram-based assessment of 40th and 80th percentiles over the time. According to this result, a significant increase of both indices was observed before antifibrotic initiation followed by a stabilization. Bronchial alveolar lavage was performed in 8 cases before immunosuppressive therapy demonstrating a predominant neutrophilia (12%) in 4 cases, a notable eosinophilia (10%) in one case and no significant lymphocytosis.
Table 1.
Baseline characteristics of patients
| Mean ± SD or n (%) |
Median (range interquartile) | |
|---|---|---|
| Age at diagnosis (years) | 62.09 ± 12.80 | 63 (50, 76) |
| Age at initiation of antifibrotic therapy | 63.72 ± 12.72 | |
| Male | 8 (72.72) | |
| Former smoker | 5 (45.45) | |
| Never smoker | 6 (54.54) | |
| Cryobiopsy | 9 (81.81) | |
| Follow-up time (days) since diagnosis | 903 (381, 1489) | |
| Follow-up time (days) since antifibrotic initiation | 333 (156, 421) | |
| FVC% pred at baseline | 62.82 ± 22.30 | 52.7 (49, 77) |
| DLCO% pred at baseline | 35.55 ± 10.74 | 34 (29, 37.1) |
| TORVAN index (points) | 17.18 ± 5.13 | 19 (13, 21) |
| HRCT features | ||
| Reticulations | 7 (63.6) | |
| Traction bronchiectasis | 8 (72.7) | |
| Honeycombing | 2 (27.2) | |
| Ground-glass | 7 (63.6) | |
| Consolidations | 3 (27.2) | |
| Mosaic attenuation | 2 (27.2) | |
| Upper-mid lung predominance | 3 (27.2) | |
| Lower lung predominance | 4 (36.3) | |
| Prednisone | 11 (100) | |
| Daily dosage (mg) | 16.81 ± 11.18 | |
| Azathioprin | 5 (45.04) | |
| Daily dosage (mg) | 150 | |
| Methotrexate | 1 (9,09) | |
| Weekly dosage (mg) | 7.5 | |
| Cyclophosphamide | 1 (9.09) | |
| Dosage i.v. per cycle (mg) | 1000 | |
Fig. 1.

Change in the percentiles (Hounsfield units) of attenuation histogram over the time. Panel a demonstrates change in the 40th percentiles while panel b demonstrates change in the 80th percentiles
Transbronchial cryobiopsy was performed in 9 patients (81.8%). After a multidisciplinary team discussion, patients were classified as unclassifiable-ILD in 6 cases, pleuroparenchymal fibroelastosis (PPFE) in 2 cases, idiopathic-NSIP in 1 case, asbestos-related ILD in 1 case and Hermansky-Pudlak syndrome in 1 case. Of these, three patients may be also considered as interstitial pneumonia with autoimmune features (IPAF) according to established criteria (Table 2) [20]. Treatment before antifibrotic drug initiation consisted of corticosteroids (prednisone mean dosage 16.8 ± 11.1 mg/die) in all patients: 5 combined with Azathioprin (150 mg/die), 1 with either methotrexate (7,5 mg/week) or cyclophosphamide (1000 mg i.v. per cycle) (Table 1). After a proven clinical and functional progression, ten patients were treated with pirfenidone (2403 mg/die) and 1 with nintedanib (300 mg/die). Of the 11 patients, 6 continued prednisone (5 mg/die) for a mean time of 8.5 months and 2 continued Azathioprin for a mean time of 4 months after antifibrotic initiation. Median time of antifibrotic treatment was 11.1 (5.2, 14) months.
Table 2.
Detailed characteristics of patients
| Characteristics | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Case 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BMI | 24 | 37 | 23 | 19 | 30 | 23 | 21 | 31 | 25 | 21 | 31 |
| Smoking history (pack years) | Former (12 py) | Former (70 py) | Never | Never | Never | Never | Former (20 py) | Former (15 py) | Former (25 py) | Never | Never |
| ILD relevant exposure | no | no | no | Powders, detergents | no | no | no | Cement powders | powders | no | no |
| Autoantibodies | ANA 1:80; ENA negative | ANA 1:320; ENA negative | ANA 1:640; ENA negative | negative | ANA 1:80; ENA negative | ANA 1:1280; ENA negative | ANA 1:160; ANCA 1:32; ENA negative | ANA 1:320; ENA negative | ANA 1:320; ENA negative | ANA 1:160; ENA negative | ANA 1:160; ENA negative |
| FVC L (%) at baseline | 1.9 (63) | 1.82 (48) | 2.74 (77) | 1.26 (52.7) | 2.1 (49) | 2.31 (52.7) | 4.28 (101) | 3.37 (97.7) | 3 (73) | 0.93 (26.5) | 1.47 (51.5) |
| DLCO % at baseline | 38 | 35 | unable | 27.4 | 27 | 30 | 64 | 29 | 37.1 | 33 | 35 |
| TORVAN index | 19 | 21 | 19 | 22 | 14 | 24 | 13 | 17 | 7 | 12 | 21 |
| BAL findings at initial diagnosis | Macrophages 65%, Lymphocytes 13%, Neutrophils 22% | Macrophages 85%, Lymphocytes 12%, Neutrophils 3%, Eosinophils 0% | Macrophage 76%, Lymphocytes 10%, Neutrophils 9%, Eosinophils 3%, Mastcells 2% | / | Macrophage 89%, Lymphocytes 9%, Eosinophils1% |
Macrophages 89%, Lymphocytes 14%, Neutrophils 2% |
Macrophage 96%, Lymphocytes 2%, Neutrophils 2% | Macrophage 91%, Lymphocytes 9%, Neutrophils 10% | Macrophages 77%, Lymphocytes 5%, Neutrophils 8%, Eosinophils 10% | / | / |
| Histological pattern | NSIP | / | Indeterminate | Compatible with PPFE | Indeterminate | Indeterminate | Indeterminate | UIP-like | Indeterminate | / | Indeterminate |
| LTOT at initiation of antifibrotic therapy | Yes | Yes | No | No | No | No | No | Yes | Yes | No | Yes |
| Distance at 6MWT (meters) | 425 | / | 578 | 363 | 313 | 402 | 540 | 323 | 480 | 418 | / |
| MDT diagnosis | iNSIP | uILD (IPAF) | uILD (IPAF) | PPFE | uILD | uILD (IPAF) | uILD | Asbestos-associated fibrosis | ILD in Hermansky-Pudlak syndrome | PPFE | uILD |
| Treatment before antifibrotics | Pred. 30 mg; AZT 125 mg; NAC 1800 mg | Pred. 15 mg; AZT 150 mg; MTX 7,5 mg | Pred. 30 mg; AZT 150 mg | Pred. 10 mg; | Pred. 10 mg; | Pred. 7,5 mg; AZT 150 mg; NAC 1800 mg | Pred. 7,5 mg; Cycl. (6 cycles) | Pred. 15 mg; AZT 150 mg | Pred. 10 mg; | Pred. 10 mg; | Pred. 40 mg; |
| Antifibrotic treatment | Pirf 2403 mg | Nint. 300 mg | Pirf 2403 mg | Pirf 1602 mg | Pirf 2403 mg | Pirf 2403 mg | Pirf 2403 mg | Pirf 2403 mg | Pirf 1602 mg | Pirf 2403 mg | Pirf 2403 mg |
Footnotes: BMI Body mass index, FVC Forced Vital capacity, DLCO Diffusing Capacity for carbon monoxide, BAL Bronchial alveolar lavage, LTOT long term oxygen therapy, MDT multidisciplinary discussion, iNSIP idiopathic-Non specific interstitial Pneumonia, uILD unclassifiable Interstitial lung disease, PPFE pleuroparenchymal fibroelastosis, Pred. prednisone, AZT Azathioprin, MTX Methotrexate, Cycl. cyclophosphamide, Pirf. Pirfenidone, Nint. Nintedanib
Median FVC was 56% (2.29 L), 56% (2.07 L), 50% (1.95 L), at time points − 24, − 12, − 6 before initiation, 44% (1.59 L) at time of initiation and 46.5% (1.77 L) at 6 months after initiation of antifibrotic treatment. Median FVC difference was significant between − 12 and − 6 months before initiation (p = 0.004) and also between − 6 and time of initiation (p = 0.005), while no further significant decline was reported between time of initiation and + 6 months (p = 0.17). This last result was obtained comparing data of only eight patients as three patients died before this time due to respiratory failure (Fig. 2). Antifibrotic drugs were generally well tolerated. Mean dosage of pirfenidone was 2242 ± 337 mg·day− 1, while for the patient treated with nintedanib mean dosage was 300 mg·day− 1. A dose reduction was needed in 2 cases under pirfenidone treatment: one for rash (case 3) and the other for nausea (case 9), while an early termination was needed in 3 cases due to death following respiratory failure (Table 2). Only one patient experienced acute exacerbations after antifibrotics initiation. Median survival time was 11.1 months since antifibrotic drug initiation (Fig. 3).
Fig. 2.
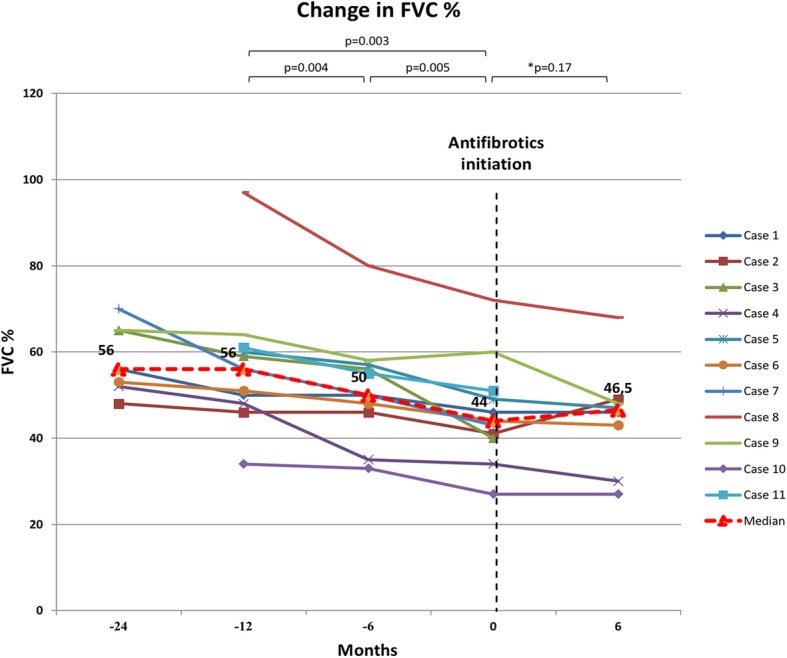
Change in FVC % predicted over the time. Footnotes: *this median difference was calculated comparing data of only eight patients as three patients died before the 6 months of follow-up since antifibrotics initiation
Fig. 3.

One-year survival of PF-ILD compared to a cohort of 257 IPF patients collected in our center
Discussion
In recent years, undeniable progress has been achieved in understanding the pathogenic mechanisms of IPF. This has progressively led to the advent of pirfenidone and nintedanib, the first two drugs able to reduce lung function decline [12–17]. Comparable to IPF, some PF-ILDs are triggered by repetitive lung parenchymal injuries and demonstrate TGFβ-mediated fibroblast activation and myofibroblast accumulation that may lead to a progressive phenotype [18–21]. However, the reasons by which some ILDs demonstrate a IPF-like behavior while some others do not are still unsolved and can be only partially justified by these simple pathogenic similarities.
Corticosteroids represent current first line therapeutic approach with the addition, in some cases, of immunomodulators. However, evidence has clearly demonstrated that a significant proportion of these patients does not benefit of these therapies [11]. Therefore, there is an emerging need to identify possible effective treatments for these specific setting. Due to their antifibrotic and anti-inflammatory activity, both pirfenidone and nintedanib, are potential therapeutic candidates for the management of PF-ILDs. In this context several trials are currently investigating the use of antifibrotic drugs in other progressive, fibrosing ILDs than IPF [19]. For example, the German RELIEF trial assesses the efficacy and safety of pirfenidone in patients with fibrosing ILDs with a progressive phenotype other than IPF [6]. After an exploratory safety trial with Pirfenidone in patients with Systemic sclerosis associated-ILD (SSc-ILD) (LOTUSS trial) has been published, currently the scleroderma lung trial III assesses the efficacy of pirfenidone on the background of Mycophenolate mofetil [7, 19]. Similarly, Pirfenidone is investigated in unclassifiable ILD and in other subgroups of PF-ILDs [19, 27]. Furthermore, nintedanib is investigated in patients with fibrosing, progressive ILDs in the INBUILD trial and results from the SENSCIS trial investigating nintedanib in SSc-ILD are expected soon [5, 8, 19]. However, reports, especially on real life data on the efficacy and safety of antifibrotic drugs in non-IPF PF-ILDs are still lacking.
To our knowledge, our report on the effects of antifibrotic drugs in non-IPF fibrosing and progressive ILDs is the largest and the most heterogeneous reported experience on the use of antifibrotics in this group of PF-ILDs [28–30]. According to our analyses, the introduction of antifibrotics demonstrated a significant reduction of lung function decline (Fig. 2) and of radiologic worsening (Fig. 4) after 6 months since their initiation. Similarly, longitudinal changes in the 40th and 80th percentiles of attenuation histogram, that represent promising radiologic parameters for monitoring the disease extent, demonstrated a progressive increase before antifibrotics initiation and a stabilization after their initiation highlighting a worsening of low density areas (40th percentiles) such as ground-glass areas and of high density areas (80th percentiles) such as fibrotic areas (Fig. 1). These results, even if derived from a limited number of patients, are promising and suggest that antifibrotics may be helpful also in this subset of patients. Interestingly, as some patients may also be classified as IPAF, antifibrotics could represent a valid therapeutic option also for this specific group [22–24]. Furthermore, as documented in IPF patients, both pirfenidone and nintedanib were generally well tolerated, with the need of a dose reduction only in a few cases [12–17].
Fig. 4.

Example of one case showing radiological worsening in the 6 months preceding antifibrotic initiation and no significant changes after 6 months since their initiation. Figure also shows the fully automatic lung parenchyma segmentation as obtained by in-house YACTA software
The one-year survival of PF-ILDs looks incredibly similar to IPF making the two diseases very similar (Fig. 3). However, the pronounced mortality in our cohort has to be also discussed in light of the already very advanced patient status and their impaired general condition. With the exception of one case who experienced a further acute exacerbation after antifibrotic initiation, no other severe adverse events were registered after the introduction of antifibrotics, confirming an acceptable safety and tolerability profile also in PF-ILDs.
This study has some strengths. Patients were evaluated in an academic center through a multidisciplinary discussion, were routinely followed every 3–6 months with pulmonary function tests and visits, and refer to a real-life setting. Moreover, even if the total number of patients is small, there is a variety in the type of PF-ILDs analyzed.
However, there are also several limitations. First of all, this is a retrospective and single-center study. This element might have created some bias of selection and lack of some data. Second, the number of patients is very small. Therefore, results have to be considered with caution and need to be confirmed by clinical trials that are still ongoing. Third, again due to the small number of patients, the statistical approach was very simple and limited to a comparison of median FVC before and after antifibrotics introduction. Finally, due to the observational and retrospective nature of the study, a comparison with an untreated control group was not possible as well as to collect also data on quality of life over the time.
Conclusions
Antifibrotic treatment after multidisciplinary team discussion and with patient consent may be a valuable treatment option in patients with progressive fibrosing non-IPF ILDs that do not benefit from corticosteroids and immunosuppressive treatments if no other treatment options exist. However, prospective, randomized clinical trials are urgently needed to assess the real impact of antifibrotic therapy in these patients.
Acknowledgements
Not applicable
Abbreviations
- BAL
Bronchoalveolar lavage
- DLCO
Diffusing capacity of the lung for carbon monoxide
- FVC
Forced vital capacity
- HRCT
High resolution computed tomography
- IPAF
Interstitial pneumonia with autoimmune features
- IPF
Idiopathic Pulmonary Fibrosis
- MDT
Multidisciplinary team
- PF-ILDs
Progressive fibrosing non-IPF interstitial lung diseases
- PPFE
Pleuroparenchymal fibroelastosis
- SSc-ILD
Systemic sclerosis associated-ILD
Authors’ contributions
SET, NK, SP, FMS, GS, DS contributed to the conception and design of the work, to data analysis and interpretation, and to the drafting and substantial revision of the work. JW, NS, MP, KL, ME have made contribution to acquisition of data. CPH, CV and MK revised the manuscript. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
The data analyzed in the current study are not publicly available but may be made available from the corresponding authors on reasonable request.
Ethics approval and consent to participate
The ethics committee of the University of Heidelberg approved this retrospective study. Due to the retrospective nature of this analysis and according to the vote of the ethics committee, written informed consent could not be obtained by the patients but patient records / information were anonymized and de-identified prior to analysis.
Consent for publication
Not applicable
Competing interests
S.E. Torrisi reports grants from Boehringer Ingelheim and F Hoffman La-Roche, outside the submitted work. J. Wälscher reports fees for consulting from Boehringer Ingelheim and Roche/InterMune, outside the submitted work. C. Vancheri reports grants and speaker’s fees from Boehringer Ingelheim and Intermune/F Hoffman La-Roche, outside the submitted work. M. Kreuter reports grants and fees for consulting from Boehringer Ingelheim and Roche/InterMune, outside the submitted work. The other authors report no conflicts of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valeyre D, Duchemann B, Nunes H, et al. Interstitial lung diseases. In: Annesi-Maesano I, Lundbäck B, Viegi G, et al., editors. Respiratory epidemiology. 2014. pp. 79–87. [Google Scholar]
- 3.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 4.Puglisi S, Torrisi SE, Giuliano R, Vindigni V, Vancheri C. What we know about the pathogenesis of idiopathic pulmonary fibrosis. Semin Respir Crit Care Med. 2016;37:358–367. doi: 10.1055/s-0036-1580693. [DOI] [PubMed] [Google Scholar]
- 5.Flaherty KR, Brown KK, Wells AU, Clerisme-Beaty E, Collard HR, Cottin V, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017;4:e000212. doi: 10.1136/bmjresp-2017-000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behr J, Neuser P, Prasse A, Kreuter M, Rabe K, Schade-Brittinger C, et al. Exploring efficacy and safety of oral Pirfenidone for progressive, non-IPF lung fibrosis (RELIEF) - a randomized, doubleblind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm Med. 2017;17(1):122. doi: 10.1186/s12890-017-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khanna D, Albera C, Fischer A, Khalidi N, Raghu G, Chung L, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. 2016;43:1672–1679. doi: 10.3899/jrheum.151322. [DOI] [PubMed] [Google Scholar]
- 8.Distler O, Brown KK, Distler JHW, Assassi S, Maher TM, Cottin V, et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS) Clin Exp Rheumatol. 2017;35(Suppl. 106):75–81. [PubMed] [Google Scholar]
- 9.Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076. doi: 10.1183/16000617.0076-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson A, Hartmann N, Schlenker-Herceg R, Wallace L. Prevalence of progressive fibrosing interstitial lung disease. Eur Respir J. 2018;52:PA3030. doi: 10.1183/13993003.congress-2018.PA3030. [DOI] [Google Scholar]
- 11.Kreuter M, Olson A, Fischer A, Bendstrup E, Mounir B, Zouad-Lejour L, et al. Current treatment of patients with non-IPF progressive fibrosing interstitial lung disease. Am J Respir Crit Care Med. 2018;197:A4273. [Google Scholar]
- 12.Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 13.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 14.Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–253. doi: 10.1183/13993003.00026-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 16.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 17.Torrisi Sebastiano Emanuele, Pavone Mauro, Vancheri Ada, Vancheri Carlo. When to start and when to stop antifibrotic therapies. European Respiratory Review. 2017;26(145):170053. doi: 10.1183/16000617.0053-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herzog EL, Mathur A, Tager AM, Feghali-Bostwick C, Schneider F, Varga J. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis Rheum. 2014;66:1967–1978. doi: 10.1002/art.38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreuter M, Walscher J, Behr J. Antifibrotic drugs as treatment of nonidiopathic pulmonary fibrosis interstitial pneumonias: the time is now (?) Curr Opin Pulm Med. 2017;23:418–425. doi: 10.1097/MCP.0000000000000408. [DOI] [PubMed] [Google Scholar]
- 20.Solomon JJ, Olson AL, Fischer A, Bull T, Brown KK, Raghu G. Scleroderma lung disease. Eur Respir Rev. 2013;22:6–19. doi: 10.1183/09059180.00005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richeldi L, Varone F, Bergna M, de Andrade J, Falk J, Hallowell R, et al. Pharmacological management of progressive-fibrosing interstitial lung diseases: a review of the current evidence. Eur Respir Rev. 2018;27(150):180074. doi: 10.1183/16000617.0074-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–987. doi: 10.1183/13993003.00150-2015. [DOI] [PubMed] [Google Scholar]
- 23.Sambataro G, Sambataro D, Torrisi SE, Vancheri A, Pavone M, Rosso R, et al. State of the art in interstitial pneumonia with autoimmune features: a systematic review on retrospective studies and suggestions for further advances. Eur Respir Rev. 2018;27(148):170139. doi: 10.1183/16000617.0139-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambataro G, Sambataro D, Torrisi SE, Vancheri A, Colaci M, Pavone M, et al. Clinical, serological and radiological features of a prospective cohort of interstitial pneumonia with autoimmune features (IPAF) patients. Respir Med. 2019;150:154–160. doi: 10.1016/j.rmed.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Colombi D, Dinkel J, Weinheimer O, Obermayer B, Buzan T, Nabers D, et al. Visual vs fully automatic histogram-based assessment of idiopathic pulmonary fibrosis (IPF) progression using sequential multidetector computed tomography (MDCT) PLoS One. 2015;10(6):e0130653. doi: 10.1371/journal.pone.0130653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torrisi SE, Ley B, Kreuter M, Wijsenbeek M, Vittinghoff E, Collard HR, et al. The added value of comorbidities in predicting survival in idiopathic pulmonary fibrosis: a multicenter observational study. Eur Respir J. 2019;53(3):1801587. doi: 10.1183/13993003.01587-2018. [DOI] [PubMed] [Google Scholar]
- 27.Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ Open Respir Res. 2018;5(1):e000289. doi: 10.1136/bmjresp-2018-000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duarte AC, Vinagre F, Soares J, Cordeiro A. Antifibrotics in interstitial lung disease related to connective tissue diseases - a paradigm shift in treatment and outcome. Acta Reumatol Port. 2019;44(2):161–162. [PubMed] [Google Scholar]
- 29.Bennett D, Refini RM, Valentini ML, Fui A, Fossi A, Pieroni M. T al. Pirfenidone therapy for familial pulmonary fibrosis: a real-life study. Lung. 2019;197(2):147–153. doi: 10.1007/s00408-019-00203-w. [DOI] [PubMed] [Google Scholar]
- 30.O'Brien KJ, Introne WJ, Akal O, Akal T, Barbu A, McGowan MP, et al. Prolonged treatment with open-label pirfenidone in Hermansky-Pudlak syndrome pulmonary fibrosis. Mol Genet Metab. 2018;125(1–2):168–173. doi: 10.1016/j.ymgme.2018.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data analyzed in the current study are not publicly available but may be made available from the corresponding authors on reasonable request.