

Abstract
Background
We investigated the role of PD-L1 in the metabolic reprogramming of non-small cell lung cancer (NSCLC).
Methods
Changes in glycolysis-related molecules and glycolytic activity were evaluated in PD-L1low and PD-L1high NSCLC cells after transfection or knockdown of PD-L1, respectively. Jurkat T-cell activation was assessed after co-culture with NSCLC cells. The association between PD-L1 and immune response-related molecules or glycolysis were analyzed in patients with NSCLC and The Cancer Genome Atlas (TCGA).
Results
Transfecting PD-L1 in PD-L1low cells enhanced hexokinase-2 (HK2) expression, lactate production, and extracellular acidification rates, but minimally altered GLUT1 and PKM2 expression and oxygen consumption rates. By contrast, knocking-down PD-L1 in PD-L1high cells decreased HK2 expression and glycolysis by suppressing PI3K/Akt and Erk pathways. Interferon-γ (IFNγ) secretion and activation marker expression was decreased in stimulated Jurkat T-cells when co-cultured with HK2-overexpressing vector-transfected tumor cells rather than empty vector-transfected tumor cells. Immunohistochemistry revealed that PD-L1 expression was positively correlated with HK2 expression in NSCLC (p < 0.001). In TCGA, HK2 exhibited a positive linear association with CD274 (PD-L1) expression (p < 0.001) but an inverse correlation with the expression of CD4, CD8A, and T-cell effector function-related genes in the CD274high rather than CD274low group. Consistently, there were fewer CD8+ T-cells in PD-L1positive/HK2high tumors compared to PD-L1positive/HK2low tumors in squamous cell carcinoma.
Conclusions
PD-L1 enhances glycolysis in NSCLC by upregulating HK2, which might dampen anti-tumor immunity. PD-L1 may contribute to NSCLC oncogenesis by inducing metabolic reprogramming and immune checkpoint.
Keywords: Programmed cell death-ligand-1, Hexokinase 2, Glycolysis, Non-small cell lung cancer, Tumor microenvironment
Background
The programmed cell death-1 (PD-1)/ programmed cell death-ligand (PD-L) pathway acts as an immune checkpoint for tumor cells to evade host immune surveillance [1, 2]. Recently, PD-1/PD-L1 blockade has emerged as a therapeutic strategy for patients with cancer [2], and has been approved for the first-line and second-line treatment of patients with non-small-cell lung cancer (NSCLC) [3]. However, some NSCLC patients respond to PD-1/PD-L1 blockade; thus, predictive biomarkers have been identified including PD-L1 expression, tumor mutational burden, and preexisting adaptive immune responses [4]. Although immunohistochemistry for PD-L1 has been approved as a companion/complementary diagnostics for PD-1/PD-L1 blockade, the response rate to PD-1/PD-L1 blockades is approximately 30% even in patients with PD-L1positive NSCLC [5]. Thus, more studies are needed to understand the biology and the mechanism of action of PD-1/PD-L1 blockade.
Most research into the PD-1/PD-L1 pathway has focused on the PD-1/PD-L1 interaction between target cells and immune cells. However, some studies have suggested other functions for PD-1 and PD-L1 beyond immune suppression. Intrinsic PD-L1 signaling promotes tumor cell proliferation and growth in melanoma and ovarian cancer cells by regulating autophagy and the mTOR pathway, and protects tumor cells from interferon (IFN)-mediated cytotoxicity [6–8]. In addition, PD-1 signaling alters T-cell metabolism by inhibiting glycolysis and amino acid metabolism and promoting lipolysis and fatty acid oxidation, thereby inhibiting effector T-cell differentiation [9].
To meet the metabolic requirements for growth and proliferation, tumor cells predominantly use glucose through aerobic glycolysis rather than oxidative phosphorylation (metabolic reprogramming known as the “Warburg effect”) [10]. Metabolic reprogramming is also important for proliferation and effector function of T-cells [11, 12]. Upon activation, lymphocytes undergo a metabolic transition from oxidative phosphorylation to aerobic glycolysis [12–14], which indicates that immune and tumor cells utilize similar metabolic reprogramming for their survival and function. Thus, they could compete for restricted nutrients within the tumor microenvironment (TME) and increased nutrient consumption by tumor cells could lead to an immunosuppressive TME by dampening the T-cell metabolism.
Using a murine sarcoma model, Chang et al. recently reported that PD-L1 elevated glycolysis in tumor cells, which promoted tumor progression by reducing glycolytic capacity and IFN-γ production in T-cells [15]. However, the role of PD-L1 in glucose metabolism and its clinical implication in human cancer cells is unclear. Therefore, we investigated the functional relationship between PD-L1 expression and glycolysis and its regulatory mechanisms in human lung cancer.
Materials and methods
Cell culture and reagents
NSCLC cell lines, including A549 and H460, were purchased from the American Type Culture Collection (Manassas, VA, USA). Jurkat cells were purchased from the Korean Cell Line Bank (Seoul, Republic of Korea). Cells were cultured in DMEM (A549 cells) and RPMI 1640 (H460 and Jurkat cells) supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37 °C. 2-deoxy-D-glucose, LY294002, U0126, and SB203580 were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Patients and samples
Tissue microarrays were constructed from formalin-fixed paraffin-embedded tumor tissues from 393 patients who underwent surgery for NSCLC at Seoul National University Hospital (SNUH, Seoul, Republic of Korea), including 228 pulmonary adenocarcinomas (pADCs) and 165 pulmonary squamous cell carcinomas (pSqCCs). An additional cohort of 80 patients with NSCLCs who received PD-1 blockade immunotherapy was established. Response to PD-1 blockade was assessed based on RECIST v1.1 [16]. Clinicopathological features of patients were analyzed as described in Additional file 1: Supplementary Methods. This study followed the World Medical Association Declaration of Helsinki recommendations and was approved by the Institutional Review Board (IRB) of SNUH (No.: H-1404-100-572).
Transfection of plasmid vector and siRNAs
A human PD-L1-expressing plasmid (Catalog No. HG10084-UT; Beijing, China) and a human HK2-expressing plasmid Catalog No. HG17967-UT; Beijing, China) were purchased from Sino Biological Inc.. PD-L1-specific siRNA-1 and 2 complementary to PD-L1 (GeneBank accession No.: NM 014143.2) were designed and synthesized by Bioneer (Daejeon, Republic of Korea). The sequences of siRNAs were as follows: PD-L1 siRNA-1, sense 5′-CAGCAUUGGAACUUCUGAU(dTdT)-3′ and antisense 5′-AUCAGAAGUUCCAAUGCUG(dTdT)-3′; PD-L1 siRNA-2, sense 5′-GAAUCAACACAACAACUAA(dTdT)-3′ and antisense 5′-UUAGUUGUUGUGUUGAUUC(dTdT)-3′. For transfection, cells were plated in either 12-well plates or 24-well plates and allowed to adhere for 24 h. Then the cells were transfected with plasmid or siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in Opti-MEM media (Qiagen, Germantown, MD, USA). After 6 h, the media were replaced with fresh complement media and cells were harvested 24–48 h after transfection.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) and subjected to reverse transcription using a PrimeScript First Strand cDNA Synthesis Kit (Takara Bio, Otsu, Japan). PCR was performed using SYBR® qRT-PCR Kit (Clontech Laboratories, Mountain View, CA, USA) and a Step One Plus thermal cycler (Applied Biosystems, Foster City, CA, USA) in triplicate. GAPDH and β-actin were used as the internal control. The following primer sequences were used for qRT-PCR: GAPDH forward 5′-CCCTTCATTGACCTACCTCAACTACAT-3′ and reverse 5′-ACGATACCAAAGTTGTCATGGAT-3′; β-actin forward 5′- CTGGAACGGTGAAGGTGAC-3′ and reverse 5′-AAGGGACTTCCTGTAACAATGCA -3′; CD274 (PD-L1) forward 5′-TATGGTGGTGCCGACTACAA-3′ and reverse 5′-TGGCTCCCAGAATTACCAAG-3′; SLC2A1 (GLUT1) forward 5′-GATTGGCTCCTTCTCTGTGG-3′ and reverse 5′-TCAAAGGACTTGCCCAGTTT-3′; HK2 forward 5′-CAAAGTGACAGTGGGTGTGG-3′ and reverse 5′-GCCAGGTCCTTCACTGTCTC-3′; PKM2 forward 5′-CCACTTGCAATTATTTGAGGAA-3′ and reverse 5′-GTGAGCAGACCTGCCAGACT-3′; PFKP forward 5′-GGGCCAAGGTGTACTTCATC-3′ and reverse 5′-TGGAGACACTCTCCCAGTCG-3′; PFKL forward 5′-GGTGGACCTGGAGAAGCTG-3′ and reverse 5′- GGCACCCACATAAATGCC-3′; PFKM forward 5′-GCCATCAGCCTTTGACAGA-3′ and reverse 5′-CTCCAAAAGTGCCATCACTG-3′; GPI forward 5′- GGAGACCATCACGAATGCAGA − 3′ and reverse 5′-TAGACAGGGCAACAAAGTGCT-3′; PGK forward 5′-AAGTCGGTAGTCCTTATGAGC-3′ and reverse 5′- CACATGAAAGCGGAGGTTCT-3′.
Western blotting
Total cellular proteins were extracted using lysis buffer (5 mM EDTA, 300 mM NaCl, 0.1% NP-40, 0.5 mM NaF, 0.5 mM Na3VO4, 0.5 mM PMSF, and 10 μg/mL each of aprotinin, pepstatin, and leupeptin; Sigma-Aldrich). A total of 30–50 μg protein was separated using 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Then immunoblotting was performed using antibodies against PD-L1 (clone E1L3N), GLUT1, HK2, PKM2, P-Akt, Akt (Cell Signaling Technology, Danvers, MA, USA), P-Erk, Erk, P-p38MAPK, p38MAPK, and β-actin (Santa Cruz Biotechnology, Dallas, TX, USA). Most images of western blots were from parallel gels and actin images were obtained from the stripped and re-probed blots. The immunoblots were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Uppsala, Sweden).
Glycolysis assessment: lactate production, hexokinase activity, and extracellular acidification rate (ECAR) assays
Glycolysis was evaluated using lactate production, hexokinase activity, and ECAR assays, as detailed in the Additional file 1: Supplementary Material and Methods.
Co-culture assay
Direct co-culture and transwell co-culture system were performed. Co-culture experiments were performed in 24-well plates without or with pore size 0.4 μm insets (Corning Costar, Corning, NY, USA). A549 cell (5 × 104) were seeded and cultured in the outer wells of 24-well plates in DMEM supplemented with 10% FBS for 24 h. A549 cells were transfected with empty or HK2-expressing vectors, as mentioned above. After 24 h, when fully upregulated HK2 expression, medium was changed to RPMI supplemented with 10% FBS and 1% penicillin/streptomycin. Incubating tumor cells for another 24 h, Jurkat cells (4 × 105) were added to directly to tumor cells or added to inner wells in transwell system. After 1 h of stabilization time, final concentration of 2 μg/ml soluble anti-CD3 (eBioscience, San Diego, CA, USA), 1 μg/ml soluble anti-CD28 (eBioscience) and 5 μg/ml anti-mouse Ig (SouthernBiotech, Birmingham, AL, USA) were added. 24 h later, media was harvested for IFN-γ ELISA assay and Jurkat cells were harvested for flow cytometry.
Enzyme-linked immunosorbent assay (ELISA) for IFN-γ
IFN-γ level in cell-free media was estimated using Human IFN-γ ELISA kit (R&D system, #DY285–05, Minneapolis, MN, USA) according to the manufacturer’s protocol.
Flow cytometry
Cells were harvested and washed with FACS buffer (0.5% BSA and 0.05% sodium azide in PBS). For discriminating dead cells, cells were first stained with zombie aqua (#423101, Biolegend, San Diego, CA, USA) in PBS for 10 min at room temperature in dark. Cells were stained with the PE-cy7 conjugated anti-CD69 antibody (#310911, Biolegend), PE-conjugated PD-L1 antibody (#329705, Biolegend) in FACS buffer for at least 30 min at 4 °C in dark. Flow cytometry was carried out on the LSRFortessa X-20 (BD Biosciences). Data were analyzed using FlowJo v10.1 (Treestar).
Immunohistochemistry (IHC)
IHC was performed for PD-L1 (clone E1L3N), HK2, GLUT1, PKM2, and CD8, as detailed in Additional file 1: Supplementary Material and Methods. PD-L1 positivity was defined as ≥10% of tumor cells with moderate-to-strong membranous intensity. H-scores for GLUT1, HK2 and PKM2 were estimated by integrating the intensity and proportion of staining [17]. CD8+ TILs per mm2 were automatically enumerated, as described in Additional file 1: Supplementary Material and Methods.
RNA seq and gene set enrichment analyses (GSEAs)
A549 cells transfected with PD-L1-expressing plasmid and H460 cells transfected with PD-L1 siRNA were submitted to RNA seq and GSEAs, as detailed in Additional file 1: Supplementary Material and Methods.
The Cancer Genome Atlas (TCGA) data analyses
The level 3 data of TCGA were downloaded from the UCSC Cancer Browser (https://genome-cancer.ucsc.edu) including pADC (N = 513) and pSqCC (N = 502) datasets and genome analysis was performed, as described in Additional file 1: Supplementary Material and Methods.
Statistical analyses
Statistical analyses were performed using SPSS v.21 (IBM Corp., New York, NY, USA), as detailed in Additional file 1: Supplementary Material and Methods. Two-sided p values < 0.05 were considered statistically significant in all analyses.
Results
PD-L1 upregulates HK2 expression and aerobic glycolysis in lung cancer cells
GLUT1 is the most common glucose transporter in humans. HK2 and PKM2 catalyze the first and final step of glycolysis, respectively [10, 18, 19]. To investigate whether PD-L1 affects the glycolysis in human NSCLC, we screened basal expression of PD-L1 and glycolysis-related molecules and glycolytic activity in several NSCLC cell lines (Additional file 3: Figure S1A-E). PD-L1low A549 cells showed lower HK2 expression and decreased aerobic glycolysis, as determined using lactate production, hexokinase activity, and ECAR assays, whereas PD-L1high H460 cells showed higher HK2 expression and increased glycolysis (Additional file 3: Figure S1F-J). Thus, we overexpressed PD-L1 in PD-L1low A549 cells and knocked-down PD-L1 in PD-L1high H460 cells for further experiments.
Upon PD-L1 overexpression in A549 cells, HK2 expression and hexokinase activity were increased (Fig. 1a, b). Moreover, PD-L1 overexpression significantly increased lactate production and ECAR, which was suppressed by 2-deoxy-D-glucose, a glucose analog that inhibits hexokinase (Fig. 1c, d). By contrast, downregulation of PD-L1 in H460 cells decreased HK2 expression, hexokinase activity, lactate production, and ECAR (Fig. 2). Oxygen consumption rate (Additional file 3: Figure S2) and transcription of other glycolytic genes were minimally affected by PD-L1 expression (Fig. 2a-c; Additional file 3: Figure S3). Consistently, gene set enrichment analyses revealed that the glycolytic pathway was enhanced in A549 cells after PD-L1 transfection and downregulated in H460 cells after PD-L1 knockdown (Additional file 3: Figure S4).
Fig. 1.

PD-L1 overexpression increases the expression and activity of HK2 and glycolysis in lung cancer cells. PD-L1low A549 cells were transfected with empty or PD-L1-expressing plasmid. Twenty-four hours after transfection, cells were submitted to Western blotting using antibodies against PD-L1, GLUT1, HK2, PKM2, and β-actin (a) or hexokinase activity assay (b). Lactate production was measured in the absence (ddH2O) or presence of 2-deoxy-glucose (2-DG) in A549 cells after PD-L1 overexpression (c). ECAR assays were performed in A549 cells transfected with empty or PD-L1-expressing plasmid upon adding glucose, oligomycin, and 2-DG to the culture media (d). Histograms represent values normalized to control. Data represent the means ± SEMs of at least three independent experiments or are representative of three independent experiments. All p values were calculated using unpaired Student’s t-tests and one-way ANOVA. **p < 0.001
Fig. 2.

PD-L1 knockdown decreases the expression and activity of HK2 and glycolysis in lung cancer cells. H460 cells were transfected with scrambled control siRNA (sc), PD-L1-specific siRNA1, or 2. Forty-eight hours after transfection, mRNA and protein levels of GLUT1, HK2, and PKM2 were evaluated using qRT-PCR and Western blotting (a-d). Hexokinase activity assays (e), lactate production assays (f), and ECAR assays (g) were also performed. Histograms represent values normalized to control. Data represent the means ± SEMs of at least three independent experiments or are representative of three independent experiments. All p values were calculated using unpaired Student’s t-tests and one-way ANOVA. *p < 0.05; **p < 0.001
To explore signaling pathways involved in PD-L1-mediated HK2 up-regulation, we evaluated several pathways that control glucose metabolism. PD-L1 knockdown decreased Akt and Erk phosphorylation but minimally altered p38MAPK phosphorylation in H460 cells (Fig. 3a). Consistently, HK2 expression and lactate production were decreased by a PI3K/Akt inhibitor (LY294002) and an Erk inhibitor (U0126) but not by a p38MAPK inhibitor (SB203580) (Fig. 3b, c). Together, these results suggest that PD-L1 upregulates HK2 expression via the PI3K/Akt and Erk pathways, thereby increasing glycolytic activity in human lung cancer cells.
Fig. 3.

PD-L1 upregulates the expression of HK2 via the PI3K/Akt and Erk signaling pathways. a H460 cells were transfected with scrambled control siRNA (sc), PD-L1-specific siRNA1, or 2. Forty-eight hours after transfection, cells were analyzed by Western blotting using antibodies against P-Akt, Akt, P-Erk, Erk, P-p38MAPK, p38MAPK and β-actin. b H460 cells were treated with LY294002 (a PI3K inhibitor, 10 μM), U0126 (an Erk inhibitor, 10 μM), or SB203580 (a p38MAPK inhibitor, 10 μM) and analyzed by Western blotting for HK2 and β-actin. c H460 cells were treated with LY294002, U0126, or SB203580 and submitted to lactate production assays. Histograms represent the values normalized to control. Data represent the means ± SEMs of at least three independent experiments or are representative of three independent experiments. All p values were calculated using one-way ANOVA. **p < 0.001
PD-L1 expression is positively correlated with HK2 expression in NSCLCs from patients
Next, the correlation between PD-L1 expression and glycolysis in human NSCLCs was investigated by immunohistochemistry and FDG-PET in 393 patients with NSCLC (Additional file 2: Table S1). Consistent with previous reports [20], all PET indices and expression of GLUT1 and HK2 were higher in pSqCC than pADC (all p < 0.001) (Additional file 3: Figure S5). Of note, only HK2 expression was significantly higher in PD-L1positive NSCLC compared to PD-L1negative NSCLC (p < 0.001) (Fig. 4). This positive correlation between the expression of PD-L1 and HK2 was observed in NSCLCs with wild-type EGFR and in those with mutated EGFR (p = 0.001 and p = 0.023, respectively) (Fig. 4b, c), and in both pSqCC and pADC (p < 0.001 and p = 0.009, respectively) (Additional file 3: Figure S6). In TCGA analyses, HK2 mRNA levels also exhibited a significant positive correlation with CD274 mRNA levels in NSCLC (Spearman rho = 0.189, p < 0.001; data not shown).
Fig. 4.

Positive correlation between the expression of PD-L1 and HK2 in human lung cancer tissues. Immunohistochemistry (IHC) analyses for GLUT1, HK2, and PKM2 were performed using tumor tissues from NSCLC patients (N = 393). a The expression (H-score) of these molecules was compared between PD-L1negative and PD-L1positive NSCLCs and statistical differences were analyzed using Mann Whitney U-tests. The expression of these molecules was also compared between PD-L1negative and PD-L1positive cases in patients with b wild-type EGFR (N = 230) and c mutated EGFR (N = 126). Statistically differences were analyzed using Mann Whitney U-tests. The whiskers are drawn from the 10th percentile to the 90th percentile. The midline of the box is the median and “+” denotes the mean. Points below and above the whiskers are individual points. d Representative IHC images are as follows: D-1, a pSqCC case that is PD-L1negative and expresses low levels of GLUT1, HK2, and PKM2; D-2, a pSqCC case that is PD-L1positive and expresses high levels of GLUT1, HK2, and PKM2; D-3, a pADC case that is PD-L1negative and expresses low levels of GLUT1, HK2, and PKM2; D-4, a pADC case that is PD-L1positive and expresses low level of GLUT1 and high levels of HK2 and PKM2. Cases were separated into low and high expression group using the median H-score of each molecule as a cutoff. Cases with PD-L1 score 2 and 3 (moderate to strong membranous staining in ≥10% of tumor cells) was designated as PD-L1positive. (original magnification × 400, bar = 100 μm). Abbreviations: N, negative; P, positive; L, low; H, high
Increased tumoral HK2 expression impairs T-cell effector function
To explore the biological and clinical relevance of the PD-L1-mediated upregulation of HK2 and glycolysis in NSCLC, Jurkat cells were co-cultured with A549 cells. As previously demonstrated, PD-L1 upregulates tumoral glycolysis/HK2 expression and therefore, we tried to find the outcomes of upregulated tumoral HK2 expression in anti-tumor immunity. We transfected HK2 low A549 cell lines with an empty vector and a HK2 vector and then, co-cultured with Jurkat cells using direct and transwell co-culture systems. After 24 h CD3/CD28 activation, we analyzed the IFN-γ secretion and activation marker expression on Jurkat cells for assessing the effector T-cell function (Fig. 5). In both of direct and transwell co-culture systems, IFN-γ secretion and CD69 expression, an activation marker were highest in Jurkat cells without tumor cell co-culture (“Jurkat only”). When co-culture with tumor cells, IFN-γ secretion was lower in Jurkat cells co-cultured with HK2 overexpressing-A549 cells (“HK2 vector”) than in Jurkat cells co-cultured with empty vector transfected-A549 cells (“empty vector”) in both direct and transwell co-culture systems (p < 0.05; Fig. 5a). In addition, CD69 showed similar tendency in both systems (Fig. 5b). These results suggested that tumoral HK2 upregulation, probably subsequent glycolysis upregulation, might impair effector T-cell functions in tumor microenvironment.
Fig. 5.

Upregulated HK2 expression in tumor cells impairs effector T-cell function. Direct co-culture and transwell co-culture system were adopted to investigate effector T-cell function impairment. Jurkat cells were cultured only (“Jurkat only”) or co-cultured with either empty vector transfected A549 cells (“empty vector”) or HK2 vector transfected A549 cells (“HK2 vector”). IFN-γ secretion was measured using ELISA (a) and CD69 expression using flow cytometry (b) in the absence or the presence of anti-CD3/CD28 stimulation. Data represent the means ± SEMs of at least three independent experiments or are representative of three independent experiments. Statistical significance was calculated using Two-way ANOVA with Tukey multiple comparisons test. *p < 0.05; **p < 0.001
HK2 expression and glycolytic signature are inversely related to the expression of T-cell effector molecules in PD-L1high human NSCLCs
To validate above findings, we analyzed the relationships between the expression of HK2 and T-cell effector response-related genes using TCGA data. HK2 expression was inversely correlated with immune-related genes (CD4, Pearson r = − 0.304, p < 0.001; CD8A, r = − 0.170, p < 0.001; GZMA, r = − 0.116, p < 0.001; PRF1, r = − 0.130, p < 0.001) (data not shown). Next, we compared the expression of immune-related genes according to the combined expression status of CD274 (PD-L1) and HK2. Notably, the expression of all immune-related genes exhibited a significant inverse correlation with HK2 in the PD-L1high group rather than the PD-L1low group (Fig. 6). A trend toward an inverse correlation between the expression of SLC2A1 (GLUT1) or PKM and immune-related genes was also observed in PD-L1high NSCLCs, but less consistently than that observed with HK2 (Additional file 3: Figure S7).
Fig. 6.

HK2 expression is inversely correlated with the expression of T-cell effector molecules in human lung cancers, particularly in those with high PD-L1 expression. a The expression levels of T-cell effector molecules, including CD4, CD8A, GZMA, GZMB, IFNG, CXCL9, CXCL10 and PRF1, were assessed according to CD274 (PD-L1) and HK2 expression status in NSCLCs from TCGA dataset (N = 1015). Cases were dichotomized into PD-L1low and PD-L1high groups based on median values and then trichotomized into HK2low (<25th percentile), HK2intermediate (25-75th percentile), and HK2high (>75th percentile) groups. Statistical differences were analyzed using Kruskal-Wallis tests. b and c The expression levels of T-cell effector molecules were compared according to HK2 expression status in NSCLCs with low CD274 (PD-L1) mRNA levels (b) and with high CD274 (PD-L1) mRNA levels (c) in NSCLCs from TCGA dataset. Statistical significance was calculated using Pearson’s correlation analyses. *p < 0.05; **p < 0.001. Abbreviations: L, low; I, intermediate; H, high
In our patient cohort, high numbers of CD8+ TILs were observed in patients with PD-L1high NSCLC (p < 0.05, data not shown), whereas the number of CD8+ TILs tended to be inversely correlated with HK2 expression in PD-L1positive NSCLCs. Furthermore, the number of CD8+ TILs was highest in PD-L1positive/HK2low pSqCCs (Fig. 7a), and HK2 expression was significantly higher in PD-L1positive/CD8+ TILlow pSqCCs compared to PD-L1positive/CD8+ TILhigh pSqCCs (Fig. 7b). In TCGA data, CD8A expression was also higher in CD274high/glycolytic-signaturelow NSCLCs compared to CD274high/glycolytic-signaturehigh NSCLCs (Fig. 7c, d). The glycolytic signature and HK2 expression were highest in NSCLCs of so-called tumor microenvironment immune type (TMIT) III (Fig. 7c, d and Additional file 3: Figure S8), which express high levels of PD-L1 but low levels of CD8, thus being considered poor responders to anti-PD-1/PD-L1 immunotherapy [21]. Together, these data indicate that HK2 expression was inversely correlated to the expression of T-cell effector molecules and the number of CD8+ TILs in PD-L1positive NSCLCs. This suggests that increased HK2 expression might negatively affect the T-cell effector function in PD-L1positive NSCLC.
Fig. 7.
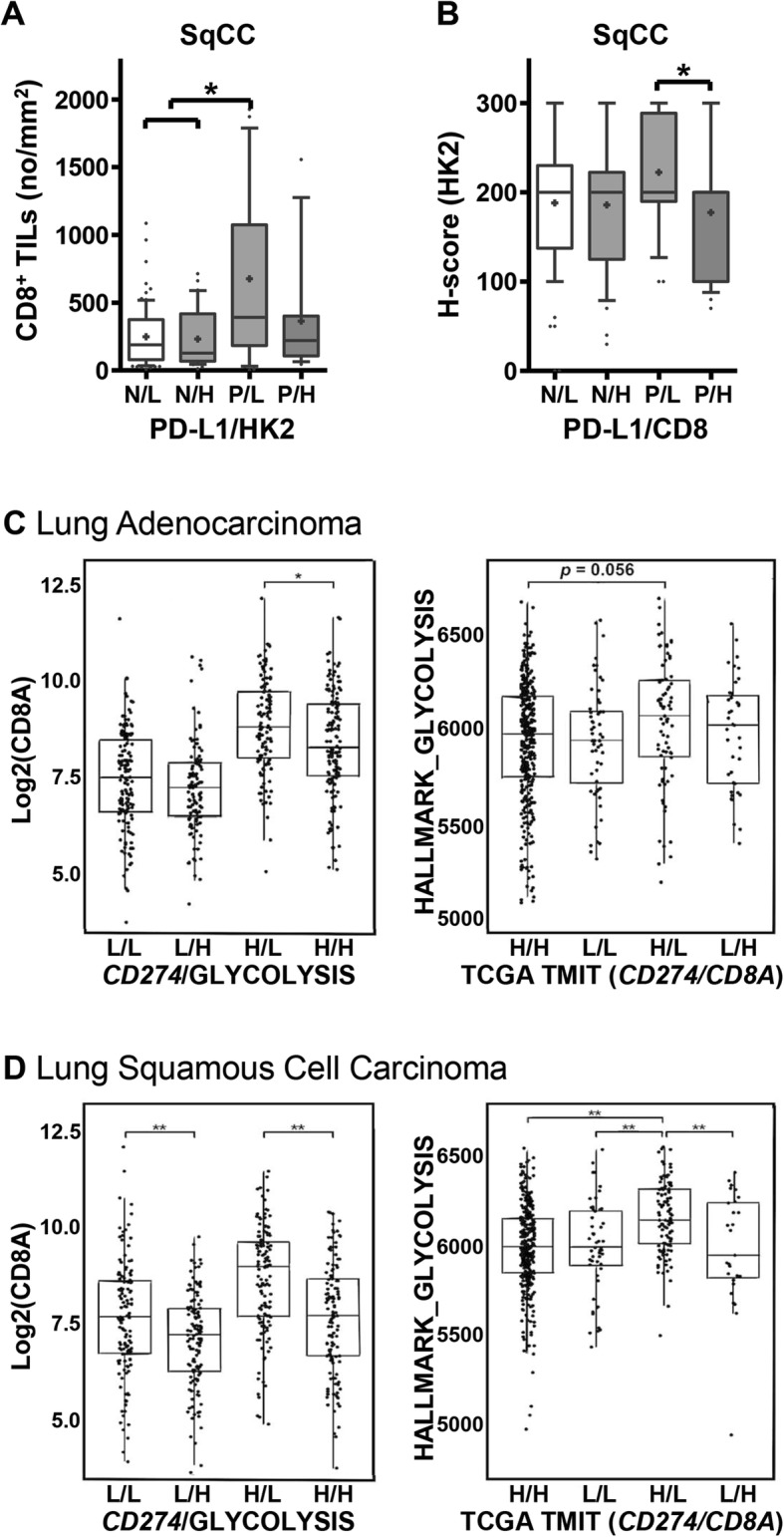
High HK2 expression is inversely correlated with the number of CD8+ TILs in patients samples and high glycolysis signature is inversely related with CD8A expression in TCGA dataset. a The numbers of CD8+ TILs were evaluated according to PD-L1 and HK2 expression status using tumor tissues from patients with pSqCC. Statistical significance was calculated using Kruskal-Wallis tests. b HK2 expression was evaluated according to PD-L1 expression and the numbers of CD8+ TILs using tumor tissues from patients with pSqCC. Statistical significance was calculated using Kruskal-Wallis tests. c & d CD8A transcript levels were compared according to CD274 (PD-L1)/glycolysis signature and the glycolysis signature was compared according to the tumor microenvironment immune type (TMIT) based on CD274 (PD-L1)/CD8A expression status c in pADC and d in pSqCC from TCGA dataset. Statistical differences were analyzed using Kruskal-Wallis tests. *p < 0.05; **p < 0.001. Abbreviations: N, negative; P, positive; L, low; H, high
High HK2 expression in NSCLC tended to be associated with poor response to anti-PD-1 immunotherapy in patients
Finally, to evaluate the effects of HK2 overexpression on the response to PD-1/PD-L1-targeted immunotherapy, we analyzed 80 NSCLC patients who were treated with nivolumab (N = 63) or pembrolizumab (N = 17). The characteristics of the patients are detailed in Additional file 2: Table S2. The median duration of follow-up was 5.2 months (1.0–49.9 months) and the overall response rate (ORR) was 16.3% (13/80). Tumors that exhibited the lowest and highest quartile of HK2 expression were designated HK2low and HK2high cases, respectively. The ORR was higher in the PD-L1positive group (26.5% [9/34]) than the PD-L1negative group (8.7% [4/46]). In addition, the ORR was higher in the PD-L1positive/HK2low group than the PD-L1positive/HK2high group, although statistically insignificant (Additional file 3: Figure S9). There was no significant difference in patient survival according to PD-L1 and HK2 status (data not shown). These findings suggest that patients with HK2high NSCLC might show poor response to anti-PD-1 immunotherapy despite PD-L1 expression. However, further validation is required.
Discussion
Several studies have found that HIF-1α enhances glycolysis by upregulating glycolytic enzymes [22, 23], and upregulates PD-L1 [24], which suggests a potential link between PD-L1 and glycolysis in cancer. The current study demonstrates that PD-L1 upregulates aerobic glycolysis in NSCLC cells by enhancing HK2 expression. This is the first study to demonstrate the intrinsic effects of PD-L1 on HK2-mediated glycolysis in human lung cancer cells. Moreover, we found a significant positive correlation between PD-L1 and HK2 expression in human NSCLC tissues. Although oncogenic mutations affect tumor cell metabolism [10, 25], the relationship of PD-L1 and HK2 expression was not affected by EGFR mutation status in NSCLCs. However, it remains unclear whether mutations in other genes affect link between PD-L1 and HK2 in NSCLCs. Meanwhile, TCGA analyses showed a positive correlation between PD-L1 and HK2 expression in hepatocellular carcinoma (rho = 0.461, p < 0.001), sarcoma (rho = 0.188, p < 0.001), and stomach adenocarcinoma (rho = 0.225, p < 0.001), but not in renal cell carcinoma and urothelial carcinoma (data not shown). These findings suggest that a functional link between PD-L1 and HK2 might occur in various cancers in a histologic type-dependent manner.
In this study, we confirmed that the changes in surface PD-L1 as well as total PD-L1 expression after PD-L1 overexpression and knockdown (Figs. 1 and 2 and Additional file 3: Figure S2A). Therefore, there is a potential that surface PD-L1 expression might give an intrinsic signal to modulate metabolism of tumor cells. By treating murine tumor cells with anti-PD-L1 blocking antibodies, Chang et al. previously showed that surface PD-L1 might be involved in tumor cell glycolysis [15]. In this study, we demonstrated that upregulation of total PD-L1 expression raised glycolysis/HK2 expression of lung cancer cells in the absence of PD-1 ligation. However, we did not compare the actual effect of surface and intracellular PD-L1 expressions on HK2 expression and glycolysis of human lung cancer, and thus it is unclear if both surface and cytoplasm PD-L1 or only surface PD-L1 might be involved in the PD-L1-mediated increase in HK2 overexpression and glycolysis in tumor cells. This is an intriguing issue and remains to be determined in further studies incorporating biochemical assays.
Of note, we also found that upregulated tumoral HK2 expression impaired an effector T-cell function (i.e. IFN-γ production), in a co-culture system. These findings were further validated using TCGA data and IHC for lung cancer tissues. HK2 expression was inversely correlated with the expression of T-cell effector response-related genes and the number of CD8+ TILs in PD-L1high NSCLCs but not PD-L1low NSCLCs. These findings suggest that the expression status of PD-L1 and HK2 in tumor cells might be involved in the regulation of immune response in the TME of NSCLCs. However, since the correlations between variables were often weak to moderate despite statistical significance in analysis of TCGA and patients tissue data, the cause-effect relationship between variables needs to be interpreted with caution. Despite of this limitation, our results regarding the relationship between PD-L1, HK2, and T-cell effector function suggests that combination analyses of HK2 and PD-L1 expression in tumor cells might provide useful information to predict the TME immune signature in NSCLC.
When classifying TME immune types (TMIT) based on PD-L1 expression and CD8+ T-cell, tumors can be divided into four groups: PD-L1high/CD8+ T-cellhigh (TMIT I), PD-L1low/CD8+ T-celllow (TMIT II), PD-L1high/CD8+ T-celllow (TMIT III), and PD-L1low/CD8+ T-cellhigh (TMIT IV). Patients with TMIT I tumors are considered the best candidates for PD-1/PD-L1-targeted therapy [21, 26]. By contrast, those with TMIT III tumors exhibit a poor response to PD-1/PD-L1 blockade despite high PD-L1 expression [21]. The current study demonstrates that PD-L1high/CD8+ T-celllow TMIT III NSCLCs could be characterized by high HK2 expression and glycolytic signature.
Regarding functional aspects of the immune responses, our results suggest that PD-L1-mediated HK2/glycolysis upregulation of tumor cells might dampen T-cell function in the TME of NSCLCs. In previous studies, tumor-derived lactate, a metabolite of glycolysis, inhibits T-cell function [27], and upregulates PD-L1 expression via the lactate receptor-TAZ pathway in lung cancer, thereby contributing to T-cell suppression [28]. Taken together, these and the current findings suggest a potential positive feedback loop of the glycolysis/lactate-PD-L1-HK2-glycolysis axis in the TME. Thus, it is feasible that tumor cells might survive and outcompete surrounding TILs in the TME by expressing PD-L1 through metabolic reprogramming for survival/proliferation, dampening TIL functions by rendering a metabolically harmful microenvironment, and giving a direct inhibitory signal to PD-1 on adjacent immune cells, as schematically summarized in Additional file 3: Figure S10. Thus, the metabolic status of tumor cells could affect the responsiveness of patients to anti-PD-1/PD-L1 immunotherapy. To address this issue, we evaluated the effects of HK2 overexpression in tumor cells on the clinical response to PD-1 blockades. We observed a trend toward a better response in patients with PD-L1positive/HK2low NSCLC compared to those with PD-L1positive/HK2high NSCLCs, but no difference in the survival time between these groups. These results might be attributable to small number of patients, variable performance status and previous treatment history, laboratory-developed PD-L1 test, limitations of retrospective evaluation, and a possible occurrence of resistance mechanisms other than HK2 overexpression. Therefore, the clinical value of HK2 as a potential metabolic biomarker for anti-PD-1/PD-L1 therapy needs further validation.
Moreover, HK2 itself could be a therapeutic target in cancer therapy. HK2 participates in aerobic glycolysis and can affect many other metabolic processes through its product, glucose-6-phosphate. Besides metabolic process, HK2 has been demonstrated to prohibit apoptosis. HK2 binds to a voltage-dependent anion channel (VDAC) and the HK2/VDAC interaction stabilizes mitochondrial membrane and prevents binding pro-apoptotic factors to VDAC [29–31]. Therefore, HK2 inhibitors or in vivo deletion by CRISPR/Cas9 [32] could affect tumor cells by inhibiting tumor metabolism and promoting apoptosis. Applying the concept of combinational therapy for cancer [33, 34], HK2 inhibition could enhance the response rate of PD-1/PD-L1 therapy by releasing immune cells from metabolic competition and directly increasing tumor cell apoptosis. This hypothesis needs to be validated by future studies.
Conclusions
In summary, this study demonstrates that PD-L1 increases glycolysis in NSCLC cells by upregulating HK2, which is associated with reduced expression of T-cell effector genes. These findings suggest that PD-L1 expression might enhance tumor cell survival by increasing glycolysis intrinsically and providing inhibitory signals to neighboring T-cells extrinsically, through interaction with PD-1 and metabolic competition. Thus, HK2 expression and glycolysis could be potential biomarkers or therapeutic targets in NSCLC patients in the era of cancer immunotherapy.
Supplementary information
Supplementary material and methods. More detailed information about Patients and samples, FDG-PET/CT image analysis, Glycolysis assays, Oxygen consumption rate (OCR) assay, RNAseq and Gene set enrichment analyses, TCGA analyses, Immunohistochemistry (IHC) and Statistical analysis was addressed. (DOCX 45 kb)
Table S1. Clinicopathological parameters of patients with NSCLCs. Table S2 Clinicopathological features of NSCLC patients with PD-1 blockade. (DOCX 21 kb)
Figure S1. The expressions of HK2 and glycolytic activity are elevated in PD-L1high lung cancer cell lines. Figure S2. PD-L1 overexpression or knockdown does not affect oxidative phosphorylation. Figure S3. PD-L1 overexpression or knockdown does not affect the mRNA level of glycolysis-related genes, other than HK2. Figure S4. PD-L1 expression is positively correlated with glycolysis signature in NSCLC cells. Figure S5. Glycolysis-related parameters analyzed by PET scanning and immunohistochemistry in NSCLC patients. Figure S6. Basal expression of glycolysis-related molecules according to PD-L1 expression in pSqCC and pADC patients. Figure S7. Expression levels of T-effector immune response-related genes according to CD274 (PD-L1) and SLC2A1 (GLUT1) or PKM expression status in NSCLC from TCGA data. Figure S8. HK2 mRNA is higher in TIMT III (CD274high/HK2low) than in TIMT I (CD274high/HK2high) NSCLC from TCGA data. Figure S9. High HK2 expression is related to a lower response rate to PD-1 blockade in patients with NSCLC. Figure S10. A model figure of this study. (PDF 1278 kb)
Acknowledgements
We’d like to thank Hyun Kyung Ahn for her technical assistance.
Abbreviations
- ECAR
Extracellular acidification rate
- IFN
Interferon
- IHC
Immunohistochemistry
- NSCLC
Non-small cell lung cancer
- OCR
Oxygen consumption rate
- pADC
Pulmonary adenocarcinoma
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death-ligand 1
- pSqCC
Pulmonary squamous cell carcinoma
- sc
Scramble
- siRNA
Small interfering RNA
- TCGA
The Cancer Genome Atlas
- TME
Tumor microenvironment
- TMIT
Tumor microenvironment immune type
Authors’ contributions
Designed the study and experiments: DHC, YKJ. Performed the experiments and analyzed the data: SK, JYJ, JK, DK, YAK, JCP. Performed statistical analysis: SK, CYO. Performed acquisition of clinical data: CYO, BK, MK, TMK, DSH. Wrote the paper: SK, JYJ, DHC, YKJ. All authors read and approved the final manuscript.
Funding
This work was supported by the Seoul National University Hospital Research Fund (grant No. 800–20160023), the Seoul National University (SNU) Invitation Program for Distinguished Scholar (2017. 3. to YKJ) (grant No. 0320170160 [2017–1312]), and the Basic Science Research Program (grant No.: NRF-2016R1D1A1B01015964) through the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was performed in accordance with the World Medical Association Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of SNUH (H-1404-100-572). Informed consent for participation was waivered by the IRB of SNUH on the basis that this study was a retrospective study using archived material, and did not increase risk to the patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sehui Kim and Ji-Young Jang contributed equally to this work.
Contributor Information
Sehui Kim, Email: sulparangi@naver.com.
Ji-Young Jang, Email: jiyoung.jang@bioinfra.co.kr.
Jaemoon Koh, Email: tarda@naver.com.
Dohee Kwon, Email: kwondh11@gmail.com.
Young A. Kim, Email: pathgirl@daum.net
Jin Chul Paeng, Email: paengjc@snu.ac.kr.
Chan-Young Ock, Email: ock.chanyoung@gmail.com.
Bhumsuk Keam, Email: bhumsuk@snu.ac.kr.
Miso Kim, Email: misokim85@gmail.com.
Tae Min Kim, Email: gabriel9@snu.ac.kr.
Dae Seog Heo, Email: heo1013@snu.ac.kr.
Doo Hyun Chung, Email: doohyun@snu.ac.kr.
Yoon Kyung Jeon, Phone: +82-2-740-8323, Email: ykjeon@snu.ac.kr, Email: junarplus@chol.com.
Electronic supplementary material
The online version of this article (10.1186/s13046-019-1407-5) contains supplementary material, which is available to authorized users.
References
- 1.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann Oncol. 2016;27(8):1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 3.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 4.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32–38. doi: 10.1016/j.coph.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark CA, Gupta HB, Sareddy G, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016;76(23):6964–6974. doi: 10.1158/0008-5472.CAN-16-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gato-Canas M, Zuazo M, Arasanz H, et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. 2017;20(8):1818–1829. doi: 10.1016/j.celrep.2017.07.075. [DOI] [PubMed] [Google Scholar]
- 8.Gupta HB, Clark CA, Yuan B, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther. 2016;1:16030. doi: 10.1038/sigtrans.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herbel C, Patsoukis N, Bardhan K, et al. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. 2016;5(1):29. doi: 10.1186/s40169-016-0110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearce EL, Poffenberger MC, Chang CH, et al. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CH, Qiu J, O'Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Koh J, Go H, Keam B, et al. Clinicopathologic analysis of programmed cell death-1 and programmed cell death-ligand 1 and 2 expressions in pulmonary adenocarcinoma: comparison with histology and driver oncogenic alteration status. Mod Pathol. 2015;28(9):1154–1166. doi: 10.1038/modpathol.2015.63. [DOI] [PubMed] [Google Scholar]
- 18.Dong G, Mao Q, Xia W, et al. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett. 2016;11(3):1980–1986. doi: 10.3892/ol.2016.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patra KC, Wang Q, Bhaskar PT, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24(2):213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodwin J, Neugent ML, Lee SY, et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat Commun. 2017;8:15503. doi: 10.1038/ncomms15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teng MW, Ngiow SF, Ribas A, et al. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139–2145. doi: 10.1158/0008-5472.CAN-15-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noman MZ, Desantis G, Janji B, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makinoshima H, Takita M, Matsumoto S, et al. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J Biol Chem. 2014;289(30):20813–20823. doi: 10.1074/jbc.M114.575464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ock CY, Keam B, Kim S, et al. Pan-cancer immunogenomic perspective on the tumor microenvironment based on PD-L1 and CD8 T-cell infiltration. Clin Cancer Res. 2016;22(9):2261–2270. doi: 10.1158/1078-0432.CCR-15-2834. [DOI] [PubMed] [Google Scholar]
- 27.Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 28.Feng J, Yang H, Zhang Y, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36(42):5829–5839. doi: 10.1038/onc.2017.188. [DOI] [PubMed] [Google Scholar]
- 29.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277(9):7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 30.Gall JM, Wong V, Pimental DR, et al. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 2011;79(11):1207–1216. doi: 10.1038/ki.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shulga N, Wilson-Smith R, Pastorino JG. Hexokinase II detachment from the mitochondria potentiates cisplatin induced cytotoxicity through a caspase-2 dependent mechanism. Cell Cycle (Georgetown, Tex) 2009;8(20):3355–3364. doi: 10.4161/cc.8.20.9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh D, Venkataramani P, Nandi S, et al. CRISPR–Cas9 a boon or bane: the bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019;19(1):12. doi: 10.1186/s12935-019-0726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharjee Sonali, Nandi Saikat. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers. 2018;10(9):298. doi: 10.3390/cancers10090298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh D, Nandi S, Bhattacharjee S. Combination therapy to checkmate glioblastoma: clinical challenges and advances. Clin Transl Med. 2018;7(1):33. doi: 10.1186/s40169-018-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material and methods. More detailed information about Patients and samples, FDG-PET/CT image analysis, Glycolysis assays, Oxygen consumption rate (OCR) assay, RNAseq and Gene set enrichment analyses, TCGA analyses, Immunohistochemistry (IHC) and Statistical analysis was addressed. (DOCX 45 kb)
Table S1. Clinicopathological parameters of patients with NSCLCs. Table S2 Clinicopathological features of NSCLC patients with PD-1 blockade. (DOCX 21 kb)
Figure S1. The expressions of HK2 and glycolytic activity are elevated in PD-L1high lung cancer cell lines. Figure S2. PD-L1 overexpression or knockdown does not affect oxidative phosphorylation. Figure S3. PD-L1 overexpression or knockdown does not affect the mRNA level of glycolysis-related genes, other than HK2. Figure S4. PD-L1 expression is positively correlated with glycolysis signature in NSCLC cells. Figure S5. Glycolysis-related parameters analyzed by PET scanning and immunohistochemistry in NSCLC patients. Figure S6. Basal expression of glycolysis-related molecules according to PD-L1 expression in pSqCC and pADC patients. Figure S7. Expression levels of T-effector immune response-related genes according to CD274 (PD-L1) and SLC2A1 (GLUT1) or PKM expression status in NSCLC from TCGA data. Figure S8. HK2 mRNA is higher in TIMT III (CD274high/HK2low) than in TIMT I (CD274high/HK2high) NSCLC from TCGA data. Figure S9. High HK2 expression is related to a lower response rate to PD-1 blockade in patients with NSCLC. Figure S10. A model figure of this study. (PDF 1278 kb)
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author on reasonable request.