

Abstract
Induced pluripotent stem cells (iPSCs) have shown promise in investigating donor‐specific phenotypes and pathologies. The iPSC‐derived cardiomyocytes (iPSC‐CMs) could potentially be utilized in personalized cardiotoxicity studies, assessing individual proarrhythmic risk. However, it is unclear how closely iPSC‐CMs derived from healthy subjects can recapitulate a range of responses to drugs. It is well known that QT‐prolonging drugs induce subject‐specific clinical response and that all healthy subjects do not necessarily develop arrhythmias or exhibit similar amounts of QT prolongation. We previously reported this variability in a study of four human ether‐a‐go‐go‐related gene (hERG) potassium channel–blocking drugs in which each subject underwent intensive pharmacokinetic and pharmacodynamic sampling such that subjects had 15 time‐matched plasma drug concentration and electrocardiogram measurements throughout 24 hours after dosing in a phase I clinical research unit. In this study, iPSC‐CMs were generated from those subjects. Their drug‐concentration‐dependent QT prolongation response from the clinic was compared with in vitro drug‐concentration‐dependent action potential duration (APD) prolongation response to the same two hERG‐blocking drugs, dofetilide and moxifloxacin. Comparative results showed no significant correlation between the subject‐specific APD response slopes and clinical QT response slopes to either moxifloxacin (P = 0.75) or dofetilide (P = 0.69). Similarly, no significant correlation was found between baseline QT and baseline APD measurements (P = 0.93). This result advances our current understanding of subject‐specific iPSC‐CMs and facilitates discussion into factors obscuring correlation and considerations for future studies of subject‐specific phenotypes in iPSC‐CMs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Somatic cells from healthy or diseased subjects can be reprogrammed by transfecting a cocktail of transcription factors to induce pluripotency, and these pluripotent cells can be differentiated into various cell types. Induced pluripotent stem cells (iPSCs) differentiated into cardiomyocytes retain subjects’ genetic information and can be used to study personalized responses in the laboratory.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Do subject‐specific iPSC‐cardiomyocytes (CMs) responses to two QT‐prolonging drugs, dofetilide and moxifloxacin, correlate with individual responses from the same subjects in the clinic?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Immaturity and inherent variability of iPSC‐CMs can significantly obscure subject‐specific drug response prediction in the clinic.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Our study emphasizes a need for standardization of quality, protocol optimization, and best practices to recapitulate donor‐specific phenotype in iPSC‐CMs. This will facilitate prediction of individual susceptibility to drugs from the laboratory bench to the clinic.
Direct reprogramming of somatic cells to a state of pluripotency was a landmark development that allowed stem cells to be used in a multitude of fields, including toxicology, tissue engineering, and regenerative medicine, among many other applications.1 Since this discovery, chemically defined differentiation protocols have improved the efficiency and purity of human induced pluripotent stem cell–derived cardiomyocyte (iPSC‐CM) generation, extending their utility in pharmacological and translational applications.2, 3, 4, 5 The iPSC‐CM‐based assays have been studied for the prediction of drug‐induced torsade de pointes ventricular cardiac arrhythmias under the Comprehensive in vitro Proarrhythmia Assay initiative.6, 7, 8, 9, 10 Commercially available cell lines that are being tested within Comprehensive in vitro Proarrhythmia Assay generally originate from a few healthy donors and are being evaluated for a prediction of proarrhythmic potential of drugs in a general population. However, iPSC‐CMs generated from somatic cells retain genomic information of the original donor. This trait establishes them as potentially useful in precision medicine practices.11, 12 Although these potential precision medicine applications have been shown to be effective when evaluating disease states and treatments,11 their capacity to recapitulate variation within a healthy population is less frequently addressed.
Methods
Clinical study design and personalized concentration‐corrected QT response
This study (http://www.clinicaltrials.gov, NCT02308748) was approved by the US Food and Drug Administration Research Involving Human Subjects Committee and the local institutional review board. All subjects gave written informed consent, and the study was performed at a phase I clinic (Spaulding Clinical, West Bend, WI). Healthy subjects between 18 and 35 years of age, weighing between 50 and 85 kg, and without any family history of cardiovascular disease or unexplained sudden cardiac death were eligible for participation in the study. In addition, the subjects had to have <12 ventricular ectopic beats during a 3‐hour continuous recording, as well as a baseline corrected QT (QTc) of <430 ms, using the Fridericia correction.13 Twenty‐two healthy subjects were enrolled into a five‐period, randomized, crossover trial studying drug‐induced multicurrent channel block. Each treatment period was separated by 1 week, and during each period, subjects were dosed three times a day with placebo, a selective human ether‐a‐go‐go‐related gene (hERG) channel blocker (moxifloxacin or dofetilide), a late sodium current blocker (mexiletine or lidocaine), or a calcium channel blocker (diltiazem), administered either alone or in combinations thereof. This current study focuses solely on the selective hERG channel blockers, of which 20 subjects received doses of dofetilide or moxifloxacin administered alone.
Personalized QT response to drug was defined as the slope of an individual subject's placebo‐controlled concentration‐dependent QT prolongation from baseline, corrected for heart rate using the Fridericia correction.14 These slopes were calculated using mixed linear effect modeling by inputting subject‐specific ΔΔQTc (time of day‐matched placebo and predose baseline QTc change) and plasma drug concentrations into PROC MIXED in SAS version 9.3 (SAS Institute, Cary, NC), with the concentration as a fixed effect and the subject as a random effect.
Cell reprogramming and differentiation into cardiomyocytes
For the 20 subjects dosed with dofetilide and moxifloxacin administered alone, cell reprogramming and differentiation into iPSC‐CMs using their peripheral blood mononuclear cells (PBMCs) was done at Stem Cell Theranostics (Redwood City, CA). Commercially available Sendai virus vectors encoding for Oct4, Sox2, Klf4, and c‐Myc (Life Technologies, Invitrogen) were transfected into PBMCs for iPSC reprogramming.15 Recombinant Sendai virus vectors replicate only in the cytoplasm without integrating into the host genome.16 CD71+ cells were obtained from PBMCs, in the presence of FLT‐3, IL‐6, SCF, and TPO, per ThermoFisher Scientific guidelines for PBMC reprogramming using the CytoTune‐iPS Sendai reprogramming kit. All somatic cell samples were tested and cleared for the presence of mycoplasma. Cells were transitioned to the Essential 6 medium containing basic fibroblast growth factor on day 7 and to the Essential 8 media on days 18–25. A minimum of six colonies were picked from each line, and one colony per well was cultured on Matrigel‐coated plates in the presence of the Essential 8 medium. The reprogramming efficiency ranged between 0.05% and 0.1%. Based on cell morphology and growth kinetics, one clone was further expanded from each parental sample. Pluripotency characterization was carried out by immunostaining (SSEA4 and Tra‐1‐60 were extracellular markers, whereas nanog and Oct‐4 were intracellular markers) prior to expansion and cryopreservation of all iPSC lines. The iPSC lines were successfully derived for 17 of 20 subjects. Identical reprogramming and differentiation methods generated iPSC‐CMs for 16 of 17 subjects as well as for 1 donor with congenital long QT syndrome (subject SCVI22, woman, white race, 46 years old, disease: long QT type 1 (LQT1), KCNQ1 G269S); LQTS iPSC‐CMs were obtained from the Board of Trustees of the Leland Stanford Junior University biobank (Palo Alto, CA) through the Uniform Biological Material Transfer Agreement. The iPSC‐CM lines used in experiments were 100% CMs and predominately constituted ventricular‐like cells, with a smaller proportion of atrial‐like and nodal‐like cells.
Cell culture
Six‐well tissue culture plates were prepared for cell plating by pipetting 1 mL of 1:60 Matrigel (Corning, 356231) in RPMI 1640 (Gibco, 22400‐089) solution into each well and incubating 3–4 hours at 37°C and 5% CO2. The iPSC‐CMs previously stored in liquid nitrogen were thawed and plated at 2.5 × 106 cells per well using plating media (Stem Cell Theranostics) followed by incubation at 37°C and 5% CO2. At 24 hours after thawing, the full volume of the plating medium was gently aspirated and replaced with warm maintenance medium.
At 72 hours after thawing, the cells formed a uniform, beating monolayer and were dissociated and replated for voltage‐sensitive dye applications. For each subject, a 96‐well glass bottom plate (MatTek, P‐96G‐1.5‐5‐F) was prepared by overnight incubation with 100 μL per well of 1:200 Fibronectin (Sigma, F1141) in Dulbecco's Phosphate‐Buffered Saline without Ca2+ and Mg2+ (Gibco, 14040‐133). For each six‐well plate of preplated cells, the maintenance medium was aspirated, and the monolayer was washed with ~5 mL warm Dulbecco's Phosphate‐Buffered Saline without Ca2+ and Mg2+. After washing and gentle aspiration, 1 mL of Accutase (Gibco, A11105‐01) was added to each well, and the plate was incubated at 37°C and 5% CO2 for 12 minutes. One milliliter of warm 10 μg/mL DNAse1 (Millipore, 260913‐10MU) in TrypLE Express (Gibco, 12604‐013) was added to each well. Following incubation for 5 minutes, the TrypLE/cell suspension was briefly and gently pipetted up and down to mechanically lift cells. This suspension was then deposited into ~5 mL of cold neutralization buffer (Stem Cell Theranostics, 10% fetal bovine serum in Dulbecco's Modified Eagle's Medium (DMEM)) in a 50‐mL centrifuge tube. The cells were then spun down at 200g for 5 minutes and resuspended using 2 mL of warm plating medium. Cells were counted using an automatic cell counter, and 30 μL of the cell suspension (~45,000 cells/well) was seeded onto the glass portion of a 96‐well plate. The plate was then allowed to rest for 20 minutes at room temperature (20–25°C), after which 70 μL of warm plating medium was slowly added to each well. At 24 hours after replating, the plating medium was aspirated and replaced with warm maintenance medium. The cells were maintained at 37°C and 5% CO2 with medium changes occurring every 48 hours. After replating, iPSC‐CMs were kept in culture for 5–6 days before drug testing. This time in culture was determined via pilot experiments that found optimal recording conditions 5–6 days after dissociation.
Action potential recordings
For action potential recordings, the maintenance medium in each well of the 96‐well plate was replaced with the 6 μM di‐4‐ANEPPS (Life Technologies) prepared in serum‐free medium (FluoroBrite DMEM, Gibco A18967),17 and then replaced again with dye‐free FluoroBrite solution, ensuring that the exposure time to di‐4‐ANEPPS was 1–2 minutes. The cells were allowed to recover from staining for ~2 hours before baseline action potential duration (APD) data were collected. Optical assaying of APD was conducted using CellOPTIQ (Clyde Biosciences, Glasgow, UK) hardware and software. All data collection was done at 37°C and 5% CO2. Each well was evaluated to find an area of uniformly beating monolayer to measure from; these areas were kept constant for each well throughout the experiment. For each drug concentration and baseline, 20‐second recordings of action potential were taken from each well, in four replicates for the three lower concentrations and eight replicates for the two highest concentrations.
Drug dilutions and dosing
Drug concentration selection was informed by clinical maximum concentration (Cmax), limitations on dimethylsulfoxide concentration, and pilot experimentation to determine minimum effective concentrations to observe arrhythmia‐like events. For dofetilide (SelleckChem, S1658), the Cmax was 2.14 nM, determined by a previous clinical trial,18 and concentrations in iPSC‐CM experiments varied from 0.5 to 8 nM. This range was informed via optimization experiments conducted to determine a maximum concentration beyond which all spontaneous beating would cease. This was found to be ~10 nM. For moxifloxacin (SelleckChem, S1465), the Cmax was 3.5 μM, and concentrations varied from 7 to 200 μM.
The morning of each assay, serial drug dilutions in Fluorobrite DMEM were prepared fresh for both drugs. For both drugs, a 96‐well “dosing plate” was prepared. This dosing plate enabled one‐to‐one delivery of each well's drug concentration. To deliver the drug, the experimental plate was kept on the CellOPTIQ system, and a multichannel pipette was used to deliver 20 μL from each well of the dose plate to its corresponding well in the experimental plate immediately after baseline APD recording. The drug‐induced changes in action potential and arrhythmia‐like events were recorded 30 minutes after dosing. This 30‐minute equilibration period was implemented for wo reasons: first, to mirror the protocol used to gather electrocardiogram (ECG) data after dosing in the clinical study, in which subjects were exposed to drugs for an incubation period before ECG recordings were made, and second, to allow the cells to equilibrate after exposure to ambient conditions.
Data analysis
Offline data analysis was done using CellOPTIQ software. The 20‐second recording of beats was first evaluated for arrhythmic character. Arrhythmia‐like events from the recording window of each well, including runs of tachyarrhythmia, ectopic beats, and early after depolarizations, were noted and tabulated. Every complete beat from the 20‐second recording window was averaged together and modeled to determine the total profile of the average APD. Rate correction was conducted using two approaches. In the first, APD90 vs. beating interval at baseline was plotted for each subject‐specific iPSC‐CM line. Each individual's baseline data were then fitted with a linear model (Table S3 and Figure S1 ). The slopes of each model were then applied to a formula (below) to remove dependence on beating rate for all postdrug data.
| (Formula 1) |
B = Subject‐specific slope from the linear model of baseline APD vs. beating interval.
In the second approach, APD90 was corrected for rate using the inverse cube‐root of the beating interval, as described by the Fridericia correction formula (APD90cF, below).
| (Formula 2) |
After rate correction, exclusion criteria were imposed on the well‐by‐well data set to limit intrasubject variability introduced by outliers. Wells with beating rates outside of six SDs above and below the plate‐wide mean or with a rate‐corrected APD90c value outside of six SDs above and below the plate‐wide mean were excluded from analysis. Subsequently, changes from baseline corrected for vehicle control and beat‐rate‐corrected APD90 (ΔΔAPD90c) were calculated for each well. ΔΔAPD90c was used for comparison with the clinical metric ΔΔQTc. Variables were tested for normality using Shapiro‐Wilk tests. All statistical analyses were performed in R 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria) unless noted otherwise.
Personalized iPSC‐CM response
Personalized iPSC‐CM response to drug was defined as the slope of regression fit of drug‐induced ΔΔAPD90c prolongation vs. drug concentration, as determined by mixed linear effects modeling. The dofetilide‐induced APD prolongation became saturated by the highest concentration (8 nM); therefore, only the linear portion of dofetilide concentrations (0.5–4 nM) was used to estimate the slope. A hierarchical modeling approach was used to determine if subject‐specific slopes better explained the variance when assessing personalized iPSC‐CM response to drug as the slope of regression fit. For both moxifloxacin and dofetilide, a regression model with a single slope as level one and model with subject‐specific slopes as level two were used, and Bayesian Information Criterion and Aikake Information Criterion were used to assess model fit to determine whether a regression model with subject‐specific slopes better explains the variance. For both drugs, Bayesian Information Criterion and Aikake Information Criterion were higher when using a single slope as compared with subject‐specific slopes, verifying that subject‐specific slopes better explained the variance. The association between in vitro ΔΔAPD90c and clinical concentration‐dependent QTc prolongation was assessed using Spearman's rank correlation coefficient.
Results
Subject‐specific iPSC‐CM response to dofetilide and moxifloxacin
For spontaneously beating iPSC‐CMs derived from each subject, moxifloxacin and dofetilide both induced concentration‐dependent APD90c prolongation when corrected for beating rate (APD90c, Tables S1 and S2 ). Figure 1 shows representative action potential recording traces from one iPSC‐CM line for all tested concentrations. The apparent morphological discrepancies can be attributed to the drug‐induced changes, augmented by the variability between wells. The average maximum change from baseline and vehicle control for moxifloxacin and dofetilide was 175 ± 129 ms and 319 ± 111 ms, respectively.
Figure 1.
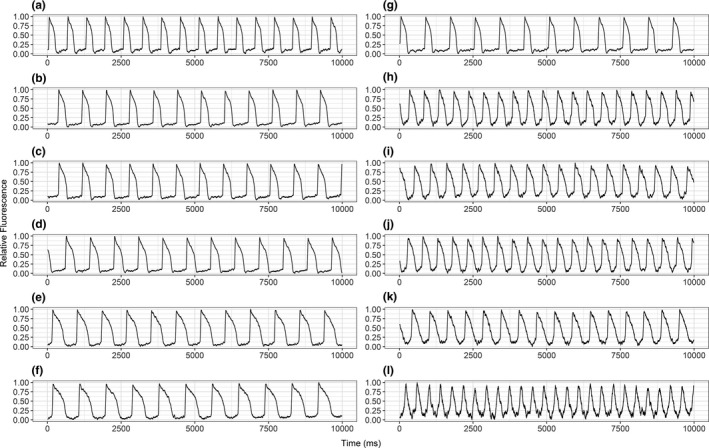
Sample raw action potential duration recordings. Representative 20‐second action potential recordings from a healthy subject's induced pluripotent stem cell cardiomyocytes (a, g) at the baseline and after addition of (b–f) 10, 21, 70, 140, and 200 μM moxifloxacin and (h–l) 0.5, 1, 2, 4, and 8 nM dofetilide. Each subfigure represents the action potential traces from a unique well, collected at a parallel timepoint, ~30 minutes after drug administration.
Baseline‐controlled and vehicle‐controlled rate‐corrected action potential duration measurements (ΔΔAPD90c) for each iPSC‐CM line are shown in Figures 2 and 3 for moxifloxacin and dofetilide, respectively. The range of moxifloxacin‐induced ΔΔAPD90c prolongation, observed at the highest concentration, was 165 ms to 321 ms. The range of dofetilide‐induced ΔΔAPD90c prolongation for the cohort, taken at the highest concentration, was 25 ms to 118 ms.
Figure 2.
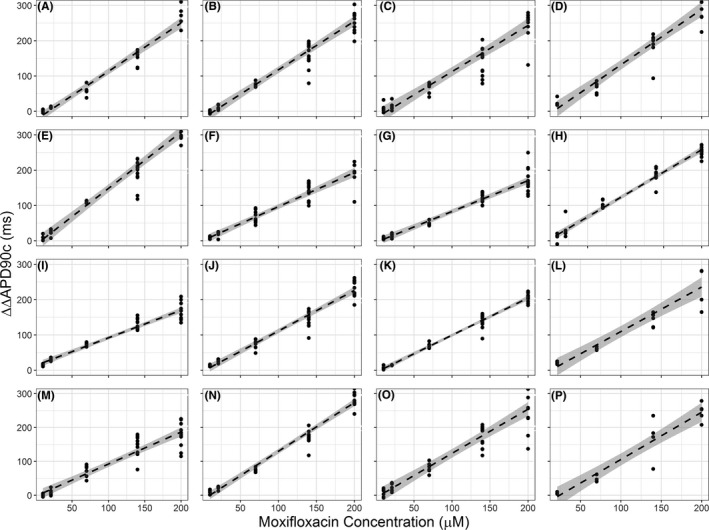
Individual induced pluripotent stem cell cardiomyocytes (iPSC‐CMs) drug response—moxifloxacin. The baseline action potential duration (ΔΔAPD90c) response to the concentration range of moxifloxacin for all lines of subject‐specific iPSC‐CMs (subjects A through P). A linear model, represented by the dashed line, was generated to describe the ΔΔAPD90c response to moxifloxacin across the whole concentration range. The 95% confidence intervals for the model are shaded in gray.
Figure 3.

Individual induced pluripotent stem cell cardiomyocytes (iPSC‐CMs) drug response—dofetilide. The action potential duration (ΔΔAPD90c) response to the concentration range of dofetilide for all lines of subject‐specific iPSC‐CMs. A linear model, represented by the dashed line, was generated to describe the APD response to dofetilide from 0.5 to 4 nM. The 95% confidence intervals for the model are shaded.
Correlative results between clinical and in vitro response
At baseline, the cohort had an average rate‐QTc interval of 397 ± 15.5 ms and an average APD90c of 292 ± 32.2 ms. No significant correlative relationship between APD90c and QTc (ρ = −0.025; P = 0.93; Figure 4) was found. Baseline‐controlled and vehicle‐controlled rate‐corrected QT measurements (ΔΔQTc) were calculated for each subject's response to the drug. Using mixed linear effects modeling, subject‐specific ΔΔQTc concentration‐response slopes were also calculated.
Figure 4.
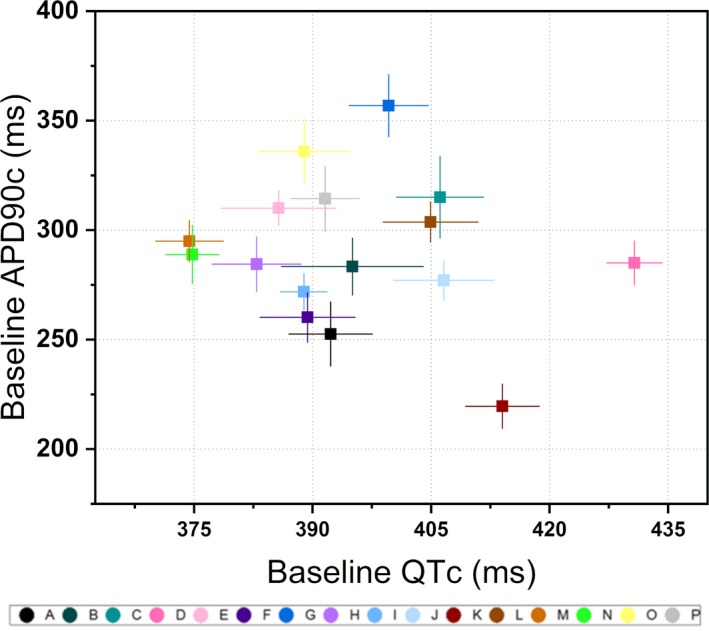
Baseline correlation plot. Average rate‐corrected QT (QTc) interval at baseline, as calculated as the average of 18 measurements collected over 6 days of dosing, plotted against action potential duration (APD90c) at baseline, calculated as the plate‐wide baseline averages for each line of subject‐specific cells. Error bars on both axes represent an SD above and below the mean.
The cohort's subject‐specific iPSC‐CM and clinical responses to moxifloxacin and dofetilide are graphically summarized in Figure 5. For moxifloxacin, no correlation (ρ = −0.089; P = 0.75) was found between ΔΔAPD90c and ΔΔQTc response slopes. For dofetilide, there was also no correlation (ρ = −0.106; P = 0.70). This analysis was also conducted using ΔΔAPD90cF, calculated with Fridericia's rate correction formula Figure 5 c,d. No correlation was identified between ΔΔAPD90cF and ΔΔQTc response slopes for either moxifloxacin or dofetilide (P = 0.47 and P = 0.97, respectively).
Figure 5.

Correlation between induced pluripotent stem cell cardiomyocyte (iPSC‐CM) and clinical drug response. (a) Action potential duration (ΔΔAPD90c) response slope vs. clinical ΔΔQTc response slope for each subject as determined via linear models of both baseline QTc (ΔΔQTc) vs. moxifloxacin concentration and ΔΔAPD90c vs. moxifloxacin concentration. (b) ΔΔAPD90c response slope vs. ΔΔQTc response slope for each subject vs. dofetilide concentration. (c) ΔΔAPD90cF moxifloxacin response slope vs. ΔΔQTc moxifloxacin response slope for each subject. (d) ΔΔAPD90c dofetilide response slope vs. ΔΔQTc dofetilide response slope for each subject. (e) The ΔΔAPD90c moxifloxacin response slopes vs. ΔΔAPD90c dofetilide response slopes, comparing the clinical responses to each drug for each subject. (f) The ΔΔQTc moxifloxacin response slopes vs. ΔΔQTc dofetilide response slopes, comparing the iPSC‐CM responses to each drug for each subject.
As summarized graphically in Figure 5 f, no correlation was observed between dofetilide and moxifloxacin ΔΔQTc responses by subject (ρ = 0.096; P = 0.73). When considering the ΔΔAPD90c response by subject to both moxifloxacin and dofetilide, no correlation was observed (Figure 5 e; ρ = −0.288; P = 0.28).
Arrhythmia‐like events
Of the 16 iPSC‐CM lines derived from the healthy cohort, no drug‐induced arrhythmia‐like events were observed at the studied drug concentration range. For comparison, iPSC‐CMs generated from an LQT1 patient showed arrhythmic beating events at 0.5, 4, and 8 nM dofetilide in 1 of 6 wells, 5 of 11 wells, and 11 of 12 wells, respectively. No arrhythmia‐like events were observed with the administration of 2 nM dofetilide. Likewise, 1 of 6 wells at 21 µM and 70 µM, 3 of 12 wells at 140 µM and all 12 of 12 wells at 200 µM moxifloxacin induced arrhythmia‐like events with no events observed at the lowest (10 µM) moxifloxacin concentration (Figure 6).
Figure 6.
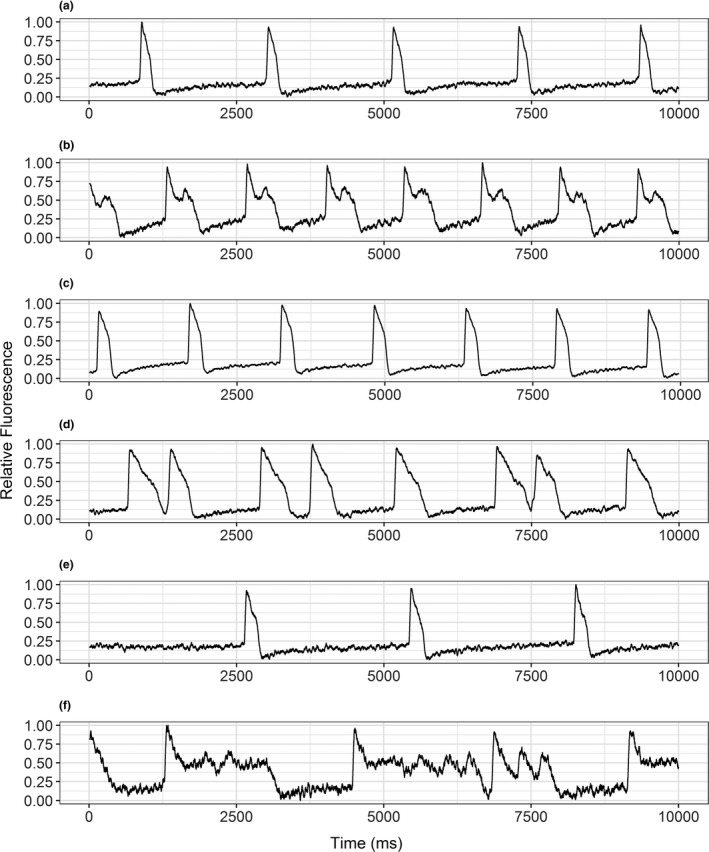
LQTS example traces with arrhythmias. Representative 20‐second action potential recordings of induced pluripotent stem cell cardiomyocytes (iPSC‐CMs) derived from a patient with long QT syndrome. (a, c, e) Show wells beating spontaneously at baseline. (b, d, f) Show those same wells with arrhythmic beating events after dosing with 4 nM dofetilide, 140 μM moxifloxacin, and 200 μM moxifloxacin, respectively.
Discussion
In this study, subject‐specific iPSC‐CM responses to dofetilide and moxifloxacin are compared with individual clinical responses to the same drugs for a cohort of 16 subjects. These two drugs delay cardiac repolarization by blocking the hERG potassium channel. The delay in repolarization can be identified as QT interval prolongation on an ECG, as well as APD prolongation at a cellular level in iPSCs. Both biomarkers are surrogate end points of proarrhythmic risk. Voltage‐sensitive dye imaging was used to measure APD in these subject‐specific iPSC‐CMs. Comparative analysis was then conducted on APD and previously collected QT measurements to assess the correlation between the slope of drug concentration vs. QTc prolongation in the clinic and the slope of drug concentration vs. APD prolongation in the laboratory. This detailed assessment of pharmacokinetics and pharmacodynamics went beyond prior similar studies; however, no correlation was observed between baseline QT measurements and baseline APD measurements or between clinical QTc prolongation and in vitro APD prolongation to dofetilide or moxifloxacin. Our findings are in contrast to recent publications.19, 20 The lack of observed correlation between in vitro and in vivo subject‐specific response to the studied drugs may be related to several factors worthy of further discussion.
Although iPSC‐CMs carry the genetic information specific to their donors,21 the expression levels of several key cardiac ion channels, including hERG (IKr current), sodium, and calcium ion channels in iPSC‐CMs differ greatly from adult CMs.13 Previous studies of iPSC‐CMs found that the expression profiles of cardiac ion channels closely resemble immature CMs, with variable hERG expression levels depending on iPSC‐CM source.22 Furthermore, electrophysiology studies of iPSC‐CMs have identified low density of the inward rectifier potassium current (IK1) and a low resting membrane potential compared with adult CMs.23, 24 These are important cellular limitations worth consideration. Because of these discrepancies, there could be differences in voltage‐gated ion channel function and sensitivity to drug‐induced changes as compared with adult cardiomyocyte.25 Generally speaking, although the genome is maintained from each individual subject, the proteome and the resultant electrophysiological properties unique to iPSC‐CMs may vary in a way that obscures subject‐specific electrophysiological response in vitro. An example of this is that dofetilide and moxifloxacin affect the beating rate of iPSC‐CMs; however, they do not have a substantial effect on heart rate in the clinic.
In addition to the proteomic source of uncertainty, it has been demonstrated that the derivation of iPSC‐CMs may itself be a highly variable process.26 Due to subjective picking of colonies for differentiation, as well as subtle variations in derivation protocols, the resultant cells may possess some amount of inherent variability that can influence the final electrophysiological assays. Thus, if multiple iPSC lines were generated from one subject, the phenotypes exhibited could vary significantly. This inherent variability might have confounded the results presented in this study. Although the hierarchical model analysis results suggest that iPSC‐CMs had subject‐specific responses, it is unclear how these unique responses might have been distorted by the sources of variability described.
Another limitation of iPSCs relates to epigenetic factors, which are not carried from the donor to the resultant iPSC‐CMs. Any epigenetic factors that may influence the QT interval are effectively “reset” in the iPSC derivation and differentiation processes, potentially complicating efforts to correlate in vitro and clinical subject‐specific responses.27 Despite the stringent subject inclusion and exclusion criteria in the clinical trial (see Experimental Procedure), epigenetic factors may have still played a role.
Beyond the characteristics of these iPSC‐CMs, it is also important to weigh how closely two biomarkers of ventricular repolarization, cellular APD and clinical QT, are related to each other. Both QT and APD are variable metrics at baseline, with more variability introduced with exposure to the drug. This variability might have played a role in obscuring subject‐specific correlations. For the cohort, the average coefficient of variance for baseline QT was 1.34 ± 0.39%, indicating that very small variations could be expected in QT day to day, but otherwise, the subject's QT remained consistent. This is in contrast to the iPSC‐CMs’ APD, where the coefficients of variance for each subject's baseline cellular APD was 4.12 ± 0.97%, demonstrating that a higher portion of the APD signal could potentially be attributed to variability, as compared with the corresponding baseline QT signal. Moreover, as with QT, the variability in these cells increases after administering the drug. As such, this analysis suggests that the signal of the cells may be too variable to parse subtle clinical differences. If this is indeed the case, further refinement in the iPSC‐CMs and electrophysiological assays may be necessary to increase accuracy and improve the chance of observing correlation between APD and QT.
One area with a clear relationship between clinical and cellular phenotype is the presence of arrhythmic events in LQTS‐derived cells. Although no statistically significant relationship was clear between APD and QT analysis, the cell lines derived from healthy subjects never exhibited arrhythmia‐like events, whereas the LQTS‐derived cells regularly exhibited drug‐induced arrhythmias. In this sense, by expanding the range of observations to include more extreme phenotypes, a relationship between cell lines and donors could become more apparent.
There have been several studies similar to our observations on congenital LQTS iPSC‐CMs showing a relationship between donor physiology and iPSC‐CM phenotype for pathological cells possessing disease‐specific phenotypes.28, 29, 30 However, these studies can largely fall into one of two categories: those investigating specific toxicities31 and those studying personalized responses in a healthy cohort.19, 20 One study positively identified correlation for a group of 20 subjects, made up of the most extreme responders within a much wider cohort of more than 80 subjects. That study found that the two extremes could be differentiated based on the response of the cells.20 In contrast, our cohort, containing 16 healthy subjects, was likely more representative of a random sampling of responders. It is possible that by studying the extremes of the healthy population, as opposed to random subjects, some of the complications described above can be overcome so that QT can be correlated with iPSC‐CM's APD. Another study found a positive correlation between the slope of QT vs. moxifloxacin response in the clinic and the slope of field potential duration response in the iPSC‐CMs among a cohort of 10 randomly selected healthy subjects.19 This study, however, only identified a positive correlation in a subset of each subject's data. By focusing only on the cellular response around the Cmax of moxifloxacin, some significant positive correlation was identified, but this disappeared when the analysis was expanded. When we assessed the cellular response around the Cmax, we found no significant correlation. Nevertheless, the results of both studies are generally in agreement; when observing drug responses across the full range of concentrations, no statistically significant correlation could be identified.
In future experiments, a larger sample size in the in vitro assay would increase the power to detect small correlations. Related to this is the limitation of the small effect size observed in APD prolongation at clinically relevant concentrations. Although this problem is partially mitigated by expanding experimental concentration ranges well beyond therapeutic concentrations, the small effect size may have contributed to obscuring correlative results and is worth consideration in future experiments. Moreover, a limiting factor in the results presented here was the use of only one run for each cell line. As addressed above, there can be variability in iPSC‐CM experiments depending on the plate. By executing more runs of each cell line, a more accurate description of each line could be developed to capture some of the inherent variability in these experiments and help improve correlative analysis.
Our results underscore a need for iPSC‐CM standards in applications involving personalized response to drugs. This would also present an opportunity to analyze how emerging methods in the maturation of iPSC‐CMs may affect correlative results, an aspect that is worth exploring in future studies. To that end, a better understanding of the cells and more clearly defined parameters will help ensure that differences observed among cell lines accurately represent the underlying donor‐specific phenotype rather than the result of protocol variability.
In conclusion, the correlation analysis of clinical QTc responses from multiple simultaneously collected pharmacokinetic and pharmacodynamic measurements and subject‐specific iPSC‐CM responses to two hERG‐blocking drugs, dofetilide and moxifloxacin, found no relationship between the clinical response observed for each subject and their corresponding cell line's response in vitro. We also did not find a relationship between baseline QTc and baseline APD90c across the cohort. Using subject‐specific iPSC‐CMs in predictive drug response assays is an emerging field, and the results described here are contrary to previously published observations. A potential cause for the differing results may be related to the variability inherent in the iPSC‐CM model system. Variations in the derivation protocols and the culturing of each cell line may obscure the subtle differences that exist between the subjects in the clinic. Standardization of protocols along with a better understanding of the factors that can affect the inherent variability of subject‐specific iPSC‐CMs will be necessary before iPSC‐CMs could serve as a predictive tool for the donor's response in the clinic.
Funding
The study was funded through the US Food and Drug Adminstration Office of Chief Scientist Challenge grant.
Conflict of Interest
The authors declared no competing interests for this work. J.C.W. is a co‐founder of Khloris Biosciences but has no competing interests, as the work presented was performed independently.
Author Contributions
D.S., K.B., D.G.S., D.P., J.V., and C.D. wrote the manuscript. K.B. and D.G.S. designed the research. K.B. and D.S. performed the research. K.B., D.S., C.D., D.G.S., J.C.W., and D.P. analyzed the data. J.C.W. contributed new reagents/analytical tools.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the US Food and Drug Administration's views or policies. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Department of Health and Human Services.
Supporting information
Figure S1. Mean beating intervals referenced in experimental procedure. Mean beat‐to‐beat interval at baseline across all wells for each subject. Error bars represent a standard deviation above and below the mean. Metrics represented here are related to formula 1 and the analysis described in the methods.
Table S1. ΔΔAPD90 data related to Figure 2. The moxifloxacin concentration and corresponding ΔΔAPD90c and ΔΔAPD90cF in ms for each subject's iPSC‐CM line, as referenced in Results and shown graphically in Figure 2.
Table S2. ΔΔAPD90 data related to Figure 3. The dofetilide concentration and corresponding ΔΔAPD90c and ΔΔAPD90cF in ms for each subject's iPSC‐CM line, as referenced in Results and shown graphically in Figure 3 .
Table S3. Metrics used in formula 1. Slopes and intercepts from the subject specific linear fitting of baseline APD and beating interval, described in Experimental Procedures. Slopes were extracted for use in calculating APD90c as shown in Formula 1.
Acknowledgments
The authors thank Marina Kondratovich and Qianyu Dang for helpful discussion on the statistical analysis.
References
- 1. Takahashi, K. & Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 140, 2457–2461 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Mordwinkin, N.M. , Burridge, P.W. & Wu, J.C. A review of human pluripotent stem cell‐derived cardiomyocytes for high‐throughput drug discovery, cardiotoxicity screening, and publication standards. J. Cardiovasc. Transl. Res. 6, 22–30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ebert, A.D. , Liang, P. & Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 60, 408–416 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith, A.S. , Macadangdang, J. , Leung, W. , Laflamme, M.A. & Kim, D.H. Human iPSC‐derived cardiomyocytes and tissue engineering strategies for disease modeling and drug screening. Biotechnol. Adv. 35, 77–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Batalov, I. & Feinberg, A.W. Differentiation of cardiomyocytes from human pluripotent stem cells using monolayer culture. Biomark Insights. 10, 71–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strauss, D.G. et al Comprehensive in vitro proarrhythmia assay (CiPA) update from a cardiac safety research consortium/health and environmental sciences institute/FDA meeting. Ther. Innov. Regul. Sci. 53, 2168479018795117 (2018). [DOI] [PubMed] [Google Scholar]
- 7. Blinova, K. et al International multisite study of human‐induced pluripotent stem cell‐derived cardiomyocytes for drug proarrhythmic potential assessment. Cell Rep. 24, 3582–3592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel, D. , Stohlman, J. , Dang, Q. , Strauss, D.G. & Blinova, K. Assessment of proarrhythmic potential of drugs in optogenetically paced induced pluripotent stem cell‐derived cardiomyocytes. Toxicol. Sci. 170, 167–179 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Millard, D. et al Cross‐site reliability of human induced pluripotent stem cell‐derived cardiomyocyte based safety assays using microelectrode arrays: results from a blinded CiPA pilot study. Toxicol. Sci. 164, 550–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ando, H. et al A new paradigm for drug‐induced torsadogenic risk assessment using human iPS cell‐derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 84, 111–127 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Itzhaki, I. et al Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Matsa, E. et al Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 32, 952–962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johannesen, L. et al Late sodium current block for drug‐induced long QT syndrome: results from a prospective clinical trial. Clin. Pharmacol. Ther. 99, 214–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fridericia, L.S. The duration of systole in an electrocardiogram in normal humans and in patients with heart disease. 1920. Ann. Noninvasive Electrocardiol. 8, 343–351 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ban, H. et al Efficient generation of transgene‐free human induced pluripotent stem cells (iPSCs) by temperature‐sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 108, 14234–14239 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li, H.O. et al A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J. Virol. 74, 6564–6569 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schocken, D. et al Comparative analysis of media effects on human induced pluripotent stem cell‐derived cardiomyocytes in proarrhythmia risk assessment. J. Pharmacol. Toxicol. Methods 90, 39–47 (2018). [DOI] [PubMed] [Google Scholar]
- 18. Johannesen, L. et al Differentiating drug‐induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Pharmacol. Ther. 96, 549–558 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Shinozawa, T. et al Recapitulation of clinical individual susceptibility to drug‐induced QT prolongation in healthy subjects using iPSC‐derived cardiomyocytes. Stem Cell Reports 8, 226–234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stillitano, F. et al Modeling susceptibility to drug‐induced long QT with a panel of subject‐specific induced pluripotent stem cells. Elife 6 pii: e19406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang, G. & Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: an epigenetic perspective. Cell Res. 23, 49–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blinova, K. et al Comprehensive translational assessment of human‐induced pluripotent stem cell derived cardiomyocytes for evaluating drug‐induced arrhythmias. Toxicol. Sci. 155, 234–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma, J. et al High purity human‐induced pluripotent stem cell‐derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 301, H2006–H2017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doss, M.X. et al Maximum diastolic potential of human induced pluripotent stem cell‐derived cardiomyocytes depends critically on I(Kr). PLoS One 7, e40288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gibson, J.K. , Yue, Y. , Bronson, J. , Palmer, C. & Numann, R. Human stem cell‐derived cardiomyocytes detect drug‐mediated changes in action potentials and ion currents. Paper presented at: Journal of pharmacological and toxicological methods. 11th annual focused issue on methods in safety pharmacology; 2014. [DOI] [PubMed]
- 26. Carcamo‐Orive, I. et al Analysis of transcriptional variability in a large human iPSC library reveals genetic and non‐genetic determinants of heterogeneity. Cell Stem Cell 20, e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang, G. & Zhang, Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell 13, 149–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun, N. et al Patient‐specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4, 130ra47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim, C. et al Studying arrhythmogenic right ventricular dysplasia with patient‐specific iPSCs. Nature 494, 105–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carvajal‐Vergara, X. et al Patient‐specific induced pluripotent stem‐cell‐derived models of LEOPARD syndrome. Nature 465, 808–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burridge, P.W. et al Human induced pluripotent stem cell‐derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat. Med. 22, 547–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mean beating intervals referenced in experimental procedure. Mean beat‐to‐beat interval at baseline across all wells for each subject. Error bars represent a standard deviation above and below the mean. Metrics represented here are related to formula 1 and the analysis described in the methods.
Table S1. ΔΔAPD90 data related to Figure 2. The moxifloxacin concentration and corresponding ΔΔAPD90c and ΔΔAPD90cF in ms for each subject's iPSC‐CM line, as referenced in Results and shown graphically in Figure 2.
Table S2. ΔΔAPD90 data related to Figure 3. The dofetilide concentration and corresponding ΔΔAPD90c and ΔΔAPD90cF in ms for each subject's iPSC‐CM line, as referenced in Results and shown graphically in Figure 3 .
Table S3. Metrics used in formula 1. Slopes and intercepts from the subject specific linear fitting of baseline APD and beating interval, described in Experimental Procedures. Slopes were extracted for use in calculating APD90c as shown in Formula 1.