

Abstract
Fms‐like tyrosine kinase 3 (FLT3) internal tandem duplication (ITD) mutations, common in pediatric acute myeloid leukemia (AML), associate with early relapse and poor prognosis. Past studies have suggested additional cooperative mutations are required for leukemogenesis in FLT3‐ITD+ AML. Using RNA sequencing and a next‐generation targeted gene panel, we broadly characterize the co‐occurring genomic alterations in pediatric cytogenetically normal (CN) FLT3‐ITD+ AML to gain a deeper understanding of the clonal patterns and heterogeneity at diagnosis and relapse. We show that chimeric transcripts were present in 21 of 34 (62%) of de novo samples, 2 (6%) of these samples included a rare reoccurring fusion partner BCL11B. At diagnosis, the median number of mutations other than FLT3 per patient was 1 (range 0–3), which involved 8 gene pathways; WT1 and NPM1 mutations were frequently observed (35% and 24%, respectively). Fusion transcripts and high variant allele frequency (VAF) mutants, which included WT1,NPM1,SMARCA2,RAD21, and TYK2, were retained from diagnosis to relapse. We did observe reduction in VAF of simple or single mutation clones, but VAFs were preserved or expanded in more complex clones with multiple mutations. Our data provide the first insight into the genomic complexity of pediatric CN FLT3‐ITD+ AML and could help stratify future targeted treatment strategies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Pediatric acute myeloid leukemia (AML) is a heterogeneous disease with a relatively low number of somatic mutations. Other pediatric cancers are known for the presence of chimeric transcripts. Few studies have looked at disease progression within pediatric AML.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addresses the question about the clonal heterogeneity between diagnosis and relapse in a cohort of cytogenetically normal Fms‐like tyrosine kinase 3 (FLT3)‐internal tandem duplication (ITD)‐positive pediatric AML, as well as chimeric transcripts present in this population with hopes of gaining insight into therapy shortcomings.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ There is a knowledge gap about childhood AML disease progression between diagnosis and relapse. This study specifically enhances our knowledge about the stability of pediatric FLT3‐ITD disease progression from diagnosis to relapse and lack of clonal resolution.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study highlights the therapy shortcomings in treatment of FLT3‐ITD, an aggressive form of AML. It promotes further investigation into novel therapies strategies that may be more active against this aggressive form of AML.
Pediatric acute myeloid leukemia (AML) is a rare heterogeneous disease that accounts for 30% of all childhood leukemia.1, 2 It is distinct from the adult counterpart in both their genomic alterations and therapeutic response.3 However, in both settings, pediatric and adult AML have relatively low somatic mutation burden compared with other tumor types.4, 5 Fms‐like tyrosine kinase 3 (FLT3) internal tandem duplication (ITD) mutations are among the most common mutations in AML occurring in ~10–15% of pediatric and 30% of adult AML.2 Within cytogenetically normal (CN) AML, FLT3‐ITD is present in 18% of the pediatric and ~50% of the adult cases.2 FLT3‐ITD–positive AML is considered to be an aggressive disease with high risk of early relapse and decreased survival for both the pediatric and adult populations.6, 7, 8, 9 FLT3‐ITD in cellular models leads to a proliferation and survival advantage.10 Given the poor prognostic impact and growth advantage of FLT3‐ITD, it makes it an interesting target therapeutically. A number of clinical trials have been published and others are underway investigating the prognostic outcomes of targeting FLT3 in AML.11 However, the introduction of FLT3‐ITD alone into murine models is insufficient to recapitulate the AML phenotype,12, 13 showing additional cooperative events are needed. Previously, Zwaan et al.14 revealed that in newly diagnosed pediatric AML, FLT3‐ITD mutations may co‐occur with MLL‐PTD, NUP98‐NSD1 fusions, and WT1, or NPM1 mutations. In a study of 20 children with varying subtypes of AML, of note including only one sample that was CN and FLT3‐ITD+, it was observed that the dominant variants/clones within the heterogeneous population persist from diagnosis to relapse, whereas subclonal variants, even established drivers, can be lost at relapse.15 Relapse is thought to be driven by the selection or emergence of resistant subclones.16, 17 Despite this progress, studies evaluating the underlying biology and identification of candidate genes contributing to de novo and relapsed pediatric CN FLT3‐ITD–positive AML have been largely under‐reported in the literature.
The objective of the present study was to gain a deeper understanding of the clonal heterogeneity between diagnosis and relapse in larger cohort of pediatric patients with CN FLT3‐ITD–positive AML, with hopes of gaining insight into therapy shortcomings. Given the low somatic mutational burden reported for AML, samples were sequenced by RNASeq18 and a next‐generation sequencing gene panel consisting of 80 genes19 for chimeric transcripts and co‐occurring mutations, respectively.
Methods
Please refer to Supplemental Materials and Methods for more detailed protocols.
Patients and samples
Leukemic blasts from either diagnosis or relapse were collected from 37 individual patients (Figure 1 a) with CN FLT3‐ITD–positive AML that were treated on AML02 (ClinicalTrials.gov Identifier: NCT00136084; N = 18),20 AML08 (NCT00703820; N = 13), RELHEM (NCT00908167; N = 3),21 AML02/RELHEM (N = 1), or AML08/RELHEM (N = 2) clinical trials at St. Jude Children's Research Hospital. The median age was 12.4 years (range 2.7–19.2 years) with 11 adolescents (defined as 15–21 years), 25 children (3–14 years), and 1 toddler/infant (<3 years). At diagnosis, the median FLT3‐ITD allelic ratio (AR) was 0.69 (range 0.04–18.15), median ITD length was 48 base pairs (range 21–183 bp), and median unique FLT3‐ITD sequences was 1.5 (range 1–5). A summary of patient demographics at diagnosis are listed in Table 1, and characteristics of samples and analyses performed are listed in Table S1 .
Figure 1.
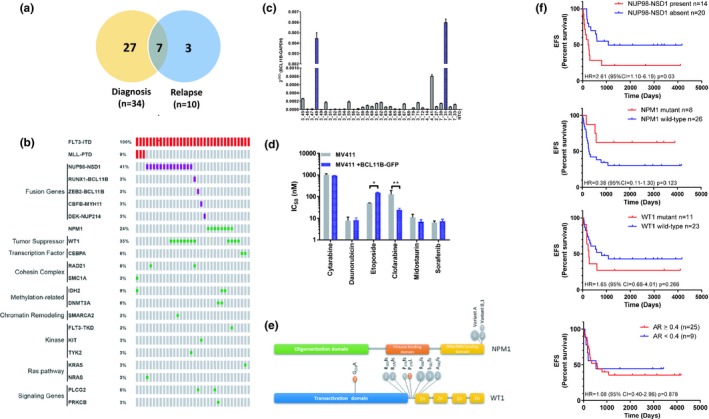
Characterization of de novo Fms‐like tyrosine kinase 3 (FLT3) internal tandem duplication (ITD)‐positive samples. (a) Venn diagram of patients’ samples analyzed in this study. Matched samples are depicted by overlapping circles. (b) OncoPrint of duplications (red), fusion genes (purple), and individual mutations (indels and missense both in green) detected by RNASeq or targeted gene panel. (c) Real‐time polymerase chain reaction quantifying BCL11B transcripts in de novo patient samples. Transcript levels are shown as after standardization to GAPDH. (d) MV411 cells or MV411 cells expressing BCL11B‐GFP were treated for 72 hours with dimethylsulfoxide or increasing concentrations of the indicated drug for 72 hours, and cell viability was measured by 3‐(4 ,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) assay. Half‐maximal inhibitory concentration (IC 50) measurements represent three experiments with six replicates each (n = 18). *P = 0.0096, **P < 0.0001. (e) Lollipop plots showing domain structure, and mutation locations within WT1 and NPM1. Grey circles denote insertions or deletions, and orange circles denote missense mutations. Number of mutations at each location is indicated within the circle. (f) Kaplan–Meier plots show the rates of event‐free survival (EFS) of 34 children and adolescents with de novo FLT3‐ITD–positive acute myeloid leukemia (AML) with or without NUP98‐NSD1,NPM1, or WT1 and allelic ratio (AR) ≥ 0.4.
Table 1.
Summary of patient demographics at diagnosis
| Variables | N |
|---|---|
| Age (yrs) | |
| <10 | 10 |
| ≥10 | 24 |
| Protocol | |
| AML02 | 18 |
| AML08 | 16 |
| Gender | |
| Female | 13 |
| Male | 21 |
| Race | |
| White | 23 |
| Black | 7 |
| Other | 4 |
| Karyotype | |
| CBF | 0/34 |
| M7 | 0/34 |
| 11q23 | 0/34 |
| t(8;21) | 0/34 |
| inv(16) | 0/34 |
| MRD (induction I) | |
| <0.1% | 8 |
| ≥0.1% | 25 |
| NA | 1 |
| (Min, Median, Max) | (0, 0.06, 0.74) |
| WBC (Min, median, Max; 109/L) | (2.5, 116, 412.2) |
| Platelet (Min, median, Max; 109/L) | (10.6, 59, 150) |
| Bone Marrow Blasts (%) (Min, Median, Max) | (2, 82, 97) |
RNA sequencing and targeted gene panel
RNA library construction and analysis for RNA sequencing (RNASeq) has been described previously.18 The RNASeq coverage average was 57X. Eighty protein‐coding genes19 were sequenced for mutation status using targeted amplicon sequencing with the MiSeq platform (Illumina). DNA libraries were prepared and analyzed as previously described by Eisfeld et al.19 The average targeted gene panel coverage was 775X.
Confirmation of fusions and breakpoints
The RNASeq data were run through CREST22 for identification of chimeric transcripts. The cDNA from samples identified to contain a chimeric transcript were analyzed by polymerase chain reaction to confirm the presence of fusions or breakpoints using the primers indicated in Table S2 .
Statistical analysis
The Kaplan–Meier method was used to estimate the distribution of event‐free survival (EFS), defined as the time elapsed from protocol enrollment until relapse, second malignancy, or death with times for subjects having none of these events censored at last follow‐up. Overall survival (OS) was defined as the time elapsed from protocol enrollment to death with times for living subjects censored at last follow‐up. The Cox proportional hazards regression model was used to evaluate the association of EFS with the NUP98‐NSD1 fusion, the WT1 mutation, NPM1 mutation, or FLT3 AR (dichomotomized as <0.4 or ≥0.4). The FLT3‐ITD AR has been previously published as a prognostic indicator, with AR ≥ 0.4 significantly affecting progression‐free survival and relapse.23
Results
Characterization of pediatric de novo CN FLT3‐ITD–positive samples
Chimeric transcripts
Fusion genes represent a distinguishing feature of pediatric cancer, specifically leukemia.3, 18 Thus, we investigated the transcriptome from newly diagnosed CN FLT3‐ITD–positive AML for potential fusion transcripts. Nineteen of 34 (56%) samples contained a previously described chimeric transcript (Figure 1 b, Table S3 ) including: MLL‐PTD (n = 3), NUP98‐NSD1 (n = 14), CBFB‐MYH11 (n = 1), and DEK‐NUP214 (n = 1), which were confirmed by real‐time polymerase chain reaction and Sanger Sequencing. Two samples (6%) contained an in‐frame fusion partner, BCL11B (B‐cell chronic lymphatic leukemia/lymphoma B) with either of the transcriptional genes RUNX1 or ZEB2 (Figure 1 b, Table S3 ). For both fusions, the breakpoint in BCL11B was at amino acid 21, thus allowing the fusions to maintain the six zinc finger domains found within BCL11B but losing the Friend of GATA (FOG) repression domain. The ZEB2‐BCL11B fusion also contain the N‐terminal ZEB2 nucleosome remodeling/deacetylase‐interaction motif. RNA expression verified that BCL11B was expressed >40‐fold when these fusion were present (Figure 1 c). It was observed that when BCL11B was overexpressed in T‐ALL cells, it triggers a chemo‐resistance phenotype to etoposide.24 Thus, we sought to determine if overexpression of BCL11B in FLT3‐ITD–positive cell lines would trigger a similar response. When BCL11B was overexpressed in a pediatric FLT3‐ITD–positive cell line, MV4‐11, there was no increase in growth rate (Figure S1 ). Cells were less sensitive to etoposide (half‐maximal inhibitory concentration (IC50) 149 nM vs. 50 nM, (P = 0.0096) but had an increase in sensitivity to clofarabine (IC50 25 nM to 127 M, P < 0.0001). No altered sensitivity was seen for cytarabine, daunorubicin, or two FLT3 inhibitors midostaurin and sorafenib (Figure 1 d, Figure S1 ).
Co‐occurring mutations
Low somatic mutation burden compared with other tumor types is also a distinguishing feature of AML.4, 5 Thus, to categorize other genomic alterations that co‐occur with FLT3‐ITD, a targeted 80‐gene panel19 was utilized. At diagnosis, 15 genes classified as tumor suppressors, transcription factors, cohesin complex genes, methylation‐related genes, chromatin remodeling genes, kinases, Ras pathway genes, and signaling genes, were observed to contain mutations with a variant allele frequency (VAF) of 0.10 or greater (Figure 1 b, Table S4 ). Per patient, the median number of mutated genes in addition to FLT3‐ITD was 1 (range 0–3). The most common co‐occurring mutations within this pediatric cohort were in WT1 and NPM1, making up 35% (n = 12) and 24% (n = 8) of the mutations observed, respectively (Figure 1 b,e, Table S5 ). The median VAF for WT1 was 0.40 (range 0.14–0.55), whereas it was 0.41 (range 0.29–0.51) for NPM1 (Table S4 ). Two alterations within WT1 were detected in three patients, which were resolved to different alleles. It is predicted that the WT1 deletions and insertions would result in truncated WT1 protein around exon 7, with none keeping the integrity of the reading frame. Additional co‐occurring mutations were seen in RAD21, CEBPα, IDH2, DNMT3A, and PLCG2, with VAF ranging from 0.43−0.55. In one FLT3‐ITD diagnosis sample, no additional co‐occurring mutations were observed using the targeted panel.
Genomic subtypes within FLT3‐ITD–positive AML and clinical outcome
It is known that pediatric FLT3‐ITD–positive AML is associated with worse prognosis,7, 8, 9 especially those that contain NUP98‐NSD1, and WT1.3 However, in these studies, the cytogenetics of the FLT3‐ITD samples were of mixed karyotypes. Thus, we evaluated our CN FLT3‐ITD–positive samples to see if this worse prognosis association held true for EFS and OS. Additionally, we also looked at NPM1, which is known to be associated with a more favorable outcome3 and FLT3 AR (<0.4 compared with ≥0.4). A FLT3 AR ≥ 0.4 has been previously reported to be associated with a worse prognosis in children.23 Of these four features, only the presence of the NUP98‐NSD1 was significantly associated with a poorer EFS (P = 0.03; hazard ratio = 2.61; 95% confidence interval = 1.10–6.19; Figure 1 f, Figure S2 ). NPM1, WT1, and AR ≥ 0.4 showed no significant association with EFS or OS in this cohort.
Characterization of pediatric relapse FLT3‐ITD–positive samples
Chimeric transcripts
In matched diagnosis‐relapse samples, fusion transcripts were maintained (Figure 2 a) during disease progression. NUP98‐NSD1 and DEK214‐NSD1 fusions were also observed in two of the relapse samples without a matched diagnosis sample. A novel relapse‐specific fusion between the 5′ untranslated region (UTR) of Leucine Zipper Protein 6 and Oxysterol Binding Protein Like 1A was observed at relapse that was not detected at diagnosis (Figure 2 a, Table S3 ).
Figure 2.
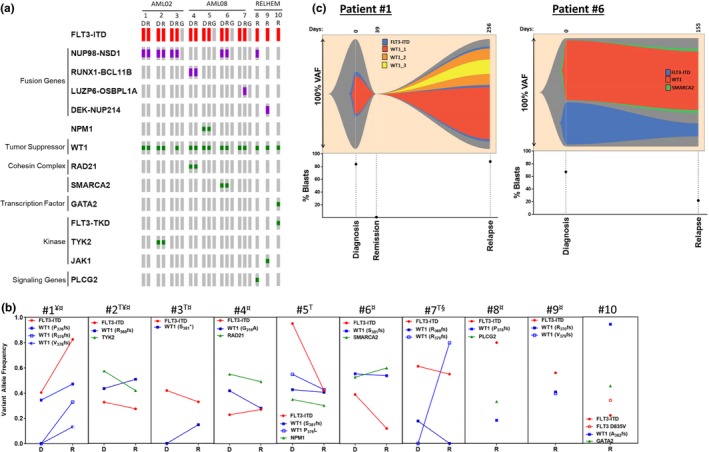
Characterization of relapse Fms‐like tyrosine kinase 3 (FLT3) internal tandem duplication (ITD)‐positive samples. (a) OncoPrint of duplications (red), fusion genes (purple), and individual mutations (indels and missense both in green). Sample identified as: D, diagnosis; R, relapse; and G, germline. (b) Mutation variant allele frequency (VAF) is shown from diagnosis to relapse. T, transplant; ¥, FLT3‐ITD VAF was determined by RNASeq; ¤, contain fusion transcripts at diagnosis and relapse; and §, contains a relapse specific fusion transcript. (c) Fishplots depict two clonal patterns identified between diagnosis and relapse.
Co‐occurring mutations
It was observed that there was no resolution of the ITD from diagnosis to relapse (Figure 2 b). For other mutations, retention was observed regardless of the variant's dominance (Figure 2 a,b) with either maintenance or increased VAF at relapse. No trend in VAF was observed in relation to transplant (Figure 2 b). For patients 3, 5, 6, and 7 with germline DNA samples, no mutations were observed by targeted gene panel analysis (Figure 2 a). A thought‐provoking observation was that all of this cohort's relapse samples contained at least one WT1 mutation. In most cases, the same WT1 variant allele was maintained between diagnosis and relapse (Figure 2 b, Table S4 ). However, in the case of patient 7, the WT1 variant at diagnosis was no longer detected at relapse, but a new variant was acquired at relapse with a greater VAF than the original mutation. Additionally, the emergence of new WT1 variants was observed in two more patients; patients 1 and 3 (Figure 2 b). When more than one WT1 variant was observed in a sample it was also resolved to different alleles. We found no evidence that the expression of WT1 was associated with mutation status, allelic status, or disease progression (Figure S3 ).
Clonal patterns of disease progression
It has been suggested that there are three major clonal patterns to relapse: (i) the regrowth of leukemic stem cells (LSCs) without additional mutations making the mutation profile (not accounting for VAF) at relapse indistinguishable from diagnosis, (ii) the outgrowth of diagnostic LSC clones with newly acquired mutations, or (iii) the outgrowth of preleukemic hematopoietic stem cells containing early acquired mutations and newly acquired mutations not present at diagnosis.25, 26 When analyzing diagnosis to relapse samples, although the mutation burden is low, a characteristic of AML, we observed all three clonal patterns (Figure 2 c, Figure S4 ). Six of the seven samples had outgrowth of the LSC, with only one exhibiting outgrowth of hematopoietic stem cells, suggesting relapse in these patients was mainly due to the outgrowth of LSC. When looking at clonal patterns from diagnosis to relapse, it should be noted that there was a reduction in mutation VAF of simple clones, whereas in more complex clones the VAF seemed to either be preserved or expanded (Figure S4 ).
Discussion
Pediatric FLT3‐ITD–positive AML is an aggressive disease linked with early relapse and worse prognosis. Deciphering whether relapse arises from the acquisition of new mutations or is the outgrowth of diagnostic clones is important for understanding gaps in current treatment regimens. In the present genomic profiling study of CN pediatric FLT3‐ITD AML, clonal stability of mutational patterns was observed from diagnosis to relapse. These observations are consistent with a report by Farrar et al.15 involving children with AML of varying subtypes and cytogenetics, where mutations with a high VAF > 0.4 persisted from diagnosis to relapse. They did note fluidity in some cases between diagnosis and relapse with resolution of diagnostic variants, emergence of relapse variants, and marked changes in VAF from diagnosis to relapse. We did not observe this fluidity in our cohort of CN FLT3‐ITD samples. In the study by Farrar et al.,15 the one CN FLT3‐ITD–positive sample did show similar clonal stability. It could be conceivable that stability in some of the variants present may be due to germline mutations. However, in the majority of cases, germline samples were not available, and this could not be evaluated. When germline samples were available, no diagnosis or relapse variants were detected suggesting pathogenic potential of these mutations and stability from diagnosis to relapse.
We observed the maintenance of WT1 mutations with relapse. Other studies carried out in adult AML populations have shown associations among WT1 mutations, relapse, and disease resistance.27, 28, 29 Taken together, one could propose a potential role for WT1 mutations in disease progression in conjunction with FLT3‐ITD. The exact mechanism would need to be studied further to delineate. The main AML mutational hot spot for WT1 centers around exon 7 for both adult and pediatric cases.3, 28 The location of the mutations in exon 7 are N‐terminal to the zinc finger domains, suggesting that these WT1 mutants would maintain the transactivation domain but be truncated or lose the integrity of the zinc finger domains. Loss of zinc finger domains in the Ikaros family of transcriptional regulators results in a classical dominant negative phenotype inhibiting DNA binding of their dimeric partners.30 Thus, given that the RNA expression levels are not altered one could hypothesize that WT1 mutants may be functioning in a dominant negative way preventing the wild‐type WT1 from accessing the DNA or altering interactions with additional proteins, such as p53 or TET2. However, the penetrance of such a dominant negative phenotype would need to be further explored.
Fusion transcripts are a common feature in pediatric AML and are known to be AML drivers.3, 18, 31, 32 A rare recurring fusion partner, BCL11B, was identified in this study, including a novel fusion RUNX1‐BCL11B. ZEB2‐BCL11B has been identified in a young adult with FLT3‐ITD AML33 with the same breakpoints reported here. Furthermore, a single ZEB2‐BCL11B fusion was also identified in the Children's Oncology Group cohort3 of unknown FLT3‐ITD status. BCL11B, both a repressor and transactivator, has been implicated in the pathogenesis of both adult hematological malignancies and lymphomas.34, 35 A HELIOS‐BCL11B fusion was identified in adult T‐cell leukemia/lymphoma by Fujimoto et al.,34 that has the same breakpoint within BCL11B as reported here. They observed that the HELIOS‐BCL11B fusion caused a decrease in transcriptional suppression, as well as altered subnuclear localization. Fuijimoto et al.,34 hypothesized that this altered localization and loss of transcriptional suppression may lead to aberrant transcription leading to leukemogenesis in adult T‐cell leukemia/lymphoma. Thus, there is potential for BCL11B to have a role in the pathogenesis of pediatric FLT3‐ITD AML.
In summary, our data represent a broad depiction of the genomic landscape of pediatric (CN) FLT3‐ITD–positive AML from diagnosis to relapse. The categorization of co‐occurring genomic lesions, as well as recurring chimeric transcripts, highlights the multifaceted biology behind FLT3‐ITD–positive pediatric AML. Genomic analysis of relapse samples provides insight into the pathogenesis of FLT3‐ITD–positive AML highlighting unseen vulnerabilities in current frontline treatment strategies given the absence of clonal resolution and dominance of WT1 mutations. In the dawn of targeted therapies, comprehensive sequencing, including RNAseq for fusion transcripts should be considered at diagnosis and throughout treatment to recognize problematic and complex clones that may require combination therapies, including chemotherapy and/or targeted therapies to achieve deep and durable remission. Optimistically, having this better understanding of the genomic heterogeneity during progression of pediatric FLT3‐ITD AML will allow for advancement in current and future treatment strategies, as well as guide us in the improved mutation testing.
Funding
The following funding sources supported this study: the American Lebanese Syrian Associated Charities, National Institutes of Health (NIH) Cancer Center Support Grant P30 CA021765, R01 CA138744 (to SDB), and R35 CA197734 (to JCB), the Ohio State University Comprehensive Cancer Center, Pelotonia Foundation, and NIH Cancer Center Support Grant P30 CA016058.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
D.R.B., S.B.P., and S.D.B. wrote the manuscript. D.R.B., S.B.P., J.S.B., J.C.B., Y‐D.W., Y.Z., L.S., and S.J.O. designed the research. D.R.B., S.B.P., Y‐D.W., L.S., Y.L., D.F., S.S., G.N., H.I., R.C.R., R.P., D.G., S.J.O., J.S.B., and K.K. performed the research. D.R.B., S.B.P., Y‐D.W., L.S., Y.L., D.F., S.J.O., J.S.B., K.K., T.A.G., J.E.R., and S.D.B. analyzed the data. S.B.P., L.S., G.N., Y‐D.W., D.F., and Y.L. contributed new reagents/analytical tools.
Supporting information
Supplementary Materials and Methods
Figure S1. Cell viability assessment for MV411 overexpressing BCL11B.
Figure S2. Association of selected molecular features on overall survival.
Figure S3. Expression of WT1 transcripts.
Figure S4. Visualizing clonal patterns from diagnosis to relapse using fishplots.
Table S1. Characteristics of diagnosis samples, paired diagnosis/relapse samples, and relapse samples.
Table S2. Primers utilized in study.
Table S3. Related to Figure 1. Validated chimeric transcripts.
Table S4. NGS‐targeted gene panel validation list.
Table S5. Related to Figure 1. NPM1 mutations.
References
- 1. Gamis, A.S. , Alonzo, T.A. , Perentesis, J.P. , Meshinchi, S. & COG Acute Myeloid Lueukemia Committee . Children's Oncology Group's 2013 blueprint for research: acute myeloid leukemia. Pediatr. Blood Cancer 60, 964–971 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Creutzig, U. et al Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120, 3187–3205 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Bolouri, H. et al The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age‐specific mutational interactions. Nat. Med. 24, 103–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network et al Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Radtke, I. et al Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 106, 12944–12949 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thiede, C. et al Analysis of FLT3‐activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99, 4326–4335 (2002). [DOI] [PubMed] [Google Scholar]
- 7. Meshinchi, S. et al Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 97, 89–94 (2001). [DOI] [PubMed] [Google Scholar]
- 8. Zwaan, C.M. et al FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102, 2387–2394 (2003). [DOI] [PubMed] [Google Scholar]
- 9. Wu, X. et al Prognostic significance of FLT3‐ITD in pediatric acute myeloid leukemia: a meta‐analysis of cohort studies. Mol. Cell. Biochem. 420, 121–128 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Kiyoi, H. et al Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 12, 1333–1337 (1998). [DOI] [PubMed] [Google Scholar]
- 11. Daver, N. , Schlenk, R.F. , Russell, N.H. & Levis, M.J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33, 299–312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelly, L.M. et al FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 99, 310–318 (2002). [DOI] [PubMed] [Google Scholar]
- 13. Li, L. et al Knock‐in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood 111, 3849–3858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zwaan, C.M. et al Collaborative efforts driving progress in pediatric acute myeloid leukemia. J. Clin. Oncol. 33, 2949–2962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farrar, J.E. et al Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Can. Res. 76, 2197–2205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greaves, M. & Maley, C.C. Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turner, N.C. & Reis‐Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 13, e178–e185 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Gruber, T.A. et al An Inv(16)(p13.3q24.3)‐encoded CBFA2T3‐GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 22, 683–697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eisfeld, A.K. et al Adult acute myeloid leukemia with trisomy 11 as the sole abnormality is characterized by the presence of five distinct gene mutations: MLL‐PTD, DNMT3A, U2AF1, FLT3‐ITD and IDH2. Leukemia 30, 2254–2258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubnitz, J.E. et al Minimal residual disease‐directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 11, 543–552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inaba, H. et al Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J. Clin. Oncol. 29, 3293–3300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang, J. et al CREST maps somatic structural variation in cancer genomes with base‐pair resolution. Nat. Methods 8, 652–654 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meshinchi, S. et al Clinical implications of FLT3 mutations in pediatric AML. Blood 108, 3654–3661 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grabarczyk, P. et al Increased expression of bcl11b leads to chemoresistance accompanied by G1 accumulation. PLoS One 5, e12532 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hackl, H. , Astanina, K. & Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 10, 51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ding, L. et al Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature 481, 506–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vosberg, S. et al Relapse of acute myeloid leukemia after allogeneic stem cell transplantation is associated with gain of WT1 alterations and high mutation load. Haematologica 103, e581–e584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Virappane, P. et al Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J. Clin. Oncol. 26, 5429–5435 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Hou, H.A. et al WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood 115, 5222–5231 (2010). [DOI] [PubMed] [Google Scholar]
- 30. John, L.B. & Ward, A.C. The Ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol. Immunol. 48, 1272–1278 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Thanasopoulou, A. , Tzankov, A. & Schwaller, J. Potent co‐operation between the NUP98‐NSD1 fusion and the FLT3‐ITD mutation in acute myeloid leukemia induction. Haematologica 99, 1465–1471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang, G.G. , Cai, L. , Pasillas, M.P. & Kamps, M.P. NUP98‐NSD1 links H3K36 methylation to Hox‐A gene activation and leukaemogenesis. Nat. Cell Biol. 9, 804–812 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Torkildsen, S. et al Novel ZEB2‐BCL11B fusion gene identified by RNA‐sequencing in acute myeloid leukemia with t(2;14)(q22;q32). PLoS One 10, e0132736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fujimoto, R. et al HELIOS‐BCL11B fusion gene involvement in a t(2;14)(q34;q32) in an adult T‐cell leukemia patient. Cancer Genet. 205, 356–364 (2012). [DOI] [PubMed] [Google Scholar]
- 35. Huang, X. , Du, X. & Li, Y. The role of BCL11B in hematological malignancy. Exp. Hematol. Oncol. 1, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods
Figure S1. Cell viability assessment for MV411 overexpressing BCL11B.
Figure S2. Association of selected molecular features on overall survival.
Figure S3. Expression of WT1 transcripts.
Figure S4. Visualizing clonal patterns from diagnosis to relapse using fishplots.
Table S1. Characteristics of diagnosis samples, paired diagnosis/relapse samples, and relapse samples.
Table S2. Primers utilized in study.
Table S3. Related to Figure 1. Validated chimeric transcripts.
Table S4. NGS‐targeted gene panel validation list.
Table S5. Related to Figure 1. NPM1 mutations.