

Abstract
T‐cell–engaging bispecific antibodies (T‐BsAbs) are an important class of antibody therapeutics in immuno‐oncology. T‐BsAbs simultaneously bind to CD3 on T cells and a tumor‐associated antigen on tumor cells, activate T cells, and redirect T cells’ cytotoxicity against tumor cells. Cytokine release syndrome (CRS), a common dose‐limiting adverse event for T‐BsAbs, is associated with T‐cell activation. A “priming” dose strategy (i.e., a lower initial dose followed by a higher maintenance dose) has been implemented in the clinic to mitigate CRS and to achieve efficacious doses with T‐BsAbs. So far, the selection of the optimal priming dosing regimen is largely empirical. A “fit‐for‐purpose” semimechanistic pharmacokinetic/pharmacodynamic model was developed to characterize the cytokine release profiles upon T‐BsAb treatment, including the priming effect observed with repeated dosing. This model can be utilized to simulate cytokine profiles following various dosing regimens and may assist the design of clinical dosing strategies for T‐BsAbs programs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Although many T‐cell–engaging bispecific antibodies (T‐BsAbs) are in clinical development, determining the optimal priming dose regimen for mitigating cytokine release syndrome (CRS) remains a major challenge.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ How can we efficiently determine optimal dosing regimen for T‐BsAbs using quantitative cytokine modeling?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The current study illustrated that a semimechanistic cytokine pharmacokinetic/pharmacodynamic model could be applied to support the determination of optimal dosing regimens for T‐BsAbs to mitigate CRS.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Empirical approaches for determining the priming dose regimen for T‐BsAbs can be resource intensive and inefficient. The semimechanistic cytokine model presented here can integrate the existing knowledge from ongoing clinical trials and make predictions to enable the conduct of more efficient clinical trials.
Immuno‐oncology has shown tremendous potential to treat various cancers by harnessing the power of the human immune system to destroy tumor cells. An important class of therapeutic antibodies, T‐cell–engaging bispecific antibodies (T‐BsAbs), was developed over the past 3 decades to exploit the ability of T cells to exert antitumor immunity.1 T‐BsAbs can simultaneously engage CD3 on T cells and a tumor‐associated antigen (TAA) on cancer cells, which activates T cells and redirects their cytotoxic response to cancer cells. During T cell activation, inflammatory cytokines (e.g., interferon‐γ, tumor necrosis factor, and interleukin (IL)‐6) are secreted, and can result in a sharp increase in the circulating cytokine concentrations.
Cytokine release syndrome (CRS) is a systemic inflammatory response driven by cytokine release.2, 3 Typical CRS symptoms may include fever, fatigue, hypotension/tachycardia, nausea, capillary leak, and organ dysfunction.2 Clinical management of CRS is critical, as severe CRS can lead to life‐threatening complications. Corticosteroids or an IL‐6 receptor blocking monoclonal antibody (tocilizumab), together with vigilant supportive care, can be used to treat and manage CRS.2
Importantly, CRS is one of the most commonly observed toxicities of T‐BsAbs and may limit the ability to achieve sufficient drug exposure for efficacy. For example, during the early clinical studies of blinatumomab in patients with relapsed or refractory non‐Hodgkin's lymphoma (NHL) or chronic lymphocytic leukemia, short 2‐hour or 4‐hour i.v. infusions were initially tested.4 These clinical trials were terminated early due to the presence of adverse events (AEs; e.g., CRS, neurologic AEs, and infections) and the absence of clinical responses.4
To mitigate toxicities, including CRS, and to achieve efficacious doses, an intrapatient “priming” dose strategy has been investigated for T‐BsAbs, where a lower initial dose is followed by higher maintenance doses. It is based on the observation that cytokine levels as well as CRS severity seem to attenuate upon repeated dosing.5, 6 This “priming” effect enables a higher maintenance dose to ultimately be reached. For example, a priming dose of 9 μg/day followed by a maintenance dose of 28 μg/day was established as the dosing regimen for blinatumomab to treat patients with relapsed or refractory B‐cell precursor acute lymphoblastic leukemia (B‐ALL).7 The priming dose strategy has been tested for other T‐BsAbs in clinical trials (e.g., anti‐CD123 × anti‐CD3 T‐BsAb, anti‐EpCAM × anti‐CD3 T‐BsAb).8, 9 The phase I study of the anti‐CD123 × anti‐CD3 T‐BsAb (flotetuzumab) investigated two predetermined priming dose regimens (100 to 500 ng/kg/day or 30 to 100 to 500 ng/kg), in order to limit infusion reaction/CRS events.8 In addition, multiple dosing regimens, including a repeated dose, a one‐step priming dose regimen, or a two‐step priming dose regimen, were tested for MT110 (anti‐EpCAM × anti‐CD3 T‐BsAb) in a phase I study to improve the tolerability.9 Despite the fact that there are >20 T‐BsAbs in clinical development1 and a number of them have adopted the priming dose strategy, the optimal dosing regimen is challenging to determine and is largely an empirical process.
Positive correlations between cytokine levels and CRS severity have been observed in clinical studies with chimeric antigen receptor T‐cell treatment.10, 11, 12, 13 However, due to large intersubject variabilities in patients’ sensitivity to develop CRS, a general threshold for cytokine levels associated with severe CRS has yet to be established. Nevertheless, serum cytokine levels were suggested as biomarkers for CRS due to the underlying pathophysiology characterized by elevated inflammatory cytokines and systemic inflammation.14 In the current report, a quantitative modeling framework was developed for characterizing the cytokine profiles upon T‐BsAb treatment, with the goal to facilitate the design of priming dose strategies to minimize CRS toxicities.
Methods
Model overview
A “fit‐for‐purpose” semimechanistic pharmacokinetic/pharmacodynamic (PK/PD) model was established for characterizing the cytokine release profiles upon T‐BsAb treatment (Figure 1). The model describes the cytokine release as results of formation of trimolecular synapse by binding to both CD3 on T cells and TAA on cancer cells. A time‐variant negative feedback loop was incorporated to account for the observed “priming effect” (i.e., attenuation of cytokine release following repeated doses). We also incorporated mechanistic considerations regarding tumor types, specifically hematological vs. solid tumors, in modeling the cytokine profiles upon T‐BsAb treatment. Mechanistically, the trimolecular synapse is considered as biologically “active” for stimulating cytokine release.15, 16 Thus, changes in synapse concentration as results of tumor cell killing can affect the cytokine release. For hematological malignancies, the depletion of target tumor cells was incorporated to account for its impact on formation of trimolecular synapse (Eqs.9 and 12). In comparison, for solid tumors, depletion of tumor target cells is deemed negligible over the time frame for cytokine release (within days cytokines return to baseline) and is omitted from the model (Eq. 10). The model parameters (Table 1) provide mechanistic information about the cytokine release process.
Figure 1.
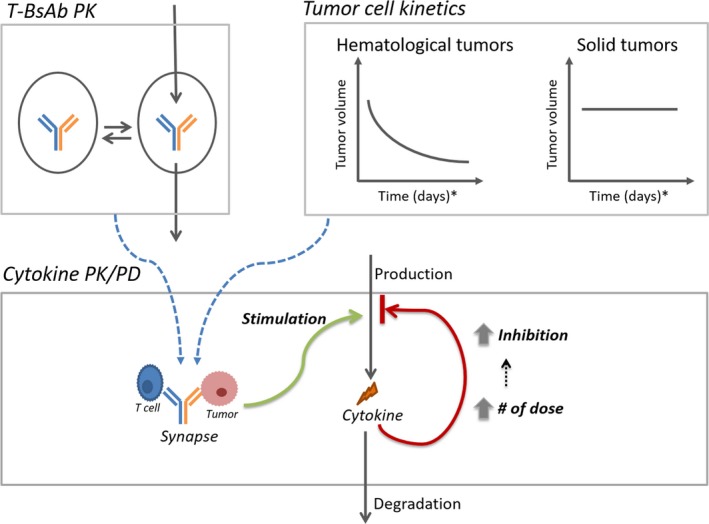
Cytokine pharmacokinetic/pharmacodynamic (PK/PD) model structure. Model details are described in the methods section. Briefly, an appropriate PK model accounts for the drug exposure. Depending on the tumor type (hematological or solid), the tumor kinetics are accounted for in the model to account for the impact of tumor burden on the active synapse concentration. For the cytokine PD model, the synapse exposure then stimulates cytokine release. A time‐variant negative feedback loop accounts for the priming effect, where the negative inhibition increases with the increasing number of doses. T‐BsAb, T‐cell–engaging bispecific antibody.
Table 1.
Parameter estimates for cytokine PK/PD model using blinatumomab human data and P‐cadherin LP DART cynomolgus monkey data
| Parameter | Description | Unit | Estimate (%CV) | |
|---|---|---|---|---|
| Blinatumomab (human) | P‐cadherin LP DART (monkey) | |||
| Emax | Maximum cytokine release rate | pg/mL/hour | 3.59 × 103 (14) | 2.10 × 105 (72) |
| EC50 | Exposure of active drug species to achieve half‐maximum cytokine release rate | (ng/mL) × (cell/μL)a or ng/mLb | 1.00 × 104 (FIXED) | 3.00 × 103 (FIXED) |
| H | Hill coefficient for cytokine release | N/A | 9.20 × 10−1 (3) | 1.08 × 100 (6) |
| Imax | Maximum inhibition of cytokine release | N/A | 1 (FIXED) | 1 (FIXED) |
| IC50 | Cytokine exposure to achieve half‐maximum cytokine inhibition | pg/mL × hour | 1.82 × 102 (12) | 1.76 × 101 (107) |
| k deg | Degradation rate for cytokine | hour−1 | 1.80 × 10−1 (13) | 2.5 × 10−1 (16) |
| K | Priming factor for cytokine release upon 2nd dose | N/A | 2.83 × 100 (36) | 7.31 × 100 (92) |
| k kill | Killing rate for tumor cells | hour−1 | 4.80 × 10−3 (FIXED)c | N/A |
| BL | Cytokine level at baseline | pg/mL | N/A | 1.66 × 101 (31) |
| MTT | Lag time for cytokine elevation in circulation | hour | N/A | 3.21 × 100 (26) |
| Res prop err | Proportional error | N/A | 3.10 × 10−1 (5) | 3.20 × 10−1 (5) |
%CV, percentage of coefficient of variation; N/A, not applicable; PD, pharmacodynamic; PK, pharmacokinetic; Res Prop Err, residual proportional error.
aFor T‐cell–engaging bispecific antibody (T‐BsAb) with hematological tumor indication (e.g., blinatumomab). bFor T‐BsAb with solid tumor indication (e.g., P‐cadherin LP DART). cFIXED: fixed parameter; estimated separately and then fixed in the cytokine model.
Theoretical: The model
The cytokine PK/PD model components are defined below (Figure 1). First, the model describes the PK of T‐BsAbs by appropriate PK models (e.g., two‐compartment PK model). The PK model equations were omitted here for conciseness.
For the cytokine PD model, the cytokine (C) profile is modeled as the result of release (RL), time‐variant negative feedback (IH), and degradation, assuming first‐order degradation with rate of k deg (Eq. 1):
| (1) |
The cytokine release (with RL representing cytokine release rate) was stimulated by T‐BsAbs. The trimolecular synapse, formed between the drug, CD3 on T cells, and TAA on tumor cells is considered as “active” to stimulate cytokine release (Eq. 2).1, 15
| (2) |
Syn represents the synapse concentration. Emax and EC50 represent the maximum cytokine release rate and the synapse concentration to achieve 50% of maximum cytokine release rate, respectively. The Hill coefficient H represents the steepness of the exposure–response relationship.
Based on quasi‐equilibrium approximation, the formation of the synapse following sequential binding among T‐BsAb, CD3, and TAA can be described by Eqs. 3, 4, and 5. The other sequence of binding (TAA followed by CD3) will result in the same final equation (Eq. 5). Here, T and Tu represent the unbound concentration of CD3 on T cells and TAA on tumor cells, respectively. D represents free drug concentration. Kd 1 and Kd 2 represent the binding affinity of T‐BsAb against CD3 and TAA, respectively. It is assumed that the binding affinity of the dimer against the targets can be approximated by that of the naked T‐BsAb.
| (3) |
| (4) |
| (5) |
Because T‐BsAbs are generally very efficacious at lower doses (e.g., 5 to 15 μg/m2/day for blinatumomab), it is assumed that the bound concentrations of CD3 and TAA are negligible so that the unbound concentrations are approximated by total concentrations (Eq. 6). Here, T tot and Tu tot represent the total concentrations of CD3 and TAA, respectively.
| (6) |
Shortly after T‐BsAb dosing, blood T‐cell counts decrease first and subsequently increase to baseline levels (e.g., to within twofold of baseline for blinatumomab) over a period of days.5 This phenomenon is related to redistribution, rather than a loss of the T cells. Therefore, T tot is regarded as a constant, assuming T‐cell number does not change significantly during the treatment. By taking out the constants (“T tot,” “Kd 1,” and “Kd 2”) from Eq. 6, synapse (Syn) concentration is approximately proportional to the product of free drug concentration (D) and total TAA concentration () (Eq. 7). For hematological tumors, such as ALL and NHL, a rapid decline on tumor burden was observed shortly after the first dose of T‐BsAb.5 For hematological tumors, it is reasonable to apply Eq. 7 to capture the impact of tumor cell kinetics on synapse concentration.
| (7) |
In contrast, solid tumors may take longer treatment (e.g., months) to have significant impact on tumor burden.17 Therefore, for solid tumors, synapse concentration is mainly determined by the drug concentration during cytokine release (Eq. 8).
| (8) |
Equation can be rewritten as Eq.9 for hematological tumors using Eq. 7:
| (9) |
and as Eq.10 for solid tumors using Eq. 8:
| (10) |
Cytokine release is tightly regulated by negative feedback mechanisms to avoid overactivation of the immune system. For example, with T‐BsAb treatment, immunomodulatory cytokines, such as IL‐10, were secreted.5 IL‐10 decreases the ability of monocyte and macrophages to produce a variety of proinflammatory cytokines.18 Thus, a negative feedback loop was incorporated by assuming inhibition on cytokine release is a function of cumulative cytokine exposure during individual dosing intervals. Furthermore, this negative feedback is time‐variant, which is related to the number of doses administered (Eq. 11).
| (11) |
IH represents inhibition effect (negative feedback) on cytokine release. represents the cumulative cytokine exposure during a dosing interval. I max and IC50 represent the maximum inhibition and the cumulative cytokine exposure to achieve 50% of the maximum inhibition, respectively. The time‐variant component of the negative feedback is described using the term K N − 1. Here, K is the “priming factor,” representing the magnitude of priming effect upon repeated dosing. N represents the number of doses. The current manuscript focused on the first two doses, when the most dramatic reduction on cytokine levels is observed. Nevertheless, the empirical function K N − 1 can be further modified to capture cytokine profiles upon multiple doses.
Case study with blinatumomab
Blinatumomab cytokine data in patients were modeled using the PK/PD model described above. Specifically, Eqs. 1, 9, and (1), (9) were applied, because blinatumomab is indicated for treating hematological cancers. In addition, Eq.12 was applied to describe the tumor cell (CD19+ B cell) kinetics observed in the patients, assuming the tumor cell killing follows first‐order kinetics upon blinatumomab treatment.
| (12) |
The clinical cytokine data were digitized from a published poster.16 It describes the observed cytokine profiles in a phase I study (NCT00274742) in patients with relapsed NHL treated with blinatumomab. Cytokine profiles of patients after blinatumomab treatment (either a step dose of 5 to 15 μg/m2/day or a single dose of 60 μg/m2/day) were presented. The B‐cell kinetics were reported in a separate publication19 and were fitted using Eq. 12 to estimate the killing rate for the B cells. For the individuals treated at 0.5–5 μg/m2/day dose, it took 22 days for B cells to decrease from 453 (± 281) to 36 (± 33) cells/μL. The estimated B‐cell killing rate was then fixed during model fitting to the cytokine data. Once the model parameters were estimated, the established model was applied to simulate cytokine profiles under regimens of interest, including short infusion, constant infusion, repeated dose, and priming dose designs.
Case study with P‐cadherin LP DART
The monkey IL‐6 data with P‐cadherin LP DART treatment were characterized using the cytokine model developed for solid tumor indications (Eqs. 10 and 11) with the following modifications. First, the pretreatment steady state was set up to account for baseline level of IL‐6. Second, a transit compartmental model with five transit compartments was implemented to account for the lag time for IL‐6 to rise in circulation. The choice of five transit compartments is supported by the report of Krzyzanski,20 where the transit compartment model approximates lifespan based indirect response models when the number of delay compartment is at least five. The model equations unique to P‐cadherin LP DART are provided below.
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
Here, C 1–C 7 represent IL‐6 concentrations in various compartments (C 1: release compartment, C 2–C 6: five transit compartments, C 7: circulation). K TR represents the transit rate, and MTT represents the mean transit time for IL‐6.
The IL‐6 data come from four nonclinical safety studies in cynomolgus monkeys. Monkeys (n = 81) received P‐cadherin LP DART intravenously (via either bolus injection or 3‐hour infusion) at a wide range of dose levels (0.3 to 30 μg/kg) and under 10 different dosing regimens (repeated dose or priming dose).
Animal studies
All animal studies were conducted in accordance with animal care and use protocols approved by the Institutional Animal Care and Use Committee. The program of humane animal care and use at Pfizer Global Research and Development has been evaluated for its compliance with the US Animal Welfare Act and The Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Research, 1996).
Serum cytokine measurements upon P‐cadherin LP DART treatment
IL‐6 was quantified in serum samples using the Milliplex MAP Non‐Human Primate Cytokine Magnetic Bead Panel reagent kit (Millipore, Burlington, MA). Briefly, fluorescently coded magnetic beads coated with specific capture antibody (MagPlex‐C microspheres, Luminex, Austin, TX) were added to serum samples. After washing, a biotinylated secondary antibody was added, followed by washing and then addition of a Streptavidin‐Phycoerythrin conjugate. After a final wash, the fluorescent reporter signal from the beads was quantified using a Luminex 100 instrument (Bio‐Rad, Hercules, CA). The limit of IL‐6 detection was 3.20 pg/mL.
Results
Case study with blinatumomab
Blinatumomab (Amgen, Thousand Oaks, CA) is an anti‐CD3 × anti‐CD19 bispecific antibody indicated for the treatment of relapsed and refractory B‐ALL.7 Blinatumomab is administered by continuous infusion to provide a sustained drug exposure (blinatumomab has a short half‐life of ~ 2 hours), which may blunt the peak plasma concentration (Cmax) and help mitigate the CRS risk.4 In addition, priming dosing regimens were investigated to further mitigate the risk of CRS and other toxicities (e.g., neurotoxicity) and to achieve higher doses, based on the observations that the severity of CRS decreases with repeated dosing.4
The clinical cytokine data from a phase I study in patients with NHL were used as a case study for hematological tumors.16 Cytokine profiles of patients after either a step dose of 5 to 15 μg/m2/day (n = 21) or a single dose of 60 μg/m2/day (n = 9) were available. When a priming dosing dose of 5 μg/m2/day followed by a dose of 15 μg/m2/day was used, the cytokine levels significantly decreased after the second dose despite a threefold increase in dose. Besides the “priming” effect of repeated dose, the decreased cytokine level is likely at least in part affected by the rapid depletion of antigen‐expressing B‐cells following the first dose (5 μg/m2/day) of blinatumomab.
Both the cytokine data and the B‐cell profiles from the blinatumomab clinical study were used to fit the model (details in “Methods”). The B‐cell killing rate was estimated to be 0.0048 hour−1 and was fixed in the model. Log‐transformed vs. normal scale data were evaluated during model fitting; log‐transformed data were deemed more appropriate based on model diagnostics. The estimated parameters are presented in Table 1. Due to the limited dose levels in the study, the EC50 for cytokine release could not be accurately estimated, and was fixed at 10,000 ng/mL·cell/μL (assuming a linear exposure‐response relationship for cytokine release in the current dose ranges). As shown in Figure 2 a,b, the model was able to reasonably describe the overall trends in cytokine profiles, particularly the attenuation of cytokine level upon a priming dose regimen. Model diagnostic plots, including the concordance plot between data and model prediction (Figure 2 c), and weighted residual error against model prediction (Figure 2 d) suggest adequate overall fitting. There seems to be minor underprediction after the first dose and overprediction after the second dose, after investigating various error structures and initial parameter estimates. The presented final model was selected based on its objective function value, ability to converge, and overall goodness‐of‐fit.
Figure 2.
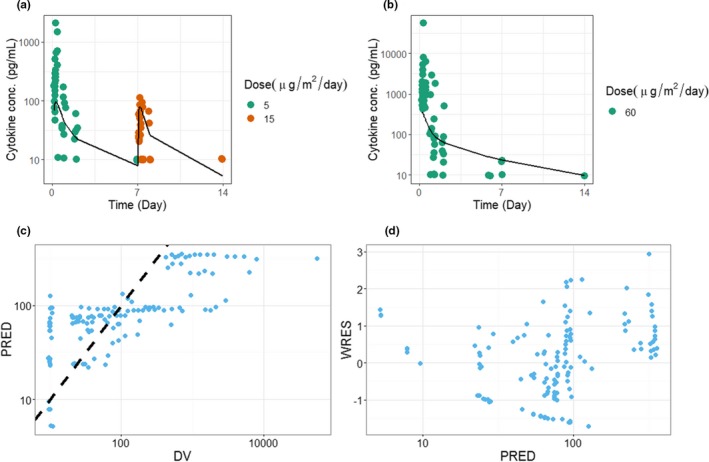
Fitting results for blinatumomab in patients. (a) Model predictions and observed data for cytokine concentration with a priming dose regimen of 5 μg/m2/day followed by 15 μg/m2/day. Symbols represent individual observations, and lines represent the model fit. (b) Model predictions and observed data for cytokine concentration with a single dose at 60 μg/m2/day. Symbols represent individual observations, and lines represent the model fit. (c) Concordance plot between the data (DV) and the model prediction (PRED). The dotted line is the line of unity. (d) Weighted residual error (WRES) against the PRED.
Case study with P‐cadherin LP DART
P‐cadherin LP DART (PF‐06671008) is an anti‐P‐cadherin × anti‐CD3 bispecific dual affinity retargeting (DART, MacroGenics, Rockville, MD) molecule with a human Fc domain fusion developed for the treatment of solid tumors. In nonclinical in vitro studies, it mediated redirected T‐lymphocyte cytotoxicity toward P‐cadherin‐expressing tumor cell lines and induced release of cytokines.21, 22 In this manuscript, it was used as a case study for solid tumor indications. P‐cadherin is a protein involved in cell‐cell adhesion and is known to be expressed in epithelial cells of some tissues. Accordingly (as indicated above in the Model Overview), P‐cadherin target concentration was assumed to be constant throughout the study.
P‐cadherin LP DART was tested in cynomolgus monkeys (n = 81) at a wide range of dose levels (0.3 to 30 μg/kg) and in 10 different dosing regimens (e.g., repeated dosing or priming dosing). The monkey IL‐6 concentrations following treatment were fitted using the PK/PD model (details in the Method section). IL‐6 was chosen because it is one of the most prominently increased cytokines in our study. It has proinflammatory properties and plays an important role in the development of CRS. Log‐transformed vs. normal scale data were evaluated during model fitting; log‐transformed data were deemed more appropriate based on model diagnostics. The estimated model parameters are presented in Table 1. The model was able to reasonably describe the IL‐6 profiles with various dosing regimens. For example, upon repeated dose regimens (Figure 3), the model was able to reasonably capture the attenuation of the cytokine upon the second dose. The model was also able to describe the observed data with priming dose regimens (Figure 4). Model diagnostic plots, including the concordance plot between data and model prediction (Figure 5 a), and weighted residual error against model prediction (Figure 5 b) were presented, suggesting sufficient overall fitting.
Figure 3.
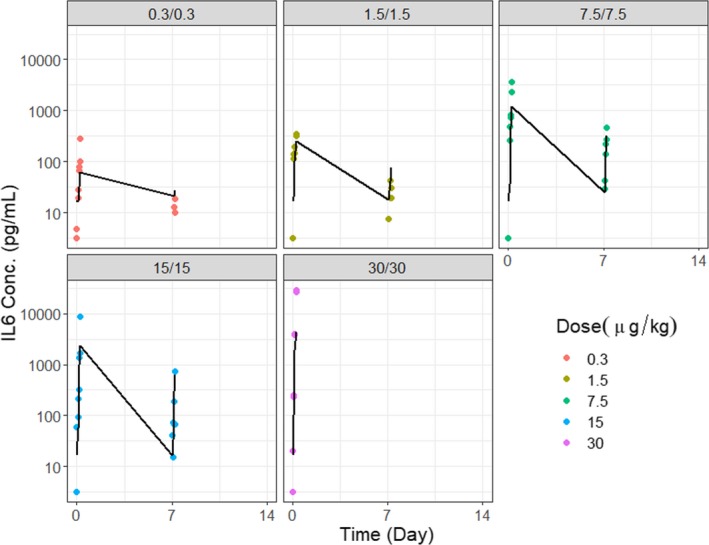
Fitting results for P‐cadherin LP DART in cynomolgus monkeys under repeated dosing regimens. Model predictions and observed data for IL‐6 concentration with repeated doses at 0.3, 1.5, 7.5, 15, and 30 μg/kg. Symbols (observations); lines (model fit).
Figure 4.
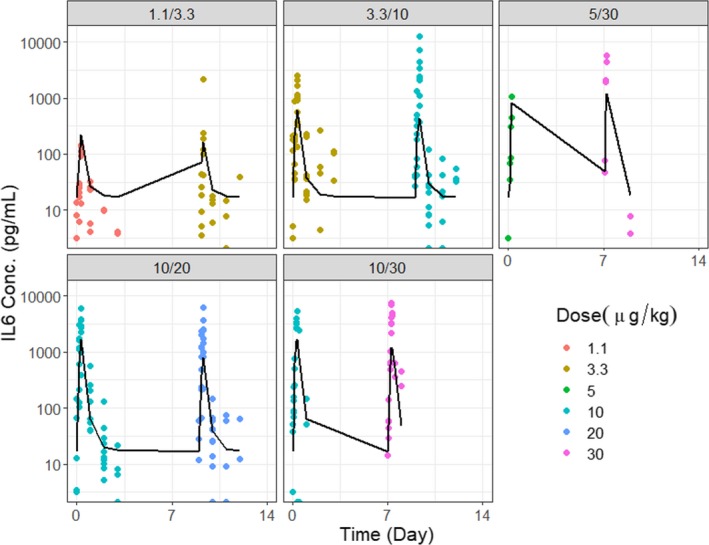
Fitting results for P‐cadherin LP DART in cynomolgus monkeys under priming dose regimens. Model predictions and observed data for IL‐6 concentration with priming dose regimens at 1.1/3.3, 3.3/10, 5/30, 10/20, and 10/30 μg/kg. Symbols (observations); lines (model fit).
Figure 5.
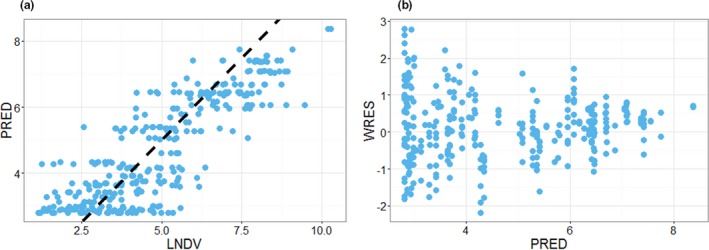
Diagnostic plots for the P‐cadherin LP DART IL‐6 data fitting. (a) Concordance plot between data (natural log transformed dependent variable (LNDV)) and model prediction (PRED). Dotted line is the line of unity. (b) Weighted residual error (WRES) against PRED.
Simulated cytokine profiles for blinatumomab
The PK/PD model for blinatumomab, established based on the clinical data in patients with NHL, was used to simulate cytokine profiles with dosing regimens of interest. First, simulated cytokine profiles were compared between short intravenous infusion (4 hours) and constant infusion at 5 μg/m2/day. Based on the simulation (Figure 6 a), 4‐hour infusion results in about sixfold higher cytokine concentration when compared with constant infusion. Next, simulated cytokine profiles were compared between single dose and priming dose regimens (Figure 6 b). Compared with a single dose of 15 μg/m2/day, the simulated cytokine peak level is lower with priming dose (5 to 15 μg/m2/day).
Figure 6.
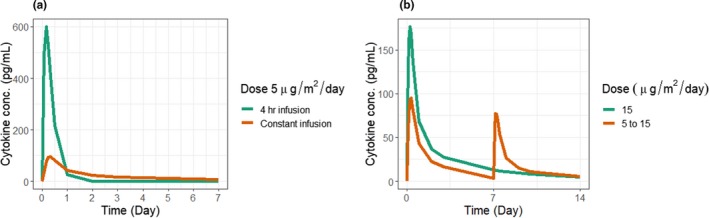
Simulated cytokine profiles for blinatumomab with different dosing regimens. (a) Simulated cytokine profile with 4‐hour infusion or constant infusion at 5 μg/m2/day. (b) Simulated cytokine profile with single dose at 15 μg/m2/day or a priming dose of 5 μg/m2/day followed by 15 μg/m2/day.
Discussion
As an important therapeutic modality in immuno‐oncology, T‐BsAbs may cause elevated cytokines and CRS in patients. Importantly, priming dose strategies were found to mitigate CRS toxicity, based on clinical experience with blinatumomab. However, the determination of the optimal priming dose regimen remains a major challenge, due to the large number of possible permutations of dose levels and priming steps. Quantitative modeling is a valuable tool that can help increase the efficiency by integrating existing knowledge to make predictions for decision making.
The current manuscript provides a quantitative framework for characterizing the cytokine release induced by T‐BsAb treatment. The model was tailored to account for differences in tumor regression between hematological malignancies and solid tumors to reflect the impact of tumor cell killing on cytokine release. The model captures the key features of cytokine kinetics with T‐BsAb treatment, including the priming effect where cytokine release was attenuated following repeated doses. It reasonably describes the cytokine data for blinatumomab in patients (Figure 2) and for P‐cadherin LP DART in cynomolgus monkey (Figures 3 and 4) across a wide range of dose levels and regimens. The model is intended to be parsimonious to allow parameter estimation in scenarios where clinical data are limited.
We hypothesize that, some model parameters, including Imax, IC50, and k deg, can be treated as system parameters as they reflect the intrinsic properties of the immune system within specific context (e.g., in certain patient populations). For example, IC50 for cytokine inhibition should be determined by immune system's intrinsic feedback mechanisms, rather than the specific T‐BsAb to trigger the cytokine. This hypothesis will need to be tested and, once confirmed, these “system parameters” can be fixed to reduce the model dimension.
There are many potential applications for the cytokine modeling framework described here. Most importantly, it can help design optimal dosing regimens to be tested in the clinical trials. Currently, identifying the optimal priming dose regimens for T‐BsAbs is highly empirical. For example, for blinatumomab, a series of pilot phase I studies were conducted to explore different dosing regimen (short intravenous infusion, constant intravenous infusion, and priming dose) before a more extensive clinical study was initiated.23 With the proposed modeling framework, cytokine data from an initial clinical study can be utilized to establish the model, which can prospectively simulate cytokine profiles under various dosing regimens. Examples are provided by simulating dosing regimens evaluated during blinatumomab's clinical development. For example, with blinatumomab, short infusions of 2–4 hours were not tolerated by patients, and drug exposure was not sufficient for achieving efficacy. Our model simulations (Figure 6 a) suggest the expected cytokine level with short 4‐hour infusion could be sixfold higher compared with constant infusion. The high level of the circulating cytokines upon short infusion likely contributed to the dose‐limiting adverse events observed. A priming dose strategy was applied for blinatumomab in B‐ALL. Simulated cytokine levels were compared between single dose and priming dose (Figure 6 b) and suggest that the approved priming dose regimen for blinatumomab with threefold escalation could help minimize cytokine release.
At least certain aspects of the current model we established for T‐BsAb might apply to other immune‐agonistic drugs that stimulate cytokine release, primarily because certain characteristics of cytokine release (e.g., negative feedback and priming) seem to be intrinsic to the immune system. For example, it is well established that endotoxin, such as lipopolysaccharide, stimulates cytokine release in mice and that cytokine release is downregulated upon repeated dosing.24 Based on the similarity in the underlying mechanisms, the current model may be adapted for characterizing other immune‐agonistic therapeutics. With future development, the model may also be used as a translational tool for predicting cytokine response in humans based on relevant animal data (e.g., cynomolgus monkey). With sufficient data, it is possible to estimate the model parameters in both species and derive a scaling factor for translation.
The current model can be further extended as needed (e.g., expanding to two or more cytokines of interest). For example, the anti‐inflammatory cytokine IL‐10 plays important roles in the negative regulation of cytokines. Although the dynamic interactions between cytokines are very complex and not completely understood, mechanistic cytokine models are available in the literature25, 26, 27, 28 and relevant model components may be adapted to this current framework.
In summary, a semimechanistic PK/PD modeling framework was established for characterizing cytokine profiles upon T‐BsAb treatment. The established model can be utilized to simulate cytokine profiles upon various dosing regimens. Therefore, the model should be of value in determining optimal dosing strategies for T‐BsAbs programs.
Funding
No funding was received for this work.
Conflict of Interest
X.C., C.K., G.W., and D.X. are employed by Pfizer and hold stock in Pfizer.
Author Contributions
X.C., C.K., G.W., and D.X. wrote the manuscript. X.C. and D.X. designed the research. X.C., C.K., and D.X. performed the research. X.C. and D.X. analyzed the data. X.C. and C.K. contributed new reagents/analytical tools.
Disclosure
DART is a registered trademark of MacroGenics.
Acknowledgments
The authors thank the P‐cad LP DART Study Team for their collaboration. They thank Justine Lam for critical review of this manuscript.
References
- 1. Wu, Z. & Cheung, N.V. T cell engaging bispecific antibody (T‐BsAb): from technology to therapeutics. Pharmacol. Ther. 182, 161–175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee, D.W. et al Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frey, N. Cytokine release syndrome: who is at risk and how to treat. Best Pract. Res. Clin. Haematol. 30, 336–340 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Nagorsen, D. , Kufer, P. , Baeuerle, P.A. & Bargou, R. Blinatumomab: a historical perspective. Pharmacol. Ther. 136, 334–342 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Nagele, V. et al Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp. Hematol. Oncol. 6, 14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mau‐Sorensen, M. et al A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother. Pharmacol. 75, 1065–1073 (2015). [DOI] [PubMed] [Google Scholar]
- 7. BLINCYTO: Package Insert and Label Information. US Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125557s008lbl.pdf Accessed 1 July 2019
- 8. Jacobs, K. et al Lead‐in dose optimization to mitigate cytokine release syndrome in AML and MDS patients treated with flotetuzumab, a CD123 x CD3 Dart® molecule for T‐cell redirected therapy. Blood 130 (suppl. 1), 3856 (2017). [Google Scholar]
- 9. Fiedler, M.W.W. & Kebenko, M. . A phase I study of EpCAM/CD3 bispecific antibody (MT110) in patients with advanced solid tumors. J. Clin. Oncol. 30 (suppl.; abstract 2504), 2012 (2012). [Google Scholar]
- 10. Teachey, D.T. et al Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T‐cell therapy for acute lymphoblastic leukemia. Cancer Discov. 6, 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turtle, C.J. , Hay, K.A. , Gust, J. & Hanafi, L.‐A. Biomarkers of cytokine release syndrome and neurotoxicity after CD19 CAR‐T cells and mitigation of toxicity by cell dose. Blood 128, 1852 (2016). [Google Scholar]
- 12. Porter, D.L. et al Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maude, S.L. et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang, Z. & Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR‐T cell therapy. Biomark. Res. 6, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen, X. et al Mechanistic projection of first‐in‐human dose for bispecific immunomodulatory P‐cadherin LP‐DART: an integrated PK/PD modeling approach. Clin. Pharmacol. Ther. 100, 232–241 (2016). [DOI] [PubMed] [Google Scholar]
- 16. Hijazi, Y. et al Blinatumomab exposure and pharmacodynamic response in patients with non‐Hodgkin lymphoma (NHL). J. Clin. Oncol. 31, 3051 (2013).23715569 [Google Scholar]
- 17. Tabernero, J. et al Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T‐cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 35, 3002 (2017).28644773 [Google Scholar]
- 18. Wu, T.T. , Li, W.M. & Yao, Y.M. Interactions between autophagy and inhibitory cytokines. Int. J. Biol. Sci. 12, 884–897 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu, M. et al Blinatumomab, a bispecific T‐cell engager (BiTE((R))) for CD‐19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin. Pharmacokinet. 55, 1271–1288 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Krzyzanski, W. Interpretation of transit compartments pharmacodynamic models as lifespan based indirect response models. J. Pharmacokinet. Pharmacodyn. 38, 179–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Root, A.R. et al Development of PF‐06671008, a highly potent anti‐P‐cadherin/anti‐CD3 bispecific DART molecule with extended half‐life for the treatment of cancer. Antibodies 5, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher, T.S. et al A CD3‐bispecific molecule targeting P‐cadherin demonstrates T cell‐mediated regression of established solid tumors in mice. Cancer Immunol. Immunother. 67, 247–259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoffman, L. & Gore, L. Blinatumomab, a bi‐specific anti‐CD19/CD3 BiTE® antibody for the treatment of acute lymphoblastic leukemia: perspectives and current pediatric applications. Front. Oncol. 4, 63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Erroi, A. et al Differential regulation of cytokine production in lipopolysaccharide tolerance in mice. Infect. Immun. 61, 4356–4359 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Waito, M. , Walsh, S.R. , Rasiuk, A. , Bridle, B.W. & Willms, A.R. A mathematical model of cytokine dynamics during a cytokine storm In Mathematical and Computational Approaches in Advancing Modern Science and Engineering. pp 331-339. (eds. Bélair J., Frigaard I., Kunze H., Makarov R., Melnik R. & Spiteri R.). (Springer, Cham, 2016). [Google Scholar]
- 26. Brady, R. et al Personalized mathematical model of endotoxin‐induced inflammatory responses in young men and associated changes in heart rate variability. Math. Model. Nat. Phenom. 13, 42 (2018). [Google Scholar]
- 27. Morel, P.A. , Lee, R.E.C. , Faeder, J.R. Demystifying the cytokine network: mathematical models point the way. Cytokine 98, 115–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roy, A. , Clermont, G. , Daun, S. & Parker, R.S. . A mathematical model of acute inflammatory response to endotoxin challenge. American Institute of Chemical Engineers (AIChE) Annual Meeting, Salt Lake City, UT, 538 (2007).