

Summary
SGIV, or Singapore Grouper Iridovirus, is a large dsDNA virus, reaching a diameter of 220 nm and packaging a genome of 140 kb. We present a 3D Cryo-EM icosahedral reconstruction of SGIV determined at 8.6Å resolution. It reveals several layers including a T=247 icosahedral outer coat, anchor proteins, a lipid bilayer, and the encapsidated DNA. A new segmentation tool, iSeg, was applied to extract these layers from the reconstructed map. The outer coat was further segmented into major and minor capsid proteins. None of the proteins extracted by segmentation have known atomic structures. We generated models for the major coat protein using three comparative modeling tools, and evaluated each model using the CryoEM map. Our analysis reveals a new architecture in the Iridoviridae family of viruses. It shares similarities with others in the same family, e.g. CIV, but also shows new features of the major and minor capsid proteins.
Keywords: Iridovirus, Cryo-EM, Icosahedral Segmentation, Comparative modeling
Graphical Abstract
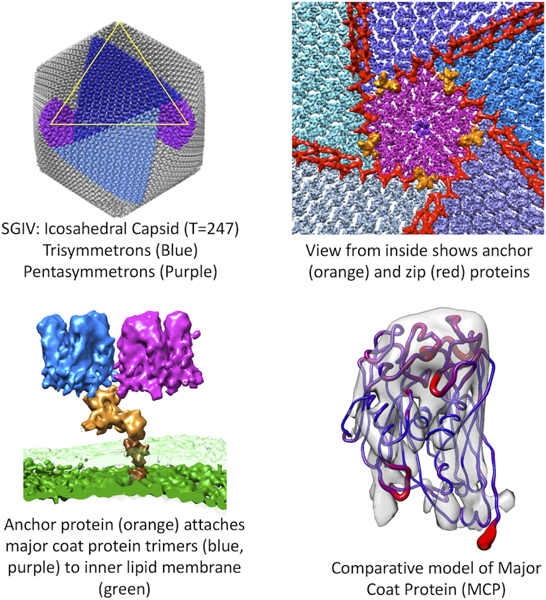
Pintilie et al. describe the architecture of the Singapore Grouper Iridovirus, revealed by CryoEM at 8.6Å resolution. Several components are identified including an outer icosahedral coat (T=247), zip proteins which run along coat proteins, and anchor proteins which attach the coat to an inner lipid membrane.
Introduction
SGIV (Singapore Grouper Iridovirus) is a large dsDNA virus in the family Iridoviridae, one of the 10 families of nucleocytoplasmic large DNA viruses (NCLDVs). It is an emerging pathogen of fish and amphibians, and hence of importance in aquaculture, as epizootics can lead to large losses of cultured fishes. Moreover, they are a source of environmental concern, as they have been implicated in the global decline of amphibian populations (Bromenshenk et al., 2010; Chinchar, 2002; Chinchar et al., 2009; Jancovich et al., 2003; Price et al., 2014).
SGIV is a species of the Ranavirus genus, one of five genera within the Iridoviridae family (Williams, 1996). The first known viruses in the Ranavirus genus were isolated in frogs (Granoff et al., 1965). Ranaviruses were later found to infect reptiles as well (Chen et al., 1999). Studies have shown however that Ranaviruses appear to have evolved from a fish virus (Jancovich et al., 2010); hence viruses in this family seem to be effective at shifting hosts.
Viruses categorized as NCLDVs tend to be very large (reaching 200nm and more across), and have similarities in genomic DNA sequence and structure. Within the Iridoviridae family, viruses include SGIV and CIV; CIV infects leafhoppers and other arthropods. The similarity between SGIV and CIV despite the difference in the hosts that they infect is remarkable. Other families of NCLDVs similarly infect hosts across a wide variety of species; they include Asfarviridae (e.g. swine flu virus), Mimiviridae (e.g. Mimivirus), Phycodnaviridae (e.g. algae-infecting PBCV, phytoplankton-infecting PpV01), and Poxviridae (e.g. smallpox in humans).
Studies have shown that NCLDVs have structural and genomic similarities to some smaller viruses such as PRD1, which infects E. coli and Salmonella, and Adenovirus, the common-cold virus, which infects humans. One of the structural similarities lies in the structure of the outer coat proteins: each protein contains two jelly roll domains, a variation of the versatile beta-barrel fold (Wimley, 2003). The outer shells in these viruses typically envelop both a lipid membrane and genomic dsDNA. In some cases outward-extending fibers are attached to coat proteins, e.g. CIV (Yan et al., 2009), and in other cases a unique 5-fold vertex consists of a large spike (PBCV).
For most NCLDVs, the outer coat layers have an icosahedral arrangement of proteins (Caspar and Klug, 1962). The smaller PRD1 and Adenovirus have T=25 icosahedral outer coats, CIV has a much larger T=147 outer coat, and PBCV a yet larger one at T=169. Here we reveal the even larger SGIV with a T=247. Our structural analysis, like the ones before, continues to show that even though the sizes of these virions can vary dramatically, as do the hosts they infect, there are significant similarities between their components.
Starting with one of the smallest related viruses, the structure of PRD1 was first revealed using Cryo-EM, showing an icosahedral outer protein coat enclosing a lipid bilayer membrane and DNA (Butcher et al., 1995). The structure of the major coat protein (MCP) P3 was then resolved using X-ray crystallography, and docked into the Cryo-EM map, creating a pseudo-atomic model of the entire shell. Phases generated from this model were then used to solve a crystal structure of the entire virion, revealing detailed arrangement and conformations of MCPs P3 and P31, and minor capsid proteins, P30 and P16 (Abrescia et al., 2004). This was a large advance in the understanding of this family of viruses, as it clearly showed the structure and interactions of the major and minor proteins in the outer coat.
The larger PBCV, with a T=169 outer protein coat, has been reconstructed using Cryo-EM. The structure of the major capsid protein (Vp54) was solved in tandem using X-ray crystallography (Zhang et al., 2011). This detailed structure showed that Vp54 is very similar to the PRD1 MCP P3, also containing two jelly-roll domains, even though the PBCV outer coat is much larger and is made up of many more proteins. Another relevant NCLDV is CIV, solved by Cryo-EM icosahedral reconstruction to 13Å resolution (Yan et al., 2009). Its outer shell is T=147. The structure of the major capsid protein (P50) is yet unknown, however based on its similar sequence to the major coat protein Vp54 from PBCV, a comparative model was built and docked into the Cryo-EM map.
In this paper, we first present a Cryo-EM icosahedral reconstruction of SGIV at 8.6Å resolution, with a yet larger T=247 outer protein coat. We apply a new method for segmenting icosahedral layers, iSeg, which improves upon the previously reported rSeg (Pintilie et al., 2016) by correcting for curvature across icosahedron faces. This allowed us to first computationally isolate several layers including the outer coat, a lipid bilayer, another inner shell of unknown composition, and the packaged DNA. The outer coat was further segmented into pentameric and trimeric protein complexes (the major coat proteins), and also zip and anchor proteins (minor coat proteins).
Second, we used comparative modeling methods, including MODELLER (Eswar et al., 2007), I-TASSER (Yang et al., 2015), and Phyre2 (Kelley and Sternberg, 2009), to build an atomic structure for Orf72, the major coat protein of SGIV. Each method produced models based on the same template (Vp54 protein) with similar double jelly-roll domain cores, though several inserted loops had varying lengths and conformations. Previous approaches to dealing with this type of modeling uncertainty include an iterative-approach (Topf et al., 2006). Our approach was to flexibly fit several resulting models into the Cryo-EM map, and then evaluate how well each model matches the map. We also modeled the uncertainty due to flexible fitting and limited resolvability of structural features in each part of the map using ProMod.
Results and Discussion
CryoEM Reconstruction
A representative micrograph of the virion is shown in Figure 1A, hinting at the icosahedral shape of SGIV. A slice through the reconstructed map is shown in Figure 1B. Several components are seen: outer icosahedral shell, anchor proteins, lipid bilayer, and an inner shell of unknown composition. A view of the surface from the outside is shown in Figures 1C,D. Asymmetric units are highlighted in Figure 1C. Adopting the terminology previously introduced to describe this type of virion (Nandhagopal et al., 2002), tri- and penta-symmetrons are shown in Figure 1D. The FSC plots in Figure 1E show self-consistency between two independent reconstructions for 1) the entire maps with no masking, 2) entire maps masking only the outer coat, and 3) between masked asymmetric units (ASUs). The masking was done using a soft mask to avoid artificial boundary related-correlations (Pintilie et al., 2016). All curves indicate a resolution of ~8.6Å using the FSC0.143 criterion. Interestingly the FSC value is higher at medium resolutions (~12Å) when comparing just the ASUs, while it dips significantly when comparing the entire maps and also just the outer coat. Such difference is likely caused by the inclusion of the non-icosahedral components of the map as a larger context.
Figure 1.
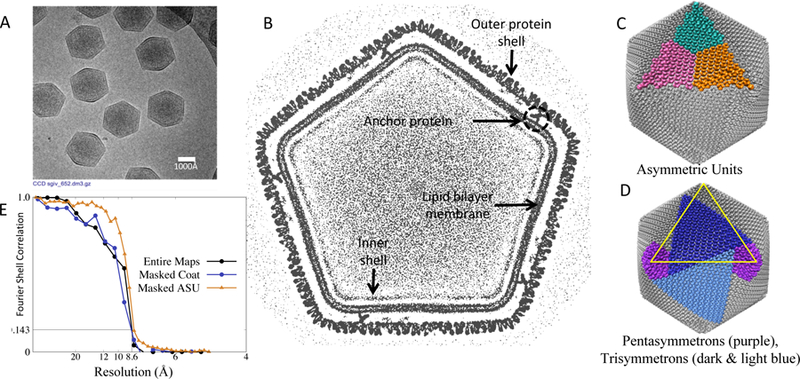
CryoEM map of SGIV. (A) Micrograph showing several SGIV particles. (B) Map cross-section showing coat, anchor proteins, lipid bilayer, and packaged DNA. (C,D) Surface of virion (gray surface), with 3 asymmetric units (cyan, magenta, and orange), pentasymmetrons (purple) and trisymmetrons (dark and light blue). (E) FSC plots of independently reconstructed maps and masked coat densities (1 ASU).
Icosahedral Segmentation with iSeg
It is very challenging to segment the molecular components in a very large, multi-component reconstruction such as this map of SGIV. We previously introduced a tool to radially segment layers in the P22 bacteriophage (Pintilie et al., 2016). We noticed for SGIV, however, that a flat icosahedral outer surface did not accurately represent the CryoEM map – the surface of each icosahedral triangle appears to curve outwards, but only in the middle of the triangle (Figure S1).
We first attempted to address this discrepancy by interpolating the icosahedral geometry towards a spherical shape, e.g. with the spherical interpolation tool in UCSF Chimera. However this does not accurately capture the curvature, as it makes both the centers and edges of each surface triangle approach a spherical shape. We thus developed a new method, whereby the icosahedral distance from center is adjusted to allow curvature across each triangle on the icosahedron surface, while holding the triangular edges rigid (this is further described in the Experimental Methods section). The new method, iSeg, supersedes the previous method rSeg, and is available as part of the Segger plugin for UCSF Chimera (Pintilie et al., 2010).
The performance of the iSeg method was evaluated in two ways: visually, and by cross-correlation score. Figure S1 shows how the deformed icosahedral surface (C) matches the outer layer of SGIV more closely than a non-deformed icosahedral surface (A), or a simple spherically-deformed icosahedral surface (B). Figure S1(D) shows a plot of cross-correlations between a non-deformed icosahedral surface and the SGIV map, and also a deformed icosahedral surface and the map. The latter has higher peaks and lower valleys, indicating a more accurate representation of the map.
In Figure 2A, the radial plot shows peaks corresponding to separable layers in the map. From left to right these include: the packaged DNA (which may contain various proteins as well), inner shell, lipid membrane, anchor proteins, zip proteins (a small inflection point is seen instead of a peak here), and the outer protein shell. A cut through the different layers is shown in Figure 2B. The outer coat is colored in with blue, zip proteins that line the boundaries between tri- and penta-symmetrons with red, anchor proteins with orange, lipid membrane with green, inner shell with light brown, and DNA with pink.
Figure 2.
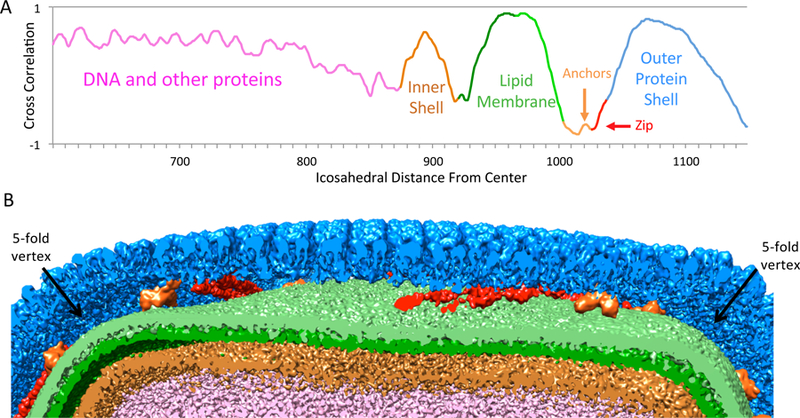
Layers in SGIV. (A) Radial plot (corrected for icosahedral & bulging geometry) showing peaks for inner shell, lipid membrane, anchor proteins, and outer protein shell. (See also Figure S1). (B) A cut-away view through the virion showing the different layers.
Two separable peaks with a peak-to-peak distance of ~20Å can be seen for the lipid membrane, confirming it is a bilayer membrane. The two layers were separated and shown in a slightly different shade of green in Figure 2B. The “inner shell” layer also has a distinct peak in the plot in Figure 2A. It looks very different from the more diffuse collection of peaks corresponding to packaged DNA. It is unclear what this layer is composed of. The extracted layer does not seem to have any resolvable features, possibly because the organization of its components does not follow icosahedral symmetry. It may be similar to scaffolding proteins partially seen in the icosahedral reconstruction of the P22 procapsid (Chen et al., 2011), and also the inner protein layer of fausto-virus (Reteno et al., 2015); the latter however was shown to have resolvable features following icosahedral symmetry.
Outer Coat
In NCLDVs such as SGIV, CIV, and PBCV, the outer coat is composed of trimers and pentamers. These morphological units are further organized into tri- and penta-symmetrons, as shown in Figure 3A for SGIV. A pentasymmetron is centered on a 5-fold vertex, with a pentameric complex at the vertex, and 5 trisymmetrons are arranged circularly around this pentasymmetron (Figure 3A). In a single virion, there are 12 pentasymmetrons and 20 trisymmetrons (see Supplementary Movie 1). In SGIV, each pentasymmetron is composed of one pentamer and 30 trimers, while each trisymmetron contains 105 trimers.
Figure 3.
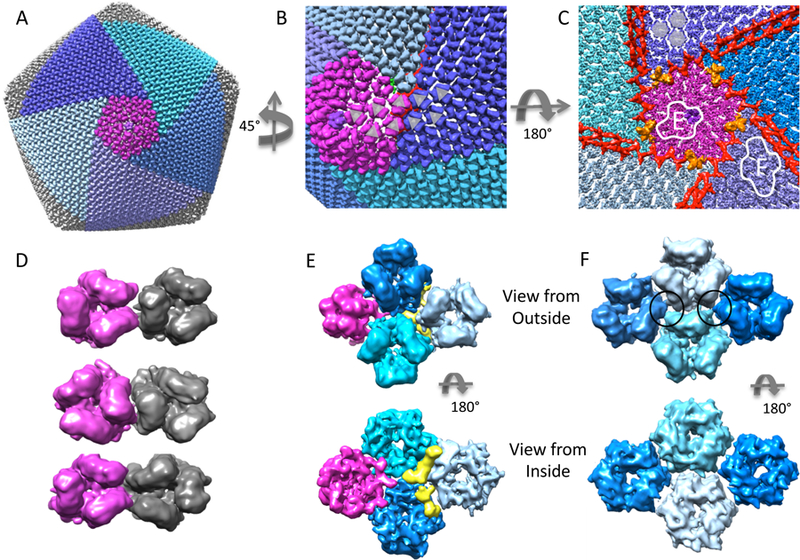
Outer coat proteins. (A) Surface of entire virion with a pentasymmetron (purple) and 5 trisymmetrons (blue/cyan) (see also Supplementary Movie 1). (B) Close up on the boundary between a pentasymmetron and trisymmetrons, showing a switch in orientation in the trimers that make up the coat. (C) View from inside, looking towards a pentasymmetron (purple). Anchor proteins are colored orange, and zip proteins are colored red. Two outlined sections are expanded in panels E and F. (D) Three different arrangements of neighbouring trimers: trisymmetrons only have the type shown at the top; within pentasymmetrons and penta-trisymmetron boundaries, all three types are seen. (E) Top and bottom views of 1 pentamer at the 5-fold vertex (purple) and 3 hexameric trimers (blue/cyan) taken from the pentasymmetron. The yellow segment may be a small protein or an unknown conformation for the N/C termini of the trimeric MCP. (F) Top and bottom view of 4 trimers taken from the center of a trisymmetron. Here, interactions between trimers (black circles) seem to occur mid-way between the inside and outside surfaces.
Each trimer is roughly hexameric in shape, which can be more easily seen when looking at it from the inside (shown in Figure 3C using transparent hexagons superimposed near the top of the image). Such hexameric shapes, along with a pentamer at the 5-fold vertex, were shown to be basic building blocks of icosahedral capsids by Caspar and Klug (Caspar and Klug, 1962). An asymmetric unit (AU) of the SGIV contains 1 protein from the pentamer at each 5-fold symmetric center, and 123 proteins in 41 hexameric trimers - 6 of those trimers occur in a pentasymmetron, and the other 35 trimers in a trisymmetron (Figure 4).
Figure 4.
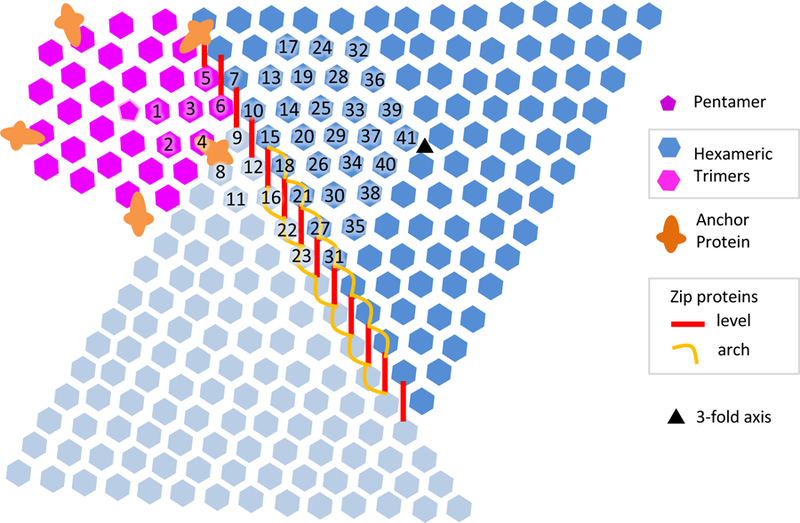
Architecture of SGIV outer coat. The components include the pentamer at the 5-fold vertex, hexameric trimers making up pentasymmetrons (purple) and trisymmetrons (blue and cyan), anchor proteins (orange), and zip proteins (red and orange). The zip proteins are made up of a ‘level’ component which is level to the inner surface, and an arch component which curves inwards towards (but not touching) the lipid membrane. The arch component is found between trimers in adjacent trisymmetrons, but not between trimers in adjacent penta and tri-symmetrons.
Interactions between Trimers
In an icosahedral particle, the contents within an asymmetric unit do not need to have identical structural conformations. Indeed, it is the case that trimers are found to have various orientations with respect to each other, as shown in Figure 3D. Within trisymmetrons, the orientations are consistently the same (Figure 3D, top), however all three orientations in Figure 3D can be seen within pentasymmetrons and at the boundary between trisymmetrons and pentasymmetrons. This suggests different interaction patterns (possibly orchestrated by other minor capsid proteins) in these two different structures.
First looking at the pentasymmetrons, a subsection of the map outlined and marked ‘1’ in Figure 3C was computationally extracted from the entire map of SGIV and shown in larger form in Figure 5A. It shows interactions between trimers and one pentamer in the pentasymmetron. Here, the bulk of the interactions between trimers seem to occur on the inside surface of the coat (Figure 5A, bottom). It is hard to say at this resolution if the connections are either 1) re-organized N or C termini from the MCP which are close to the inside surface or 2) other proteins. If the former is the case, this would be similar to what was shown with the crystal structure of the MCP of PRD1, P3 (Abrescia et al., 2004). The outer coat protein P3, which also contains 2 jelly roll domains, was shown to have different (extended or contracted) conformations at the N and C termini depending where in the coat they are positioned, allowing interactions with other P3 and other minor capsid proteins.
Figure 5.
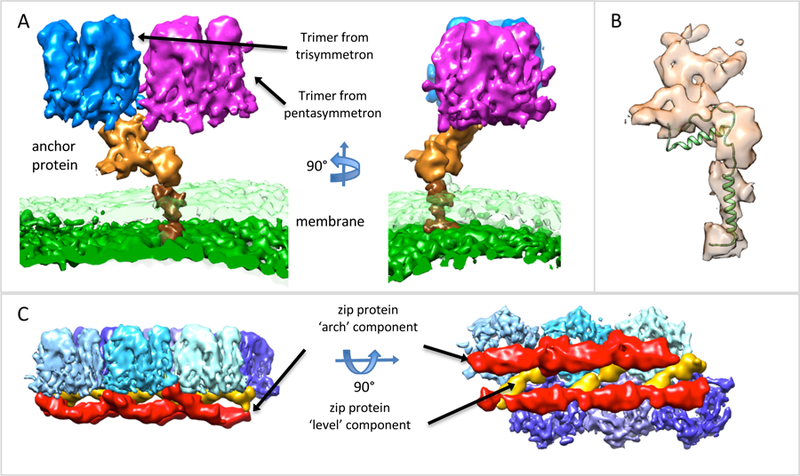
Anchor and Zip proteins. (A) Anchor protein (orange) shown next to bilayer lipid membrane, outer layer is transparent green, inner surface is solid green, and two of the capsomers it is adjacent to. The darker brown surface is the trans-membrane portion of the anchor protein. (B) Anchor protein, transparent surface, shown with the membrane protein P16 from PRD (PDB: 1W8X), showing similar architecture and trans-membrane helix. (C) Side and top view of zip proteins, red surfaces, connected to hexameric trimers, blue surfaces. The zip proteins appear to be composed of arch components, which arch away from the outer capsomeric surface, and level components, which are level with the inner surface of the capsomers.
For trisymmetrons, the portion outlined and marked ‘2’ in Figure 3C was computationally extracted and enlarged in Figure 5B. Here, the trimers appear to interact to a much lesser degree on the inside surface. Instead, connections can be seen in the middle of the trimers, marked with black solid circles in Figure 5B, top. In other NCLDVs such as CIV and PBCV, finger proteins were observed on the inside and outside of trisymmetrons; such densities are not observed for SGIV. This may be because 1) such proteins are not icosahedrally arranged in SGIV, meaning they would be blurred out in our reconstruction, or 2) changes in other components, such as the anchor and zip proteins, further described below, have made finger proteins unnecessary in stabilizing and/or assembling the trisymmetrons in SGIV.
Anchor Proteins
There are 5 anchor proteins centered around each 5-fold vertex as shown in Figures 3C and 4, and thus one anchor protein per asymmetric unit. A segmented anchor protein is shown in Figure 5A. The anchor protein is embedded in the bilayer lipid membrane at one end, and connects to two MCP trimers at the other end. One of the MCP trimers is in a pentasymmetron (purple), and the other in a trisymmetron (blue).
While protein sequences from SGIV have been previously identified (Song et al., 2004), it is still not clear what the exact sequence and molecular size of the anchor protein is. In our segmentation, it is similar to the protein P16 in PRD1 (Abrescia et al., 2004). The protein P16 also consists of a trans-membrane domain, a helix that runs parallel to the membrane and the outer shell, and a domain that attaches to trimers in the outer coat. While the anchor protein in SGIV is larger, as shown in Figure 5B superimposed on the structure of P16, it appears to have a similar architecture: a trans-membrane helix, helical-like densities that run perpendicular to the outer coat and lipid membrane, and a domain that binds to the inner surface of the outer coat.
Zip Proteins
Zip proteins line the inside surface of the outer coat, running along pentasymmetron-trisymmetron and trisymmetron-trisymmetron boundaries, as shown in Figures 3C, 4 and 5C. At penta-trisymmetron boundaries, the zip proteins appear to only have a ‘level’ component, which is parallel to the inside surface of the coat and runs directly from one trimer to another. At tri-trisymmetron boundaries, an ‘arch’ component can also be seen, which curves inwards toward, but does not contact, the lipid membrane. The zip proteins at a tri-trisymmetron boundaries are shown close-up in Figure 5C.
This architecture is similar to that of CIV, however not as much detail is seen in the reconstruction of CIV since it was at a lower resolution of ~13Å (Yan et al., 2009). For CIV, it was noted that at penta-trisymmetron boundaries, the zip proteins appeared to be monomeric, while at tri-trisymmetron boundaries they seemed to be dimeric. Compared to CIV, the SGIV trisymmetrons are larger (14 trimers on each side rather than 10), and hence a different architecture for the zip proteins is possible. Perhaps one of the most interesting observations of the SGIV architecture is that despite the increase in size in trisymmetron size, the pentasymmetrons are the same size in SGIV and CIV.
In PBCV, proteins that line trisymmetron boundaries are also seen, however in that case they appear to contact the membrane as well as the outer coat (Zhang et al., 2011), and hence they are referred to as ‘membrane proteins’. In the much smaller PRD1, a similar, long, protein which connected trimers along the inside surface was proposed to function like a ‘tape measure’ that aids in the assembly of right number of trimers in the outer coat. The zip proteins shown here for SGIV may have a similar role.
Rigid Fitting of Vp54
One hexameric trimer from the outer coat was segmented with Segger into three 3 regions. The major coat protein (MCP) of PBCV, Vp54 (PDB:1m3y), was fitted to each of the 3 regions with the Fit To Segments tool in Segger. A fit to one of the three segments is shown in Figure 6A. Each fit was evaluated for correctness by visual inspection as well as Z-score analysis (Pintilie and Chiu, 2012), as show in Figure S2. Z-scores were ~3.3, indicating reasonable confidence in the fit of the models to the CryoEM map. In the fit with the highest cross-correlation score, α-helices and β-sheets in the core part of the model (the double jelly-roll domain) match tubular and flat densities in the Cryo-EM map respectively (Supplementary Movie 2). For other fits with lower cross-correlation scores (Figure S2), this correspondence was not observed.
Figure 6.
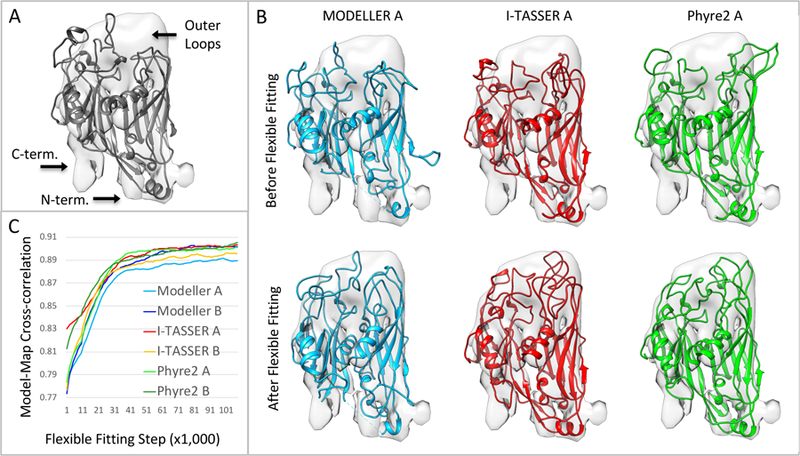
Comparative model of Orf72, the major coat protein (MCP). (A) The template protein, Vp54, gray ribbon, PDB: 1m3y, fitted to the segmented CryoEM map (See also Figure S2). (B) Three of the six comparative models built by three methods, before and after flexible fitting with MDFF. The core double jelly-roll fold is the same in each comparative model, however each comparative model has varying lengths and placements of loop insertions in the region marked ‘outer loops’. (C) Plot of model-map cross correlation between 6 comparative models (2 from each method) and the map during flexible fitting. See also Supplementary Movies 2 and 3.
Outside the core double jelly-roll domain, several parts are different between the MCPs of SGIV and PBCV. As marked in Figure 6A, two of them are close to the N and C-termini of the fitted Vp54 protein, on the inside surface of the coat. Another one is close to the outer surface of the coat, marked ‘outer loops’; the fitted model consists of several long loops in that vicinity. These differences are further explored via the building of comparative models, as discussed below.
Comparative Model of the MCP, Orf72
The sequence of Orf72 has been previously identified as the likely candidate for the major capsid protein or MCP (Song et al., 2004). This sequence was input into three comparative modeling tools: MODELLER (Eswar et al., 2007), Phyre2 (Kelley and Sternberg, 2009), and I-TASSER (Yang et al., 2015). MODELLER reported a 27% sequence identity between the sequence of Orf72 and Vp54 (PDB:1m3y), the MCP of PBCV, making it a good candidate for use as a template. Phyre2 and I-TASSER reported 23% and 24% sequence identity with the same template, respectively. Percent identities between two sequences can vary depending on the method and alignment (Raghava and Barton, 2006). MODELLER and I-TASSER produced 5 comparative models each, and Phyre2 was applied twice, producing two different comparative models.
The first two models from each method were aligned to the rigidly fitted Vp54 model using the Chimera MatchMaker tool. One of the models from each method is shown in Figure 6B, along with the segmented Cryo-EM map. The core jelly-roll domain is the same in each model, since all methods used the same Vp54 protein as a template, and hence this part aligned extremely well between all comparative models and the Vp54 protein. RMSDs between the comparative models and the Vp54 protein are ~1.0Å when considering only the core double jelly-roll domain, and ~6.0Å considering all residues. The largest differences thus occur outside the core domain, mostly in the outer loops section, where the comparative models all contain insertions of varying lengths and placements.
The comparative models also have an insertion close to the N-terminus of ~20 residues. These took the form of a long loop which does not fit well into the map. The map appears to have some secondary structure in this area, however it is not resolved sufficiently for further modeling. This part (residues 1–40) was thus removed from the model and is not shown in the figures. The C-terminus did not have a significant insertion or deletion. The map section which appears to be unoccupied close to it is also close to a long loop (residues 388–408); it has a different conformation in SGIV and hence does not fit the map well.
Flexible Fitting of the MCP
Figure 6B also shows the three comparative models, one from each method, after flexible fitting to the Cryo-EM map with MDFF (Trabuco et al., 2008); animations of the trajectories of 6 comparative models, two from each method, are also shown in Supplementary Movie 3. RMSDs between the initial and final models were 5.5Å/5.3Å for the two MODELLER models, 3.9Å/3.6Å for the two I-TASSER models, and 4.1Å/4.2Å for the two Phyre2 models. The largest changes to the model occur in the outer loops. In all models, these loops start outside the visualized map contour, and move into the contour during flexibly fitting. Figure 6C shows the model-map cross-correlation during the flexible fitting process for the 6 models. The final scores are very similar, likely because the map is not well-resolved in the region where most variation occurs, the outer loops. Thus, the CryoEM map alone may not be able to discern which model is more accurate in this case.
Uncertainty in Flexible Fitting
To model the uncertainty in the model after flexible fitting, 10 independent runs of MDFF were performed one of the comparative models produced by Phyre2. The 10 results were assessed with the ProMod tool, calculating an average model and the standard deviations at each residue position across the 10 results (Pintilie et al., 2016). The average model shown in Figure 7A, represents the most likely conformation given the starting comparative model, the observed map, and the MDFF procedure. The deviation in atomic positions arise from the combination of random (temperature-dependent) motions of the atoms as simulated with MDFF, and also gradients in the CryoEM map pulling on each atom to move it towards higher values. In areas where the map is better resolved, the final conformation typically has less variation, as atoms are more likely to move to similar positions. Similarly, where the map is less well-resolved, atoms typically assume a wider range of final positions. At the same time, deviations in the final results also depend on how rigid the model is in various regions; if the model is very rigid in some parts, the variation can also be lower in those regions. Since loops tend to be more dynamic during molecular dynamics simulation, they also usually have higher deviations across different results.
Figure 7.
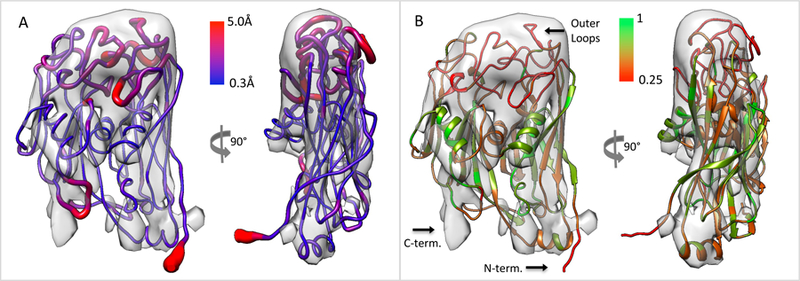
Comparative model Phyre2 flexibly fitted to segmented CryoEM map. (A) The ribbon color and thickness is varied based on the uncertainty in its position after flexible fitting. (B) The ribbon is colored based on conservation of residues across 4 models, including the template and one model from each comparative modeling method, after structure-based sequence alignment (see also Figure S3). The stereochemistry of the model was measured using Molprobity (see Figure S4).
For the SGIV MCP model, the standard deviation ranged between 0.3Å and 11.6Å; in Figure 7A, the highest deviation is capped at 5.0 (~3 standard deviations from the mean). The color coding shows that the highest deviations are indeed in the outer loop region and close to the N and C-termini. Given the map is not as well-resolved in the outer loop part, another way to interpret this modeled uncertainty is that the outer loops may adopt more than one unique conformation. This is a reasonable interpretation given that a Cryo-EM map represents many averaged particles rather than one single one, which indeed may have different conformations.
Structure-based Alignment of Comparative Models
To further compare the models to each other and to the template protein Vp54, a structure-based multiple-sequence alignment between the Vp54 protein and one model from each of the 3 comparative modeling tools was created using the MatchMaker tool in UCSF Chimera. After aligning all comparative models to the Vp54 protein as described above, a structure-based multiple sequence alignment was generated, considering residues that are within 5Å of each other. In this alignment, conservation is measured as a value between 0.25 and 1, with 0.25 meaning that all models have a different type of residue at a given position, and 1 meaning that all models have the same residue type at that position.
The Phyre2 A model is shown in Figure 7B, with the ribbon color coded by conservation. Residues that are within 5Å and more conserved across the models are colored green; these are found mostly in the core double jelly-roll domain which is the core of the protein. Other residues which are not aligned and placed differently by each method fall mostly in the outer loop region, and close to the N and C-termini. Given the higher agreement between the different comparative modeling methods in the core domain of the protein, we thus also expect that this part of the comparative model would be more accurate than the outer loop segments.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Grigore Pintilie (gregp@slac.stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Grouper embryonic cells, derived from Brown-spotted grouper Epinephelus tauvina (Chen et al., 2008; Song et al., 2004), were cultured at 27°C in the Eagle’s minimum essential (as in the cell culture_Method details), maintained and subcultured when the monolayer of cells reached 100% confluence.
Singapore grouper Iridovirus (SGIV), which belongs to Ranavirus genus, Iridoviridae family (Williams, 1996), infects the grouper fish. To maintain the source of SGIV, purified virus or cell lysate of SGIV infected GECs was kept at −80 °C.
METHOD DETAILS
Cell culture, virus infection and virus purification
Grouper embryonic cells (GECs) from the brown-spotted grouper Epinephelus tauvina (Song et al., 2004) were cultured at 27 °C in the Eagle’s minimum essential medium containing a final concentration of 10% fetal bovine serum, 162 mM NaCl, 100 IU of penicillin G per ml, 0.1 mg streptomycin sulfate per ml and 5 mM HEPES. The medium was adjusted to pH 7.4 with NaHCO3. Freshly confluent monolayers of GECs were infected with SGIV at a multiplicity of infection (m.o.i) of approximately 0.5. Infected GECs were detached at day 3 post-infection using cell scrapers and harvested by centrifugation at 500×gfor 5 min. SGIV were first purified with a discontinuous gradient of OptiPrep iodixanol as described earlier (Wu et al., 2010). These SGIV particles were further purified with CsCl gradients. Intact particles were harvested from fractions with densities of 1.3–1.4 g/ml in CsCl.
Cryo-EM and data processing of SGIV
Quantifoil grids (R2/1 in 400 mesh) were used for sample freezing. Approximately 3 μl of purified virus particles was applied onto the grids and flash-frozen into pre-cooled liquid ethane using Vitrobot Mark IV. Grids were blotted for 1 s while the humidity was 100%. Data was recorded on a Gatan Ultrascan 4k×4k CCD on a 300-kV Titan Krios with the effective magnification of 63,290× (2.37Å/pixel), the dose of 16 – 20 e/Å2 per micrograph and the defocus range from 1.5 to 3 μm.
Virus particle images were automatically picked using ethan (Kivioja et al., 2000). The boxed-out particles with the box size of 1200 × 1200 were screened manually using EMAN1 (Ludtke et al., 1999) program boxer to eliminate bad particles. About 20k particles were finally selected, and split in half to create 2 independent reconstructions. The Contrast transfer function (CTF) fitting was performed automatically using fitctf.py (Yang et al., 2009) for good CCD frames without bad drift or astigmatism and then fine-tuned manually using EMAN1 program ctfit. The dataset was divided into two subsets from the beginning and each initial model was built from EMAN1 program starticos. Orientation and center parameters for each particle image were determined by the Multi-Path Simulated Annealing algorithm (Liu et al., 2010), and were used to generate the three-dimensional (3D) icosahedral reconstruction of SGIV using EMAN1 program make3d. The resolution was assessed using the two independent reconstructions (~10k particles each) and the EMAN2 program e2proc3d.py.
Icosahedral Segmentation with iSeg
A simple way to segment radially arranged layers is to separate voxels (or watershed regions) based on their distance (radius) from the center. For icosahedral layers, the distance can be corrected for icosahedral geometry as described previously (Pintilie et al., 2016). To summarize, the vector from the center to the point being considered is projected onto the nearest vector from the center to one of the 12 triangles that make up the icosahedron. The magnitude of the projected vector represents the icosahedrally-adjusted radial distance of the point.
In larger icosahedral capsids, as in the case of SGIV, this approach fails to take into account that there is significant curvature across the icosahedron faces, as shown in Figure S1A. Interestingly, the edges of the icosahedral triangles do not curve, they remain straight. This means that a simple adjustment making the icosahedral geometry spherical does not accurately capture this effect, as shown in Figure S1B.
A more accurate way to capture this effect is to 1) hold each triangular edge fixed, 2) represent the triangular face as a deformable surface and 3) push outwards on the surface while allowing it to deform. We represent a deformable surface by splitting each triangle into smaller triangles; the new triangle vertices become nodes connected by springs, the springs being all the edges. Two parameters are then involved in the simulation of the outward force: the stiffness of the springs and the magnitude of the force applied to each node. Given these two parameters, an equilibrium state is calculated, where the outward force is balanced by the elastic springs (Reddy, 2005). We tested various values for these parameters to find the parameters where the deformed geometry matched the outer layer, as shown in Figure S1C.
The deformed geometry was then scaled to sizes ranging from 0Å to 1200Å; at 1200Å it completely encloses the outer shell. At each size, the cross-correlation between the deformed geometry and the CryoEM map was calculated, assuming uniform values of 1.0 across the surface of the scaled and deformed geometry. This plot is shown in Figures S1D and 2. The radius values corresponding to valleys or inflection points in the plot were then used to separate out watershed regions, resulting in segmented layers that can be extracted using the Extract interface in Segger. The extracted layers are shown in Figure 2.
This method has been integrated into the Segger interface, a plugin to UCSF Chimera (Pettersen et al., 2004a). The interface can be launched from the Segment Map dialog, via the iSeg button in the Shortcuts panel. The plugin and tutorial are available at the following web page: https://cryoem.slac.stanford.edu/ncmi/resources/software/segger.
Segmentation of the outer coat
To segment capsomers in the outer layer, watershed and scale space grouping was used through the Segger interface (Pintilie et al., 2010). After 6 steps of size 1, regions corresponding to each protein in the trimer were obtained. A trimer of the PBCV Vp54 protein, a homologous structure, was then fitted to each capsomer using the Fit to Segments interface in Segger. The fitted models were then used to refine the segmentation, by re-grouping watershed regions based on which fitted model they overlap the most. In the pentasymmetron part of the outer coat, extra densities (colored yellow in Figure 4) were observed, which do not occur in the trisymmetron. These were separated interactively by ungrouping and regrouping.
The other proteins in the outer coat, including zip and anchor proteins, were segmented in a similar manner. However, since no structures are available for these proteins, there was only manual refinement performed on the automatically grouped regions, i.e. 1) regions corresponding anchor proteins, including the trans-membrane part, were manually selected and grouped and 2) regions corresponding to level and arch components of the zip proteins were manually selected and grouped. The exact boundaries of zip and anchor proteins are not clear in this reconstruction, and hence these are estimated boundaries.
Comparative Modeling of MCP
The sequence of the MCP, Orf72, was used as input (the target) into three comparative modeling programs, MODELLER (Eswar et al., 2007), Phyre2 (Kelley and Sternberg, 2009), and I-TASSER (Yang et al., 2015). With MODELLER, the online tutorial was followed to generate the comparative model. The iterative refinement procedure was not applied however. For Phyre2 and I-TASSER, the sequence was simply input into their web interfaces; the resulting models were then downloaded from the result pages. Phyre2 and I-TASSER also do not take as input a CryoEM map, and hence all results are obtained independently of the Cryo-EM reconstruction.
Rigid Fitting of Vp54 and Comparative Models
The Vp54 protein and all comparative models were rigidly fitted into the segmented map of a single coat protein. An rotational search was performed using Fit to Segments (Pintilie and Chiu, 2012). This is illustrated in Figure S2.
Structure-based alignment of Comparative Models
A structure-based sequence alignment was generated using the MatchMaker tool in UCSF Chimera. The tool first aligns two sequences (the BLOSUM-62 matrix was used), then aligns one model to the other so as to minimize the RMSD between aligned residues. Residue pairs that are farther than 2Å are iteratively pruned – this is to improve the alignment of conserved domains (in this case, the double jelly-roll domain). After aligning one comparative model from each method (MODELLER, I-TASSER, and Phyre2) to the template protein Vp54, a structure-based sequence alignment was generated. In this step, residues that are within 5Å of each other from different models are aligned.
Flexible Fitting with MDFF
Comparative models were flexibly fitted into the CryoEM map using molecular dynamics flexible fitting, MDFF (Humphrey et al., 1996; Phillips et al., 2005; Trabuco et al., 2009). The simulation included 10,000 minimization steps followed by 100,000 molecular dynamics steps at a temperature of 300K. The gradient weight, which determines how much force to apply at each atom in the direction of the map gradient, was the default value of 0.3.
Probabilistic Modeling with ProMod
MDFF was applied 10 times independently to one of the comparative models obtained with Phyre2 (further details in the QUANTIFICATION AND STATISTICAL ANALYSIS below). The 10 results were then input into the ProMod tool (Pintilie et al., 2016), obtaining an average model, and standard deviation at each atom position around this average model. The standard deviations are stored in the B-factor column in the deposited PDB file (PDB: 6OJN).
Model Stereochemistry with MolProbity
The flexibly fitted model was also tested for proper stereochemistry using the MolProbity server (Chen et al., 2010). MDFF typically maintains good stereochemistry during the simulation process, and sometimes improves some of the scores, especially the clash score. The final scores (e.g. rotamer and Ramachandran favored) depend to a large degree on the initial model, in this case, the comparative model built based on the Vp54 protein as template. The phenix.geometry_minimization was also applied on the model after MDFF, which improved the scores (Afonine et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
A statistical analysis used in this work is the probabilistic modeling with ProMod detailed above. This analysis measures the mean and standard deviation in the position of each atom across 10 results of the flexible fitting of a comparative model to the Cryo-EM map. In this case, n=10, with n refering to the number of times the flexible fitting procedure was run on the comparative model. Each run was independent of each other except for the input CryoEM map and compartive model. Random motions due to implicit solvent are added by the molecular dynamics simulation; in addition, forces are applied to each atom in the direction of the gradient in the CryoEM map. The reason to use a statistical analysis here is that in each run of MDFF, the resulting model was different, and hence statistical analysis characterizes the possible outcomes.
The ProMod method is further detailed in (Pintilie et al., 2016), which describes how an average model is obtained. The method uses the numpy module in the python programming language, and is provided as a plugin to UCSF Chimera (Pettersen et al., 2004b). The input into this method are the atomic coordinates for each of the 10 resulting models. The results are an average model and a standard deviation at each atom position across each of the 10 resulting models. The average model was deposited as PDB:6OJN and the standard deviations at each atom is stored in the B-factor column. The standard deviation at each atom was averaged to obtain a standard deviation for each residue, which was then used to color code the ribbon shown in Figure 7A. This statistical analysis thus allows us to estimate the uncertainty in the position of each atom/residue in the average model, and also to observe various possible models that explain the observed CryoEM map.
Supplementary Material
Supplemental Movie 1. Related to Figure 3. The composition of the outer coat of SGIV, including 12 pentasymmetrons and 20 trisymmetrons.
Supplemental Movie 2. Related to Figure 6. Comparative model of Orf72 trimer fitted to segmented Cryo-EM map.
Supplemental Movie 3. Related to Figure 6. Comparative models from three methods, flexibly fitted to Cryo-EM segmented maps.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Singapore Grouper Iridovirus (SGIV) | NCBI | GCA_000846905.1 ViralProj14544 |
| Biological Samples | ||
| Singapore grouper iridovius | NCBI | GCA_000846905.1 ViralProj14544 |
| Grouper embryonic cell line | Chen et al., 2008 | RRID: CVCL_S009 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Minimum Essential Medium Eagle | MERCK | M2279 |
| Fetal bovine serum | MERCK | 12103C |
| NaCl | MERCK | 746398 |
| Penicillin-Streptomycin | MERCK | P4333 |
| HEPES | MERCK | H7006 |
| NaHCO3 | MERCK | S5761 |
| Deposited Data | ||
| Structure of major coat protein Vp54 from PBCV. | Nandhagopal et al., (2002). | PDB:1M3Y |
| Structure of PRD1. | Abrescia et al., 2004 | PDB: 1W8X |
| CryoEM maps (entire map, half maps, segmented maps) | This paper. | EMD:20091 |
| Comparative model of Major Coat Protein trimer (Orf72) | This paper. | PDB:6OJN |
| Experimental Models: Cell Lines | ||
| Grouper embryonic cell line | Chen et al., 2008 | RRID:CVCL_S009 |
| Software and Algorithms | ||
| Segger | Version 2.3. Includes the Segment Map, Fit to Segments, iSeg, and ProMod graphical user interfaces. Pintilie et al., 2010. | https://github.com/gregdp/segger |
| MODELLER | Version 9.21. Eswar et al., 2007. |
https://salilab.org/modeller/ |
| I-TASSER | Version 5.1. Yang et al., 2015 |
https://zhanglab.ccmb.med.umich.edu/I-TASSER/ |
| Phyre2 | Version 2.0. Kelley and Sternberg, 2009 |
http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index |
| MDFF | Version 0.4. Trabuco et al., 2009. |
https://www.ks.uiuc.edu/Research/mdff/ |
| NAMD | Vesrion 2.12. Phillips et al., 2005. |
https://www.ks.uiuc.edu/Research/namd/ |
| VMD | Version 1.9.4 Humphrey et al., 1996. |
https://www.ks.uiuc.edu/Research/vmd/ |
| UCSF Chimera | Version v1.14. Pettersen et al., 2004 |
https://www.cgl.ucsf.edu/chimera/ |
| Phenix | Version 1.15. Afonine et al., 2018. |
https://www.phenix-online.org/ |
| MolProbity | Version 4.4. Chen et al., 2010 |
http://molprobity.biochem.duke.edu/index.php |
Highlights.
8.6Å resolution Cryo-EM map of the Singapore Grouper Iridovirus (SGIV)
Major coat proteins form a T=247 icosahedral capsid with tri- and penta-symmetrons
Zip proteins attach to coat proteins, and anchor proteins to a lipid membrane
A model of the coat protein Orf72 was built using comparative modeling
Acknowledgements
This research was partially supported by grants from National Institutes of Health (R01GM079429 and P41GM103832 to W.C.), and the Mechanobiology Institute, National University of Singapore (to C.L.H). Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
Cryo-EM maps along with the comparative model of the Orf72 trimer (from Phyre2) have been deposited to EMDB and PDB with accession codes EMD-20091, PDB ID 6OJN.
The code for Segger and iSeg is available (version 2.3) as a plugin to UCSF Chimera at the following site: https://cryoem.slac.stanford.edu/ncmi/resources/software/segger
Cryo-EM maps along with the comparative model of the Orf72 trimer (from Phyre2) have been deposited to EMDB and PDB with accession codes EMD-20091, PDB ID 6OJN.
References
- Abrescia NGA, Cockburn JJB, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, Fuller SD, Martín CS, Burnett RM, Stuart DI, et al. (2004). Insights into assembly from structural analysis of bacteriophage PRD1. Nature 432, 68–74. [DOI] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD. (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. Struct. Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromenshenk JJ, Henderson CB, Wick CH, Stanford MF, Zulich AW, Jabbour RE, Deshpande SV, McCubbin PE, Seccomb RA, Welch PM, et al. (2010). Iridovirus and Microsporidian Linked to Honey Bee Colony Decline. PLoS ONE 5, e13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SJ, Bamford DH, and Fuller SD. (1995). DNA packaging orders the membrane of bacteriophage PRD1. EMBO J 14, 6078–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar DL, and Klug A. (1962). Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol 27, 1–24. [DOI] [PubMed] [Google Scholar]
- Chen D-H, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, et al. (2011). Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U. S. A 108, 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Tran BN, Lin Q, Lim TK, Wang F, and Hew C-L. (2008). iTRAQ analysis of Singapore grouper iridovirus infection in a grouper embryonic cell line. J. Gen. Virol 89, 2869–2876. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zheng J, and Jiang Y. (1999). A new iridovirus isolated from soft-shelled turtle. Virus Res 63, 147–151. [DOI] [PubMed] [Google Scholar]
- Chinchar VG. (2002). Ranaviruses (family Iridoviridae): emerging cold-blooded killers. Arch. Virol 147, 447–470. [DOI] [PubMed] [Google Scholar]
- Chinchar VG, Hyatt A, Miyazaki T, and Williams T. (2009). Family Iridoviridae: poor viral relations no longer. Curr. Top. Microbiol. Immunol 328, 123–170. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M-Y, Pieper U, and Sali A. (2007). Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci Editor. Board John E Coligan Al Chapter 2, Unit 2.9. [DOI] [PubMed]
- Granoff A, Came PE, and Rafferty KA. (1965). The isolation and properties of viruses from Rana pipiens: their possible relationship to the renal adenocarcinoma of the leopard frog. Ann. N. Y. Acad. Sci 126, 237–255. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K. (1996). VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Jancovich JK, Mao J, Chinchar VG, Wyatt C, Case ST, Kumar S, Valente G, Subramanian S, Davidson EW, Collins JP, et al. (2003). Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mortalities in North America. Virology 316, 90–103. [DOI] [PubMed] [Google Scholar]
- Jancovich JK, Bremont M, Touchman JW, and Jacobs BL. (2010). Evidence for Multiple Recent Host Species Shifts among the Ranaviruses (Family Iridoviridae). J. Virol 84, 2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, and Sternberg MJE. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc 4, 363–371. [DOI] [PubMed] [Google Scholar]
- Kivioja T, Ravantti J, Verkhovsky A, Ukkonen E, and Bamford D. (2000). Local average intensity-based method for identifying spherical particles in electron micrographs. J. Struct. Biol 131, 126–134. [DOI] [PubMed] [Google Scholar]
- Liu X, Rochat RH, and Chiu W. (2010). Reconstructing Cyano-bacteriophage P-SSP7 structure without imposing symmetry. Protoc. Exch
- Ludtke SJ, Baldwin PR, and Chiu W. (1999). EMAN: Semiautomated Software for High-Resolution Single-Particle Reconstructions. J. Struct. Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- Nandhagopal N, Simpson AA, Gurnon JR, Yan X, Baker TS, Graves MV, Van Etten JL, and Rossmann MG. (2002). The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc. Natl. Acad. Sci. U. S. A 99, 14758–14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE. (2004a). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE. (2004b). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, and Schulten K. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie G, and Chiu W. (2012). Comparison of Segger and other methods for segmentation and rigid-body docking of molecular components in cryo-EM density maps. Biopolymers 97, 742–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie G, Chen D-H, Haase-Pettingell CA, King JA, and Chiu W. (2016). Resolution and Probabilistic Models of Components in CryoEM Maps of Mature P22 Bacteriophage. Biophys. J 110, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie GD, Zhang J, Goddard TD, Chiu W, and Gossard DC. (2010). Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol 170, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price SJ, Garner TWJ, Nichols RA, Balloux F, Ayres C, Mora-Cabello de Alba A, and Bosch J. (2014). Collapse of Amphibian Communities Due to an Introduced Ranavirus. Curr. Biol 24, 2586–2591. [DOI] [PubMed] [Google Scholar]
- Raghava G, and Barton GJ. (2006). Quantification of the variation in percentage identity for protein sequence alignments. BMC Bioinformatics 7, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J. (2005). An Introduction to the Finite Element Method (New York, NY: McGraw-Hill Education; ). [Google Scholar]
- Reteno DG, Benamar S, Khalil JB, Andreani J, Armstrong N, Klose T, Rossmann M, Colson P, Raoult D, and Scola BL. (2015). Faustovirus, an Asfarvirus-Related New Lineage of Giant Viruses Infecting Amoebae. J. Virol 89, 6585–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Qin QW, Qiu J, Huang CH, Wang F, and Hew CL. (2004). Functional Genomics Analysis of Singapore Grouper Iridovirus: Complete Sequence Determination and Proteomic Analysis. J. Virol 78, 12576–12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf M, Baker ML, Marti-Renom MA, Chiu W, and Sali A. (2006). Refinement of protein structures by iterative comparative modeling and CryoEM density fitting. J. Mol. Biol 357, 1655–1668. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, and Schulten K. (2008). Flexible Fitting of Atomic Structures into Electron Microscopy Maps Using Molecular Dynamics. Struct. Lond. Engl 1993 16, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Schreiner E, Harrison CB, and Schulten K. (2009). Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods San Diego Calif 49, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T. (1996). The iridoviruses. Adv. Virus Res 46, 345–412. [DOI] [PubMed] [Google Scholar]
- Wimley WC. (2003). The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol 13, 404–411. [DOI] [PubMed] [Google Scholar]
- Wu J, Chan R, Wenk MR, and Hew C-L. (2010). Lipidomic study of intracellular Singapore grouper iridovirus. Virology 399, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Yu Z, Zhang P, Battisti AJ, Holdaway HA, Chipman PR, Bajaj C, Bergoin M, Rossmann MG, and Baker TS. (2009). The capsid proteins of a large, icosahedral dsDNA virus. J. Mol. Biol 385, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Jiang W, Chen D-H, Adiga U, Ng EG, and Chiu W. (2009). Estimating contrast transfer function and associated parameters by constrained non-linear optimization. J. Microsc 233, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xiang Y, Dunigan DD, Klose T, Chipman PR, Van Etten JL, and Rossmann MG. (2011). Three-dimensional structure and function of the Paramecium bursaria chlorella virus capsid. Proc. Natl. Acad. Sci. U. S. A 108, 14837–14842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1. Related to Figure 3. The composition of the outer coat of SGIV, including 12 pentasymmetrons and 20 trisymmetrons.
Supplemental Movie 2. Related to Figure 6. Comparative model of Orf72 trimer fitted to segmented Cryo-EM map.
Supplemental Movie 3. Related to Figure 6. Comparative models from three methods, flexibly fitted to Cryo-EM segmented maps.