

Abstract
Cystic fibrosis (CF) is both the most common and most lethal genetic disease in the Caucasian population. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and is characterized by the accumulation of thick, adherent mucus plaques in multiple organs, of which the lungs, gastrointestinal (GI) tract and pancreatic ducts are the most commonly affected. A similar pathogenesis cascade is observed in all of these organs: loss of CFTR function leads to altered ion transport, consisting of decreased chloride and bicarbonate secretion via the CFTR channel and increased sodium absorption via ENaC channel upregulation. Mucosa exposed to changes in ionic concentrations sustain severe pathophysiological consequences. Altered mucus biophysical properties and weakened innate defense mechanisms ensue, furthering the progression of the disease. Mucins, the high-molecular-weight glycoproteins responsible for the viscoelastic properties of the mucus, play a key role in the disease but the actual mechanism of mucus accumulation is still undetermined. Multiple hypotheses regarding the impact of CFTR malfunction on mucus have been proposed and are reviewed here: 1) dehydration increases mucin monomer entanglement, 2) defective Ca2+ chelation compromises mucin expansion, 3) ionic changes alter mucin interactions and 4) reactive oxygen species (ROS) increase mucin crosslinking. Although one biochemical change may dominate, it is likely that all of these mechanisms play some role in the progression of CF disease. This article discusses recent findings on the initial cause(s) of aberrant mucus properties in CF and examines therapeutic approaches aimed at correcting mucus properties.
Keywords: Mucus, Mucins, Polymeric Network, CFTR, Biochemical Interactions, Viscoelastic Properties
Introduction:
CF is a multi-organ disease with symptoms affecting tissues that express CFTR and produce mucus, among those, the lungs and the GI tract1–3. Mucus accumulation in the airways, the intestine, and the pancreatic ducts plays a critical role in the disease pathogenesis by compromising airflow and nutrient digestion. Since the resulting progressive lung disease can lead to respiratory failure and ultimately death, this review focuses on studies conducted on airway mucus, more specifically on secreted mucins lining the respiratory tree. Mucins are complex macromolecules that govern the biophysical properties of mucus. In the lungs, gel-forming mucins are secreted by goblet cells distributed throughout the conducting airways and submucosal glands located in the large airways4,5. Airway goblet cells secrete MUC5B and MUC5AC to produce a thin mucus layer that lines the epithelial surfaces. In contrast, submucosal glands secrete only MUC5B, which is expulsed from the gland ducts in the form of strands intended to sweep the large airways and remove inhaled pathogens6,7. Although both airway surfaces and glands produce mucus, the biophysical and biochemical properties of mucus produced by these two compartments may be affected differently by CFTR malfunction and therefore may play distinct roles in the progression of the CF lung disease7. In CF animal models that possess submucosal glands (e.g., pigs and rats), gland hypotrophy and plugging occur at different ages (i.e., newborn vs 6 months) and therefore lung/gland maturation may play a critical role in the progression of the CF lung disease8,9. Since the specific role of gland mucus strands in disease is still being elucidated, this review will focus on current knowledge of the impact of CFTR malfunction on the ambulatory mucus layer progressing on top of the cilia8,10.
The viscoelastic properties of the mucin gel lining the airways are critical for proper trapping and clearance of pathogens. Gel properties rely heavily on cys-rich regions scattered throughout the mucin protein backbone to organize a complex disulfide-stabilized polymeric network11. Adding to the complexity, the physicochemical properties of mucins are furthermore governed by the large O-linked oligosaccharide chains decorating the apomucin core, which can contribute up to 80% of the mucin molecular mass12. The O-glycans are designed to ensure high volume occupancy in solution as well as high water-holding capacity for mucin gels and are important for regulating mucin-mucin, mucin-pathogen, and mucin-mucosal surface interactions13. Although mucins are produced to protect the mucosa against pathogens, dysregulation of mucin secretion rate, concentration, expansion, and/or interactions can compromise the protective role of the mucus layer. In this review, we discuss how CFTR malfunction can affect mucin biochemical interactions and alter the viscoelastic properties of mucus. Mucins and mucin gel formation are briefly described herein, followed by in-depth examination of the impact of CFTR on mucin concentration, polymer compaction, mucin-mucin interactions, and the impact of oxidative stress on the mucin network. We conclude by opening the discussion on current and novel pharmacological approaches aimed at altering the mucin network (i.e., mucoactive agents).
1. Mucus and Mucins:
Healthy airway mucus is composed of approximately 90-95% water, 1-5% mucins and other proteins, 1-2% lipids and 1% salt electrolytes12. Although hundreds of proteins have been discovered in airway mucus, mucin proteins primarily govern the viscoelastic gel properties of the mucus layer11,12,14. Mucins are large (up to 50 MDa) and heavily glycosylated, but polymeric organization and intracellular packaging are tightly regulated to ensure the rapid release of high-quality viscoelastic gels12,15. Understanding the structure and organization of a polymeric mucin gel is critical to investigating how CFTR malfunction can affect its biochemical and biophysical properties.
1.1. Mucin Structure
Mucins are encoded by MUC genes, a gene family consisting of 18 different proteins16. Mucins can be divided into two groups: the secreted (or gel forming) and the tethered (or membrane-bound) mucins. MUC5B and MUC5AC are the major gel-forming mucins expressed in the airways and are predicted to possess different airway clearance functionalities6,7. In the GI tract, MUC2 is the primary gel-forming mucin expressed that, along with lesser amounts of MUC6, protects the epithelium from acidic pH, lubricates the intestine to facilitate transit, and shields the mucosa to prevent pathogen invasion17. All gel-forming mucins share a similar core protein, referred to as the apomucin. The protein core features von Willebrand factor (vWF) domains in the N- and C-terminal regions, cysteine-rich domains (CysD) scattered throughout the protein backbone including the cysteine knot (CK) at the C-terminal end, and a variable number of tandem repeat (VNTR) region that undergoes extensive glycosylation and constitutes the bulk of the protein mass (Figure 1).
Figure 1. Mucin domains govern polymeric network organization.
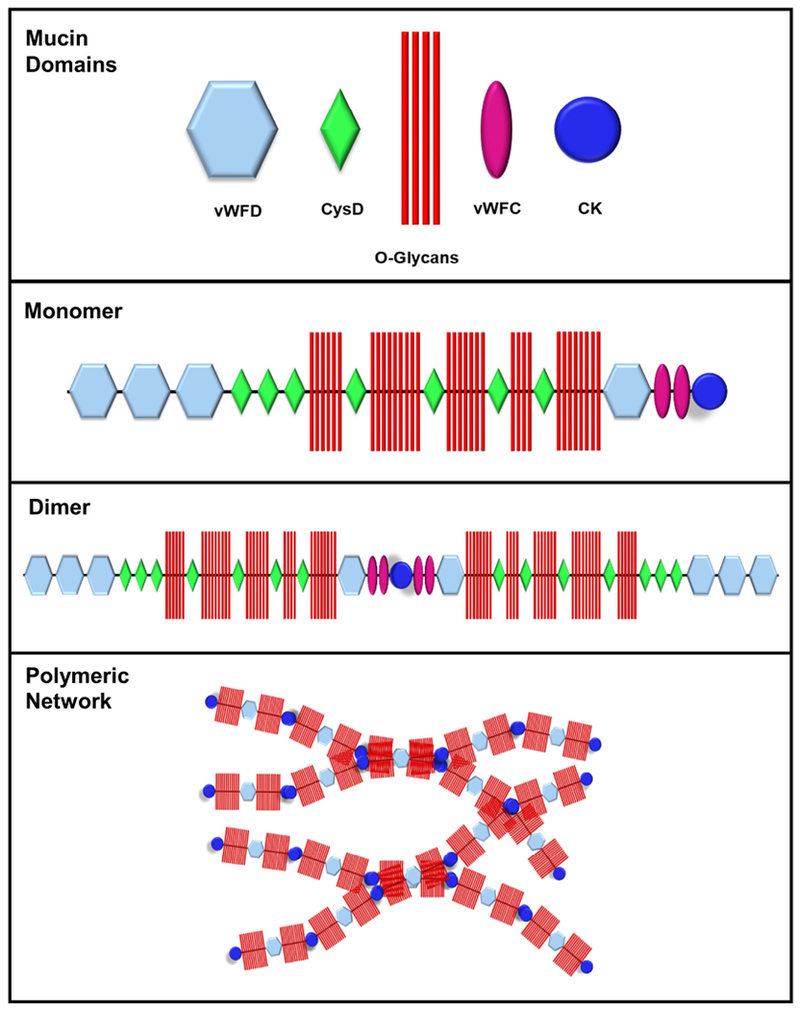
Mucin structure and assembly rely on von Willerbrand Factor (vWF) D and C domains in the N- and C-terminal regions and cysteine-rich domains (CysD) scattered throughout the protein backbone including the cysteine knot (CK) at the C-terminal end. The protein core, which is rich in serine and threonine residues, undergoes post-translational O-glycosylation (O-glycans) to form mature mucin monomers. Dimer formation ensues in the endoplasmic reticulum and is mediated by disulfide linkage of the CK domains to form linear, interwoven polymeric networks.
An important process for the biophysical properties of mucin gels is the polymeric organization of the mucus, a process dependent on the capacity of mucin monomers to form dimers and multimers via C- and N-terminus disulfide bond formation, respectively11. CK domains in the C-terminus are responsible for monomer dimerization, while vWF domains in the N-terminus are responsible for the formation of linear polymers and/or more complex multimeric networks11,15. In CF, extracellular DNA originating from dying inflammatory cells entrapped in mucus can be present in high concentrations and add to the complexity and entanglement of the polymeric mucin network, increasing gel viscosity and worsening symptoms of the disease18.
During mucin protein maturation, O-glycosylation of serine and threonine residues distributed through the VNTR occurs in the endoplasmic reticulum. This is a critical step in establishing the biological properties of mucins, as oligosaccharide composition and terminal sugar negative charges contribute to mucus swelling via electrostatic interactions and pathogen trapping via specific binding13,19,20. Glycosylation is initiated by the linkage of N-acetyl-D-galactosamine, one of three steps for mucin glycosylation that can yield eight different core structures. Following the glycan core addition, a backbone structure is linked followed by the addition of a peripheral terminal sugar, which is the source for mucin glycan variation. The number of glycans per amino acid, their distribution pattern, and their size vary from mucin to mucin, giving each mucin a unique glycosylation profile and therefore specific biological properties13.
Recent work has shown that mucin proteins interact with hundreds of other proteins present in the mucus layer, thus further affecting mucus viscoelasticity and giving rise to the term “the mucus interactome” to describe the relationship between globular proteins and mucins21.
1.2. Mucin Sources
In the lungs, gel-forming mucins are secreted by surface goblet cells and submucosal glands. Unlike goblet cells that produce both MUC5AC and MUC5B, submucosal glands only secrete MUC5B4,7. These two mucin sources may play different roles in health and disease, but additional work is needed to determine their individual functions in the lungs. Mucins at the surface epithelium are secreted constitutively at basal levels to facilitate airway maintenance but can also be acutely stimulated for extensive degranulation following exposure to a challenge like smoke, allergens, or pathogens22,23. Complex cholinergic and purinergic signaling pathways control the rate of mucin granule release from the glands and the epithelial surfaces, respectively. In addition, morphological variation has been noted between mucus released from submucosal glands and goblet cells6,7. Under cholinergic stimulation, MUC5B is slowly released from submucosal glands and forms “strands or bundles” with diameters of 5-50μm. These strands are then “coated” with thin MUC5AC sheets following their release from goblet cells, resulting in a MUC5B core bundle enveloped by an outer layer of MUC5AC6,7. It has been postulated that these unique structures are tailored for mucociliary transport of large (>300 μm) inhaled particles10. In contrast, surface goblet cells possess heterogeneous populations of MUC5AC-rich and MUC5B-rich granules within the same cell, and granule release can be regulated by different exocytotic pathways (i.e., basal vs. stimulated), suggesting that mucin secretion is a delicate event that requires accuracy and precision22.
Several signaling pathways can initiate mucin granule exocytosis, such as epidermal growth factor (EGFR), toll-like-receptors (TLRs), and cholinergic (Ach) and purinergic (P2Y2) stimulation23. Mucin production can be signaled by extracellular-regulated kinase (ERK) and activation of nuclear factor κB (NF- κB)23. Cytokines like IL-13 and IL-1 are also important regulators of mucin hypersecretion and influence goblet cell hyperplasia24,25. If airway challenges persist, goblet cell hyperplasia, metaplasia, and gland hypertrophy can ensue, substantially increasing the amount of airway mucus produced. These phenotypes are commonly observed in muco-obstructive diseases such as chronic bronchitis, COPD, asthma, and CF26.
1.3. Mucin Network Organization and Interactions
The structure and function of mucin gels lends itself to a diverse array of potential interactions. Although mucus consists of up to 95% water, its viscoelastic gel properties are the result of disulfide bonds, hydration capacity, and non-covalent interactions. The intracellular environment and extracellular milieu dictate how mucin packing, expansion, and interaction transpire. Recent investigations into intracellular arrangement and post-secretory expansion have provided key insights into how disease states can alter mucin interactions.
Mucin exocytosis from goblet cells is extremely rapid and can take only tens of milliseconds to occur once initiated20. Prior to exocytosis, these large biopolymers, which can reach up to 109 Da, are tightly packed inside granules ranging in size from 0.5 to 2 μm, and, for this reason, mucin storage requires a high level of organization for both packaging and subsequent expansion15,27. The packing of linear MUC5B polymers is governed by high Ca2+ concentration and acidic pH inside the mucin granules. The presence of calcium and hydrogen ions inside the granules shields the negatively charged glycans decorating the apomucins and prevents electrostatic repulsion, allowing the mucin polymers to organize into nematic arrangements15,20. The resulting compacted MUC5B possesses a central N-terminus core and outward facing C-termini, allowing for the formation of linear strands upon release, a feature advocated to be essential to its putative function of cleaning large inhaled particles10. The intracellular organization of MUC5AC is similar, with C-terminal dimerization occurring via disulfide bonds in the ER followed by covalent linkage of the N-termini in the Golgi and analogous packing28.
Upon exocytosis, mucin macromolecules undergo dramatic volume expansion (up to 4000-fold), a rapid and critical process that relies on Ca2+ chelation and osmotic pressure20. To ensure a fast swelling rate upon exocytosis, organized folding and unfolding of the mucins is required to avoid extreme frictional heat generation20. The extracellular environment is more alkaline, higher in bicarbonate and sodium ion concentration, and lower in calcium ion concentration. Upon opening of the granule in extracellular milieu, hydrogen bonds that stabilized the mucins intracellularly are broken due to increased pH. Simultaneously, calcium ions are chelated by bicarbonate and exchanged for sodium ions, promoting additional relaxation of the mucin network20,29. These ionic changes lead to a large internal osmotic gradient and subsequent influx of water. Swelling stops once an equilibrium between osmotic pressure and the intrinsic elastic component of the matrix is reached.
Once secreted and expanded, mucins are subject to a variety of intermolecular interactions. Acidic side chains and negatively charged polysaccharides contribute to the intermolecular formation of stabilizing salt bridges and electrostatic interactions30,31. Adding to the complexity, as the CF disease progresses mucus biophysical properties are altered by reactive oxygen species (ROS) and/or airway inflammation32.
To summarize, the noncovalent and covalent interactions, coupled with concentration-dependent interpenetration of the mucin polymers, help shape the rheological landscape for mucin gel networks. In the next section, we will discuss how CFTR dysfunction and restoration of function can impact the mucin network with regards to physical, biochemical, and biological interactions.
2. Mucus and Mucin Interactions at the CF Airway Surface:
Static mucus, as observed on CF airway surfaces, is an ideal environment for bacterial colonization as well as inflammation with the recruitment of neutrophils that results in irreversible lung damage33. Understanding the biochemical processes that occur once mucins are secreted into the airway surface liquid (ASL) layer and how they relate to the impairment of mucociliary clearance is crucial to developing effective treatments for patients with CF. Several hypotheses have been proposed to explain how CFTR malfunction affects the mucin polymeric network (see Figure 2). Aberrant CFTR-mediated Cl− and HCO3− secretion and dysregulation of ENaC-mediated Na+ transport results in ASL water hyperabsorption and subsequent dehydration of the mucus layer. Hyperconcentrated mucins in the ASL are subject to increased polymeric entanglement with new/increased solute-mucin interactions resulting from a concentration increase of non-salt molecules in the ASL. In parallel, failure of CFTR to transport bicarbonate can lead to acidification of the ASL and changes in the ionic interactions within the mucus layer. Oxidative stress and inflammation can result in atypical covalent mucin interactions and potentially change the viscoelastic properties of the gel. All of these physicochemical changes occur at the airway surface and can alter the viscoelastic properties of the mucus.
Figure 2. Model of normal vs. CF airway mucus layer illustrating changes within the mucus network (i.e., polymer entanglement, mucin compaction, and/or changes in molecular interactions) in response to altered ionic fluxes.
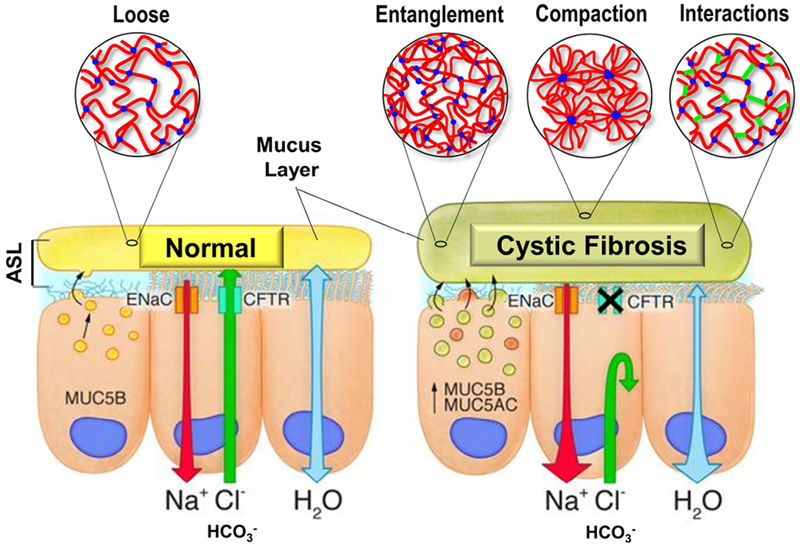
In normal individuals, CFTR function ensures proper Cl− and HCO3− secretion as well as regulates Na+ absorption via the downregulation of the ENaC channel, controlling water flux through the epithelium. A thin mucus layer is produced by airway goblet cells with optimal biophysical properties (e.g., loose transportable) for airway clearance. In CF, reduced Cl− and HCO3− secretion and increased Na+ absorption can alter the biochemical interface of the mucin network in different ways. Mucus layer hyperconcentration causes decrease in mucus mesh size (e.g., entanglement). Impaired Ca2+ sequestering prevents mucin expansion (e.g., compaction). Changes in hydration, pH, and oxidative stress can introduce additional ionic, hydrogen, hydrophobic, and disulfide bonds (e.g., interactions).
2.1. Hyperconcentration and Mucus Gels
Dysfunctional CFTR regulation of ENaC-mediated Na+ transport is accepted as the driving force behind the decreased ASL volume in CF airways, which ultimately increases the osmotic pressure of the mucus layer and collapses the cilia34,35. In addition to altered ion and fluid transport, the CF airways are characterized by goblet cell and glandular hyperplasia and subsequent overproduction of MUC5B and MUC5AC26. As a result, higher mucus concentrations have been reported in both CF patients and CF model systems9,36–38. Coupled together, mucus hypersecretion and airway dehydration produce an ASL with percent solids reaching 5-times that of normal levels37,38.
From a polymer physics point of view, two critical concentration-dependent transition points contribute to the biophysical behavior of mucins in the ASL: the semi-dilute overlap concentration (c*) and the entanglement concentration (ce)34. The transition from un-interacting oligomers to an un-entangled regime, characterized by mucin chain overlap, is reported to occur at roughly c* = 1 mg/ml for MUC5AC and MUC230,39. These mucins transition from the un-entangled overlapping regime to an entangled regime characterized by mucin chain interpenetration and reptation at ce of 25 mg/ml and 30 mg/ml for MUC5AC and MUC2, respectively39. The incident regimes under normal mucin concentration and pH values physiologically relevant to the lungs (i.e. ~2% solids and 7.0 < pH < 7.2) reflect a semi-dilute overlapping network, whereas the gastric mucus is normally an entangled gel in the normal mucin concentrations and low pH values of the stomach (pH < 2)38. MUC5AC viscosity scales with concentration and behaves as a purely viscous fluid at concentrations under 25mg/mL (~3% solids including airway salts). Above 25 mg/mL (~3.5% solids), MUC5AC solutions become viscoelastic, exhibiting both viscous and elastic behavior. Since CF mucus regularly reaches concentrations above 5% solids, it falls into the latter category, i.e., dominated by elastic gel-like behavior34,37.
The relationship between physiologically relevant mucin concentrations and rheological properties was recently examined and revealed that airway mucus complex viscosity scales with concentration38. Furthermore, macro and microrheology performed on bovine submaxillary mucus (BSM), porcine gastric mucus (PGM), and CF and non-CF human bronchial epithelial (HBE) cell-culture mucus over a range of concentrations showed consistency across mucus types38. Geometric scaling was consistent between concentration and complex viscosity, and this held true over the pH range of 6-8. This relationship to concentration also correlated to changes in mucociliary transport (MCT) and ASL osmotic properties. Hydration therapy (returning to ~2% solids) profoundly corrected CF sputum samples to near-normal viscoelasticity, reinforcing the clinical findings that administration of hydrating agents such as hypertonic saline yields beneficial results in CF patients40–42 (see section 3.1).
A secondary consequence of increased mucin concentration in CF airways is that the increased proximity of mucin polymers results in new/stronger interactions with other protein and ionic solutes that can also affect the viscoelasticity of the gel31. Indeed, while physiological mucus behavior is largely governed by the amount of available solvent, mucin chain dynamics in solution are also dependent on amount of noncovalent interactions (i.e. steric hindrance from charged residues and/or glycans, as well as intermolecular salt bridges). Addition of salt to MUC5AC networks can increase the viscosity of the gel. This has been attributed to stronger intermolecular interactions taking place as a result of reduced electrostatic repulsion of the polysaccharide side chains in the presence of salt31 (this is discussed in greater detail in section 2.3). Chaotropic agents such as guanidine HCl and urea also reduce mucus viscoelasticity30. This is due to the disruption of hydrophobic interactions and the subsequent unfolding of the mucin protein backbone. Accordingly, the next sections discuss the relevant mucin-solute interactions that contribute to airway mucus physiology and CF pathophysiology.
2.2. Ca2+ Chelation & Mucus Compaction
While predominantly a Cl− channel, CFTR also plays a general anion channel role, and therefore, mutations within CFTR can further influence the biophysical properties of mucus beyond dehydration43. In particular, much attention has been given to the channel permeability to HCO3-, both in terms of pH and polycation chelation.
Inside mucin granules, the environment is rich in Ca2+ ions and low in pH. Mucin chains are condensed in the granule as nematic arrays via counterion shielding15,20. The negatively charged glycans that would normally repel each other are shielded by Ca2+ ions. A single Ca2+ ion can neutralize two negative charges, supporting the formation of a compact intragranular mucin matrix with adjacent terminal glycans. It has also been proposed that the vWD domains in the N-terminus selectively bind calcium during granule packing15. Upon exocytosis, Ca2+ ions are exchanged for two Na+ ions, as Na+ concentration predominates by over an order of magnitude in the ASL20,26. This local increase in internal cation concentrations due to Na+ influx draws in water and causes swelling of the matrix via a Donnan effect. Simultaneously, bicarbonate, also abundant in healthy ASL, chelates the Ca2+ ions that were exchanged for Na+, effectively sequestering the Ca2+ from reforming an ionic crosslink between charged mucin glycans.
Improper calcium chelation has been noted as a possible cause for CF pathogenesis3. Irregular expansion of secreted mucins is seen in CF models and can be attributed to ionic imbalances, particularly related to the lack of bicarbonate and chloride transport through CFTR29,44,45. Incompletely unfolded MUC5B was detected in CF saliva, suggesting that terminal unpacking of secreted mucins occurs on longer timescales than previously thought46. Persistent deficiency of key counterions in CF likely contributes to keeping the secreted mucins in a semi-expanded state causing mucus stasis and plugging, though hyperconcentration in the ASL was shown to be the predominate cause of incomplete mucin maturation46.
Counterion donation during unpacking also affects MCT rates. Addition of zwitterionic HEPES buffer had no effect on MCT, whereas Tris buffer or addition of bicarbonate increased MCT in CF rats, and bicarbonate addition to ileum sections of CF mice normalized mucus properties9,45. Hence, identifying the counterions involved in the mucin unfolding and subsequent viscoelastic properties of secreted mucins is critical. Addition of calcium to pig tracheas increased ASL viscosity, but other divalent ions like magnesium and zinc had no rheological effect47. This distinction is important when considering the development of chelating compounds as therapies for CF. Although calcium chelation during mucin exocytosis may be crucial to the unfolding of the mucin, a continuous low-calcium environment is necessary to promote normal rheological properties47. If altered ionic fluxes persist after secretion, normally unfolded mucins can begin to interact in an aberrant manner, possibly causing increases in ASL viscosity.
2.3. Acidic pH & Mucus Gels:
The role of CFTR in bicarbonate transport and subsequent acidification in CF airways and models has been known for years48. Mucus pH in CF nasal airways has been measured as low as ~6.5, and in vitro models of CF have shown greater susceptibility to pH changes than controls49,50. Even small changes in hydrogen ion concentration may have important consequences given how resiliently the body regulates pH and have been implicated in the increased susceptibility of CF patients to infection51.
Recent work showed an inverse correlation between the pH and viscosity of MUC5AC gels30,31. MUC5AC begins to act more like a solid at low pH levels (pH < 4), shown rheologically by a decrease in tan (δ), the ratio of the loss modulus (G”) to the storage modulus (G’). The storage modulus increases with decreasing pH, indicating that the mucus becomes more gel-like, evidence of additional cross-linking and/or increased mucin interaction in the gel-phase. Biochemically, this can be explained by protonation of the carboxyl side chains of residues such as glutamic and aspartic acid, leading to disruption of intramolecular salt bridges and causing the mucin to unfold. Unfolding of the mucin exposes hydrophobic sites hidden in the native conformation of the protein. The newly exposed hydrophobic domains govern the interaction of the now-neutrally charged mucin molecules, reducing the elasticity and increasing the viscosity of the MUC5AC gel. This is in agreement with additional studies on microrheology of gastrointestinal mucins (MUC2) at low pH levels30. Salt bridge protonation and hydrophobic interactions are likely the driving forces behind the sol-gel transition of mucins below pH 2.
These pH ranges are physiologically relevant to the stomach and portions of the GI tract but not to human airways. Therefore, a few important distinctions must be made when comparing data from recent publications. While the protonation of carboxyl side chains in the protein backbone is a probable mechanism for increasing intermolecular interactions and gel viscosity, it is crucial to note that the pKa’s of glutamatic and aspartic acid are 4.15 and 3.71, respectively. At pH values of ~2, such as in the stomach, these side chains are ~99% protonated. However, with hydrogen ion concentrations 104-106 lower (i.e., pH 6.5), such as in the lungs, the carboxyl side chains of the acidic amino acids remain deprotonated. Examination of porcine ASL viscosity in the pH range of ~6.5-8.0, much more physiologically relevant to human airways, revealed that ASL viscosity at the small length-scales probed by fluorescence recovery after photobleaching (FRAP) assays increased slightly under acidified conditions47. However, since increased ASL viscosity as a function of acidity did not correlate with disulfide bond formation nor bicarbonate concentration, it is likely due to altered electrostatic interactions between mucin molecules. Although viscosity changes were modest (e.g., 4-6 fold) compared to the several-log fold changes with respect to concentration reported in GI and airway mucus studies, small changes in pH can have critical physiological consequences and contribute to CF pathogenesis by altering MCT30,38.
2.4. ROS & Mucus Crosslinking:
Evidence of increased levels of ROS in CF patients has been documented for decades and is thought to play a role in the progression of CF pulmonary dysfunction52. Inflammatory immune cells, notably polymorphonuclear (PMN) neutrophils, produce oxidants (such as superoxide anion, hydrogen peroxide, and hypochlorous acid) when defending against infection52. Myeloperoxidase (MPO), a pro-oxidant enzyme secreted by PMN neutrophils, is present in the ASL of CF patients and methionine sulfoxide, a byproduct of MPO oxidation, has been correlated with early CF lung disease and bronchiectasis52,53. This is of particular interest because recent findings showed that neutrophil-dominated inflammation occurs in the absence of infection36,54. Additionally, lower concentrations of the antioxidant glutathione (GSH) were measured in CF bronchoalveolar lavage fluids, furthering the potential for oxidative damage55.
Limited data exists on the effects of ROS on mucins. While cysteine residue distribution for the same mucus concentration was similar between CF and non-CF controls, disulfide bond concentration was higher in the CF population and correlated with ROS levels32. Furthermore, simulated crosslinking of healthy mucus samples with DMSO or oxygen (O2 gas mimicking treatment for hypoxia) increased the elastic modulus of the solution32. Disulfide bond formation is a crucial step to proper mucin synthesis and function; however, excessive disulfide bridging may play a role in the pathophysiology of muco-obstructive diseases. The likely explanation is that high levels of MPO induce posttranslational oxidative modifications of cysteine residues. In CF and asthma, inflammatory enzymes released by neutrophils and eosinophils, respectively, initiate the oxidation of free thiols/cysteine to form additional disulfide bridges or cystines32,56. The reaction is speculated to involve myeloperoxidase (MPO) or eosinophil peroxidase (EPO) entrapped in mucus that catalyzes the reaction of H2O2 with thiocyanate to generate potent local oxidants. The subsequent increase in mucin disulfide interactions causes polymer network crosslinking and stiffening of the mucus gel. The viscoelastic changes resulting from mucus oxidization were of a similar magnitude to that of 5-fold concentration changes. These bonds are potential therapeutic targets and can be broken by reducing agents such as NAC, dithiothreitol (DTT), and tris(2-carboxyethyl)phosphine (TCEP) (see section 3.5).
3. Pharmacological Approaches Targeting Mucus:
Improvement of mucus clearance in CF is key to preventing declines in lung function. CFTR-directed therapies have been shown to improve MCC in vivo, with ivacaftor producing a ~10% increase in MCC in patients with a G551D mutation57,58. In the age of CFTR-corrective therapies, patients with mutations not responsive to modulator compounds (e.g., nonsense mutations) will require symptomatic treatment via CFTR-independent approaches. In addition, patients treated with CFTR modulators may still benefit from therapies improving clearance as mucus plugging will persist due to residual infections and permanent lung damage. In recent years, therapeutic strategies aimed at changing the viscoelastic properties of mucus have gained momentum. Mucus provides a relevant therapeutic target for all patients regardless of genotype (unlike CFTR modulators) and/or inflammatory status (unlike dornase alfa). Compounds that change the biophysical properties of mucus are commonly called “mucoactive” but do not always work directly on mucins (e.g., dornase alfa breaks down extracellular DNA). As a result of the complexity of mucus and the intricate interactions of the mucin network, a wide range of pharmacological approaches, including osmotic agents, ion channel potentiators and inhibitors, Ca2+ chelators, surfactants, and reducing agents are currently being tested or have been approved and are discussed in this section.
3.1. Osmotic Agents
Osmotic agents, compounds that cause water to be drawn into the airway surfaces, have the potential to treat muco-obstructive diseases like CF and COPD by “reversing” the effects of water hyperabsorption via the ENaC channel. Inhalation of hypertonic saline (HS) is clinically efficacious although the precise mechanism of action has not been completely elucidated40–42. The current hypothesis is that administration of HS causes fluid influx into the airway lumen. Once water flows into the lumen, it hydrates and swells the ASL, with the magnitude of swelling being governed by mucus concentration (i.e., the higher the mucus concentration, the longer the duration of ASL swelling)42. Hence, mucus hyperconcentration in CF provides an additional osmotic driving force, potentially via counterions on the mucin glycans. Interestingly, ENaC inhibition increased the response to HS in vitro, indicating that sodium reabsorption likely diminishes the effect of aerosolized HS, which establishes another therapeutic target in CF (see section 3.2).
Supported by a straightforward mechanism of action, many clinical trials have tested the effect of osmotic agents on lung function and MCC in patients with obstructive lung disease. Hypertonic saline (7% NaCl) was first shown to be clinically effective over a decade ago40,41. Administration of HS was shown to increase lung function (FVC and FEV1) and decrease pulmonary exacerbations in CF patients41. HS was also shown to increase MCC in a sustained fashion in vivo and expedited symptom resolution during hospitalization40,41.
Another osmotic agent, mannitol, has also shown clinical potential to treat obstructive airway diseases and has been approved in the E.U., Australia, Israel, and recently the USA. Mannitol is an inert sugar that is not absorbed by the GI tract, not metabolized, and does not cross the blood-brain barrier. The mechanism of action is postulated to be similar to HS, exerting its effects by creating an osmotic gradient and drawing water into the airway lumen. In contrast to the Na+ in HS, mannitol has no transcellular absorption pathway. While Na+ is hyperabsorbed in CF, mannitol absorption rates are unaffected by the ion transport defects in CF, possibly explaining why it contributes to decreased paracellular small-molecule probe absorption rates as measured using gamma scintigraphy59. Inhalation of dry powder mannitol increased FEV1 in CF patients and improved MCC in healthy, asthmatic, bronchiectasis, and CF subjects60,61. Since mannitol can be administered as a dry powder, there is no need for nebulization. This added convenience may reduce treatment time and improve adherence to treatment plans.
3.2. Non-CFTR Ion Channel Agents
In addition to therapies that directly affect the osmolarity of the ASL, ENaC blockers have the potential to increase ASL hydration by inhibiting active Na+ absorption via the ENaC channel. While inhibition of ENaC has been shown to positively affect ASL hydration, ENaC-blocking small molecule therapies have, in general, been unsuccessful at producing long-term benefits in CF patients62. Lack of efficacy of these drugs can be attributed to their short half-life, while effects on renal ion transport is the key dose-limiting safety concern63,64. Currently, several ENaC modulators are being tested in clinical trials. Compound AZD5634 from AstraZeneca recently completed two Phase I trials. AZD5634 was well tolerated both intravenously and inhaled at all dose levels during a Phase Ia pharmacokinetic evaluation, however, data regarding its effect on MCC has not yet been released from the completed Phase Ib trial65. VX-371 from Vertex (formerly P-1037) is under investigation as well, being tested alone, in combination with HS, or in combination with orkambi for CF patients and has also been tested in patients with primary ciliary dyskinesia66.
In CF, ENaC upregulation is partly due to the proteolytic degradation of SPLUNC1, an inhibitory defense protein responsible for cellular internalization of the ENaC protein67,68. By this mechanism, SPLUNC1 reduces active sodium absorption via ENaC and, in turn, prevents ASL dehydration. Neutrophil elastase, present in high concentrations in CF sputum, degrades SPLUNC1 and therefore favors ENaC upregulation and/or fluid hyperabsorption69. SPX-101, an ENaC blocker created by Spyryx Biosciences, is a peptidomimetic compound that resembles the active ENaC-inhibiting region of the SPLUNC1 protein and is not degraded by proteases found in CF sputum70. SPX-101 has been tested in clinical trials in patients with CF, and non peer-reviewed data from a Phase II study shows a 5.2% increase in FEV1 in SPX-101 treated patients compared to the placebo control group71.
Another approach is to potentiate other chloride channels already expressed in the lungs. TMEM16A or anoctamin-1 (ANO1) is a voltage-sensitive calcium-activated chloride channel (CaCC) expressed in epithelial tissues such as the gut and the airways. Like CFTR, TMEM16A has the capacity to regulate Cl− currents directly or indirectly via the control of CFTR-mediated Cl- secretion72. Counterintuitively, inhibition of TMEM16A is speculated to promote clinical benefits, as TMEM16A expression was linked to goblet cell metaplasia73,74. In parallel, TMEM16A potentiators are currently being tested in vitro by Enterprise Therapeutics; the drugs were reported to positively stimulate anion conductance and fluid secretion in CF cultures75. With the same objective of increasing Cl- secretion, denufosol, a P2Y2-receptor agonist that stimulates Ca2+ elevation and activates CaCC channels, made its way to clinical trials in 2008. However, inhalation of denufosol for 48 weeks failed to improve pulmonary function in CF patients76,77. TMEM16A may provide an alternate route to address muco-obstructive lung disease but warrants further investigation. Another anion channel, SLC26A9, is also being studied as a potential therapeutic target in CF, although limited data exists at this time78,79.
3.3. Chelating Agents
In CF, the secretion of bicarbonate, an alkalinizing but also a chelating agent, is diminished80. As a result, calcium chelation, a critical step during mucin granule exocytosis, may be hindered and mucin maturation (i.e. transition from a compacted to an expanded form) may be compromised46. The addition of high concentrations of bicarbonate to CF airway model systems like the CF rat and pig trachea has been shown to increase MCT rates and change the proportion of condensed/expanded mucins9,46. Conversely, the addition of free calcium ions to pig tracheal surfaces was found to increase ASL viscosity, supporting that calcium sequestering is necessary for healthy airway mucus rheology47. Zinc and magnesium, both also divalent cations, do not change the ASL viscosity; therefore, chelation therapies specifically targeting calcium are of particular interest47.
Ethylenediaminetetraacetic acid (EDTA), a hexadentate ligand capable of chelating metal ions, has been used in CF models to bind calcium and normalize mucus properties45. Compared side by side on mucus from the ileum of CF mice, EDTA induced rheological changes that were similar to bicarbonate at roughly 6-fold lower concentrations (20 vs. 115 mM)29. A similar compound more selective for calcium chelation, ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), has also been used in CF research to provide calcium chelation at even lower concentrations than EDTA47. Unfortunately, tissue integrity was found to be compromised by both 20 mM EDTA and 115 mM bicarbonate, suggesting that these compounds are likely not suitable for human treatment29.
OligoG, a guluronate-rich alginate with a high affinity for calcium ions, facilitated the removal of adherent mouse ileum mucus at concentrations as low as 1.5%, while showing no effect on tissue integrity at concentrations as high as 6%81. It has been speculated that concentrations of 1.5% can be reached in the lungs via dry powder inhalation81. One Phase II clinical trial was recently completed and another trial is currently underway for the treatment of CF patients with OligoG82. Preliminary data from a recent Phase II trial showed that although there was not a significant difference in MCC, a strong trend towards a more peripheral deposition of tracer particles was observed in subjects treated with OligoG, suggesting the opening of previously occluded airways82.
Recently, a polycationic biopolymer, poly (acetyl, arginyl) glucosamine (PAAG) was used in in vitro and in vivo models of CF to displace Ca2+ and promote optimal expansion and linearization of mucins upon exocytosis83. The high-molecular-weight polyglucosamine significantly improved the viscoelastic properties of CF sputum and CF HBE mucus. In addition, PAAG increased ciliary beat frequency and MCT in CF HBE cultures, which correlated with the alteration of the MUC5B network ultrastructure towards a more linear organization. Treatment of CF mice via oral gavage and CFTR-KO ferrets via aerosolization resolved intestinal and airway plugging, respectively. Furthermore, PAAG-treated ferrets revealed significantly lower levels of inflammatory markers, suggesting Ca2+-chelating agents may be a valid therapeutic approach to treat patients with CF.
3.4. Surfactants
Surfactants are surface-active agents that reduce the surface tension between a liquid and another substance (e.g., liquid, gas or solid) and work by interfacing between the hydrophilic and hydrophobic components of a solution. Relevant to polymer gels, the addition of surfactants can interact with the hydrophobic regions of the mucin network as well as the interface of the ASL. The disruption of hydrophobic interactions thus disperses the mucin-rich fraction of the gel, creating a more homogenous or a single phase gel with lower viscosity31. Adding surfactant to an electrostatically charged system like a mucin gel initiates the formation of polymer-surfactant aggregates and, at a critical concentration, a single phase is obtained. Mucin polymers then no longer interact with each other via hydrophobic interactions, reducing surface affinity and increasing water solubility. Hence, adding surfactant increases the homogeneity of a mucin-rich solution and decreases hydrophobic crosslinking of the gel.
Nonionic surfactants such as 1,2-hexanediol effectively reduce both the storage and loss moduli of MUC5AC gels and prevent assembly of mucins via hydrophobic interactions31. The elastic modulus decreases with increasing concentrations of surfactant until a single population is governed by Brownian motion31.
Surfactant interaction with the mucus layer could be relevant as both a treatment option for muco-obstructive diseases as well as a pertinent area of research for drug delivery through mucus gel systems. For example, combining proteolytic enzymes such as papain-palmitate with surfactant compounds in the form of self-emulsifying drug delivery systems increased enzyme activity likely due to improved mucus permeability84. Clinical data on surfactants is conflicting, with studies showing positive results in patients with chronic bronchitis, negative results in healthy patients with rhinosinusitis, and neither benefit nor harm in CF patients85–87.
3.5. Reducing Agents
Another approach different from hydration therapies is fragmentation of the mucin network itself. Therapies targeted at chemically breaking down mucus (mucolytics) have been used for decades. The most common and most effective “mucolytic” used in CF is inhaled dornase alfa, a recombinant human DNase that works by enzymatically digesting extracellular DNA released by dying neutrophils entrapped in mucus88. Due to its target, dornase alfa was shown to be most effective in CF patients presenting with inflammation and was found to be ineffective in patients with other obstructive airways diseases (e.g., COPD)89,90. Despite targeting a macromolecule other than mucins, the clinical benefits of rhDNase confirmed the notion that an inhaled drug affecting the rheology of airway secretions could improve health outcomes. However, NAC, a thiol-based compound that directly affects the mucin network by cleaving intra- and inter-molecular disulfide bonds, showed only limited in vivo efficacy91,92. Nevertheless, the concept that reducing mucin disulfide bonds decreases the viscoelastic properties of mucus has been broadly demonstrated and can be explained by basic polymer physics. To facilitate clearance or lubrication, mucins interact by forming loosely interwoven mucus networks comprised of linear disulfide-linked mucin polymers able to reptate20,30. Reducing disulfide bonding was shown to lower the viscoelastic properties of the mucus because network stability is dependent on the second power of the length of the polymers that form the network30,93. Breaking mucin polymers into smaller oligomers therefore significantly decreases the random walk time and allows for greater axial diffusion30.
The concept of a stiffer mucus dominated by disulfide bridges is of great interest as it unveils a commonly overlooked target for therapeutic intervention. Diseased mucus may require greater reducing power than healthy mucus to achieve similar results. Despite the lack of clinical efficacy, NAC has been the only reducing agent approved for inhalation since the 1960’s.
Our recent study described the limitations of NAC as an inhaled reducing agent94. In brief, we showed that NAC possesses a low intrinsic reducing activity, as it mostly remains in its inactive protonated form at physiological pH and cycles slowly between active and inactive forms. As a result, NAC requires long periods of time to react to completion and the drug is rapidly cleared and/or absorbed from the epithelial surfaces. Consequently, the limited potency and slow kinetics of NAC, coupled with the off-target irritation effects including cough and bronchospasm, appear responsible for its failure in clinical pulmonary medicine as an inhaled mucolytic. Identifying the deficiencies of NAC is the first step towards preclinical testing of the next generation of reducing agents.
Other reducing agents are commonly used in laboratories but have limited or poor toxicity data. Dithiothreitol (DTT) exhibited a faster reaction time and increased potency compared to NAC. However, due to its cell toxicity, DTT cannot be used in vivo for the treatment of CF patients95. Similarly, Tris(2-carboxyethyl)phosphine (TCEP) is also used in laboratories and has the advantage of being odorless, but limited toxicological data exists on this compound.
TCEP was tested for MCT activity ex vivo and in vivo in newborn pigs following stimulation of submucosal gland secretion with methacholine. Mucus clearance rate was assessed by tracking the velocity of large metal particles (>300μm) across the airway surface. Although TCEP did not affect the overall velocity of the particles, it decreased the percentage of particles in movement, which correlated with a delay in clearance via the action of mucus strands originating from submucosal ducts10. Hence, designing novel reagents will be a balancing act between increasing potency, improving kinetics, and preventing off target effects and cell toxicity.
Parion Sciences designed several new thiol reagents (e.g., P-3001, P-2062, and P-2119) that were tested in in vitro and in vivo models. Compared to NAC, P-3001 showed superior reducing activities including faster reaction rates94. P-3001 required lower concentrations to alter the viscoelasticity of patient sputa and reduced mucus burden in βENaC mice, a model of CF lung disease. Drug effects were achieved without evidence of in vitro or in vivo toxicity. Similarly, P-2062 was more effective than NAC and rhDNase at dissolving mucus flakes collected via bronchoalveolar lavages performed on CF preschoolers36. Mucus flake integrity is also affected by treatment with TCEP (Figure 3). Although TCEP treatment of pig trachea delayed the mobilization of microdisks, P-2062 treatment accelerated tracheal mucus velocity in a sheep model of muco-obstructive lung disease, suggesting that the overall MCT is not negatively affected36.
Figure 3. Images of mucus flakes collected via bronchoalveolar lavage (BAL) from a CF preschooler and treated ex vivo with PBS or TCEP.
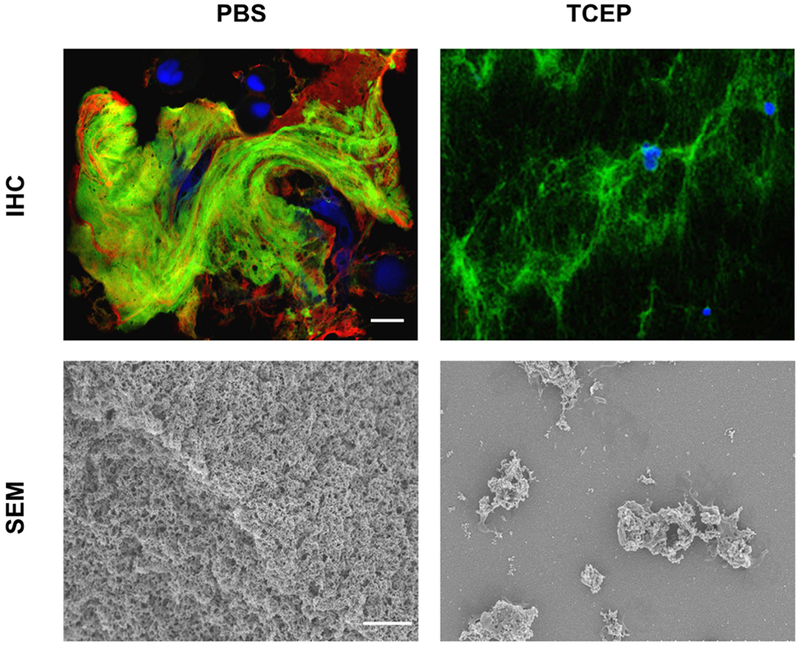
Top images show the mucin polymeric network of a mucus flake treated with PBS or TCEP (5 mM for 10 min). BAL samples were stained with anti-MUC5B (green) and anti-MUC5AC (red) antibodies. DAPI (blue) was used to stain the nuclei of entrapped inflammatory cells. Bottom images reveal the organization of the mucin mesh via scanning electron microscopy (SEM) following treatment with PBS or TCEP. Scale bar is 4 μm.
MUC5B overexpression has been shown to be a risk factor for developing idiopathic pulmonary fibrosis (IPF)96. In the bleomycin-challenged Muc5b-overexpressing mouse model of IPF, P-2119 restored MCT and minimized fibrosis following lung injury97.
These studies demonstrated that reducing the viscoelasticity of airway mucus with reducing agents may be an achievable therapeutic goal and provides unique insights into new mucolytic agents as inhaled therapies to treat a broad range of muco-obstructive diseases.
4. Conclusion and Future Directions:
Mucus is a complex polymeric gel that serves as a critical defense mechanism in multiple organ systems. In muco-obstructive diseases like CF, restoring proper mucus viscoelasticity and clearance in the lungs remain major goals. The advancement of CFTR correctors and modulators provide an exciting glimpse at how CFTR correction can change the course of CF pathogenesis. However, some patients remain ineligible for modulator therapies and rely on symptomatic treatments to control their disease. While knowledge surrounding mucin biochemistry, ASL interaction, and MCT has expanded rapidly in the past decade, there is still much to learn. Understanding the precise biochemical and biophysical mechanisms of both normal and aberrant mucus will help guide treatment efforts and ensure that all patients suffering from muco-obstructive diseases receive effective therapies.
Acknowledgements:
Supported by grants from the Cystic Fibrosis Foundation (EHRE16XX0, MARKOV18F0), Vertex Pharmaceuticals (Ehre RIA Award), and the NIH/NIDDK (2 P30 DK 065988-11).
Footnotes
The authors have no conflict to disclose
References:
- 1.Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13(6):231–240. [DOI] [PubMed] [Google Scholar]
- 2.Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol. 2014;52:136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372(9636):415–417. [DOI] [PubMed] [Google Scholar]
- 4.Okuda K, Chen G, Subramani DB, et al. Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am J Respir Crit Care Med. 2019;199(6):715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Widdicombe JH, Wine JJ. Airway Gland Structure and Function. Physiol Rev. 2015;95(4):1241–1319. [DOI] [PubMed] [Google Scholar]
- 6.Ermund A, Meiss LN, Rodriguez-Pineiro AM, et al. The normal trachea is cleaned by MUC5B mucin bundles from the submucosal glands coated with the MUC5AC mucin. Biochem Biophys Res Commun. 2017;492(3):331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostedgaard LS, Moninger TO, McMenimen JD, et al. Gel-forming mucins form distinct morphologic structures in airways. Proc Natl Acad Sci U S A. 2017;114(26):6842–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoegger MJ, Fischer AJ, McMenimen JD, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345(6198):818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birket SE, Davis JM, Fernandez CM, et al. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight. 2018;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer AJ, Pino-Argumedo MI, Hilkin BM, et al. Mucus strands from submucosal glands initiate mucociliary transport of large particles. JCI Insight. 2019;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol. 2008;70(1):459–486. [DOI] [PubMed] [Google Scholar]
- 12.Bansil R, Turner BS. The biology of mucus: Composition, synthesis and organization. Adv Drug Deliv Rev. 2018;124:3–15. [DOI] [PubMed] [Google Scholar]
- 13.Corfield AP. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim Biophys Acta. 2015;1850(1):236–252. [DOI] [PubMed] [Google Scholar]
- 14.Joo NS, Evans IA, Cho HJ, Park IH, Engelhardt JF, Wine JJ. Proteomic analysis of pure human airway gland mucus reveals a large component of protective proteins. PLoS One. 2015;10(2):e0116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trillo-Muyo S, Nilsson HE, Recktenwald CV, et al. Granule-stored MUC5B mucins are packed by the non-covalent formation of N-terminal head-to-head tetramers. J Biol Chem. 2018;293(15):5746–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rousseau K, Swallow DM. Mucin Methods: Genes Encoding Mucins and Their Genetic Variation with a Focus on Gel-Forming Mucins In: McGuckin MA, Thornton DJ, eds. Mucins: Methods and Protocols. Totowa, NJ: Humana Press; 2012:1–26. [DOI] [PubMed] [Google Scholar]
- 17.Johansson ME, Sjovall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci U S A. 1990;87(23):9188–9192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehre C, Worthington EN, Liesman RM, et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci U S A. 2012;109(41):16528–16533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verdugo P Supramolecular dynamics of mucus. Cold Spring Harb Perspect Med. 2012;2(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radicioni G, Cao R, Carpenter J, et al. The innate immune properties of airway mucosal surfaces are regulated by dynamic interactions between mucins and interacting proteins: the mucin interactome. Mucosal Immunol. 2016;9(6):1442–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaramillo AM, Azzegagh Z, Tuvim MJ, Dickey BF. Airway Mucin Secretion. Ann Am Thorac Soc. 2018;15:S164–S170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin BK, Priftis KN, Schmidt HJ, Henke MO. Secretory hyperresponsiveness and pulmonary mucus hypersecretion. Chest. 2014;146(2):496–507. [DOI] [PubMed] [Google Scholar]
- 24.Boucher RC. Muco-Obstructive Lung Diseases. N Engl J Med. 2019;380(20):1941–1953. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogers DF. Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care. 2007;52(9):1134–1146; discussion 1146-1149. [PubMed] [Google Scholar]
- 27.Kesimer M, Makhov AM, Griffith JD, Verdugo P, Sheehan JK. Unpacking a gel-forming mucin: a view of MUC5B organization after granular release. Am J Physiol Lung Cell Mol Physiol. 2010;298(1):L15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheehan JK, Kirkham S, Howard M, et al. Identification of molecular intermediates in the assembly pathway of the MUC5AC mucin. J Biol Chem. 2004;279(15):15698–15705. [DOI] [PubMed] [Google Scholar]
- 29.Ermund A, Meiss LN, Gustafsson JK, Hansson GC. Hyper-osmolarity and calcium chelation: Effects on cystic fibrosis mucus. Eur J Pharmacol. 2015;764:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Georgiades P, Pudney PD, Thornton DJ, Waigh TA. Particle tracking microrheology of purified gastrointestinal mucins. Biopolymers. 2014;101(4):366–377. [DOI] [PubMed] [Google Scholar]
- 31.Wagner CE, Turner BS, Rubinstein M, McKinley GH, Ribbeck K. A Rheological Study of the Association and Dynamics of MUC5AC Gels. Biomacromolecules. 2017;18(11):3654–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan S, Hollinger M, Lachowicz-Scroggins ME, et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med. 2015;7(276):276ra227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21):1963–1970. [DOI] [PubMed] [Google Scholar]
- 34.Button B, Cai LH, Ehre C, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science. 2012;337(6097):937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. [DOI] [PubMed] [Google Scholar]
- 36.Esther CR Jr., Muhlebach MS, Ehre C, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med. 2019;11(486):eaav3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson AG, Ehre C, Button B, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest. 2014;124(7):3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill DB, Long RF, Kissner WJ, et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J. 2018;52(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Georgiades P, di Cola E, Heenan RK, Pudney PD, Thornton DJ, Waigh TA. A combined small-angle X-ray and neutron scattering study of the structure of purified soluble gastrointestinal mucins. Biopolymers. 2014;101(12):1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354(3):241–250. [DOI] [PubMed] [Google Scholar]
- 41.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354(3):229–240. [DOI] [PubMed] [Google Scholar]
- 42.Goralski JL, Wu D, Thelin WR, Boucher RC, Button B. The in vitro effect of nebulised hypertonic saline on human bronchial epithelium. Eur Respir J. 2018;51(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang TC, Kirk KL. The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb Perspect Med. 2013;3(1):a009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambort D, Johansson ME, Gustafsson JK, et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci U S A. 2012;109(15):5645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustafsson JK, Ermund A, Ambort D, et al. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med. 2012;209(7):1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdullah LH, Evans JR, Wang TT, et al. Defective postsecretory maturation of MUC5B mucin in cystic fibrosis airways. JCI Insight. 2017;2(6):e89752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang XX, Ostedgaard LS, Hoegger MJ, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126(3):879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coakley RD, Boucher RC. Regulation and functional significance of airway surface liquid pH. JOP. 2001;2 Suppplement 4:294–300. [PubMed] [Google Scholar]
- 49.Coakley RD, Grubb BR, Paradiso AM, et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A. 2003;100(26):16083–16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garland AL, Walton WG, Coakley RD, et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci U S A. 2013;110(40):15973–15978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP, Marelich GP, Cross CE. Myeloperoxidase and protein oxidation in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L537–546. [DOI] [PubMed] [Google Scholar]
- 53.Chandler JD, Margaroli C, Horati H, et al. Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur Respir J. 2018;52(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosen BH, Evans TIA, Moll SR, et al. Infection Is Not Required for Mucoinflammatory Lung Disease in CFTR-Knockout Ferrets. Am J Respir Crit Care Med. 2018;197(10):1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dickerhof N, Pearson JF, Hoskin TS, et al. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic Biol Med. 2017;113:236–243. [DOI] [PubMed] [Google Scholar]
- 56.Dunican EM, Elicker BM, Gierada DS, et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest. 2018;128(3):997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donaldson SH, Laube BL, Corcoran TE, et al. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight. 2018;3(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennett WD, Zeman KL, Laube BL, et al. Homogeneity of Aerosol Deposition and Mucociliary Clearance are Improved Following Ivacaftor Treatment in Cystic Fibrosis. J Aerosol Med Pulm Drug Deliv. 2018;31(4):204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corcoran TE, Huber AS, Myerburg MM, et al. Multiprobe Nuclear Imaging of the Cystic Fibrosis Lung as a Biomarker of Therapeutic Effect. J Aerosol Med Pulm Drug Deliv. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aitken ML, Bellon G, De Boeck K, et al. Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. Am J Respir Crit Care Med. 2012;185(6):645–652. [DOI] [PubMed] [Google Scholar]
- 61.Daviskas E, Anderson SD, Brannan JD, Chan HK, Eberl S, Bautovich G. Inhalation of dry-powder mannitol increases mucociliary clearance. Eur Respir J. 1997;10(11):2449–2454. [DOI] [PubMed] [Google Scholar]
- 62.Burrows EF, Southern KW, Noone PG. Sodium channel blockers for cystic fibrosis. Cochrane Database Syst Rev. 2012(3):CD005087. [DOI] [PubMed] [Google Scholar]
- 63.Hirsh AJ, Molino BF, Zhang J, et al. Design, synthesis, and structure-activity relationships of novel 2-substituted pyrazinoylguanidine epithelial sodium channel blockers: drugs for cystic fibrosis and chronic bronchitis. J Med Chem. 2006;49(14):4098–4115. [DOI] [PubMed] [Google Scholar]
- 64.O’Riordan TG, Donn KH, Hodsman P, et al. Acute hyperkalemia associated with inhalation of a potent ENaC antagonist: Phase 1 trial of GS-9411. J Aerosol Med Pulm Drug Deliv. 2014;27(3):200–208. [DOI] [PubMed] [Google Scholar]
- 65.To Assess the Safety, Tolerability and Pharmacokinetics of AZD5634 Following Inhaled and Intravenous (IV)Dose Administration, November 5, 2018. www.clinicaltrials.gov
- 66.Pipeline of Investigational Medicines for CF, April 27, 2016. www.investors.vrtx.com
- 67.Garcia-Caballero A, Rasmussen JE, Gaillard E, et al. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci U S A. 2009;106(27):11412–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Webster MJ, Reidel B, Tan CD, et al. SPLUNC1 degradation by the cystic fibrosis mucosal environment drives airway surface liquid dehydration. Eur Respir J. 2018;52(4):1800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang D, Wenzel SE, Wu Q, Bowler RP, Schnell C, Chu HW. Human neutrophil elastase degrades SPLUNC1 and impairs airway epithelial defense against bacteria. PLoS One. 2013;8(5):e64689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sesma JI, Wu B, Stuhlmiller TJ, Scott DW. SPX-101 is stable in and retains function after exposure to cystic fibrosis sputum. J Cyst Fibros. 2019;18(2):244–250. [DOI] [PubMed] [Google Scholar]
- 71.Phase 2 Study Shows Promising Results for Potential Mucus Clearance Drug, June 8, 2018. www.cff.org
- 72.Benedetto R, Ousingsawat J, Cabrita I, et al. Plasma membrane-localized TMEM16 proteins are indispensable for expression of CFTR. J Mol Med (Berl). 2019;97(5):711–722. [DOI] [PubMed] [Google Scholar]
- 73.Kunzelmann K, Ousingsawat J, Cabrita I, et al. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Front Pharmacol. 2019;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scudieri P, Caci E, Bruno S, et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J Physiol. 2012;590(23):6141–6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Danahay H, Lilley S, Charlton H, Fox R, Button B, Gosling M. TMEM16A potentiators: a new therapeutic opportunity for treating Cystic Fibrosis-Related Lung Disease. In: ECFS Basic Science Conference, 27-30 Mar 2019, Dubrovnik, Croatia Vol 18 2019. [Google Scholar]
- 76.Moss RB. Pitfalls of drug development: lessons learned from trials of denufosol in cystic fibrosis. J Pediatr. 2013;162(4):676–680. [DOI] [PubMed] [Google Scholar]
- 77.Ratjen F, Durham T, Navratil T, et al. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J Cyst Fibros. 2012;11(6):539–549. [DOI] [PubMed] [Google Scholar]
- 78.Balazs A, Mall MA. Role of the SLC26A9 Chloride Channel as Disease Modifier and Potential Therapeutic Target in Cystic Fibrosis. Front Pharmacol. 2018;9:1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol. 2009;133(4):421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bridges RJ. Mechanisms of bicarbonate secretion: lessons from the airways. Cold Spring Harb Perspect Med. 2012;2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ermund A, Recktenwald CV, Skjak-Braek G, et al. OligoG CF-5/20 normalizes cystic fibrosis mucus by chelating calcium. Clin Exp Pharmacol Physiol. 2017;44(6):639–647. [DOI] [PubMed] [Google Scholar]
- 82.Donaldson SH, Bennett WD, Zeman K, et al. A First in Class Mucus Conditioning Drug - Effect of an Inhaled Alginate Oligosaccharide (Oligo-G) on Mucus Clearance in Subjects with Cystic Fibrosis. In: NACFC, 18-20 Oct 2018, Denver, Colorado, USA Vol 52 2017:S305–S305. [Google Scholar]
- 83.Fernandez-Petty CM, Hughes GW, Bowers HL, et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight. 2019;4(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Efiana NA, Phan TNQ, Wicaksono AJ, Bernkop-Schnurch A. Mucus permeating self-emulsifying drug delivery systems (SEDDS): About the impact of mucolytic enzymes. Colloids Surf B Biointerfaces. 2018;161:228–235. [DOI] [PubMed] [Google Scholar]
- 85.Anzueto A, Jubran A, Ohar JA, et al. Effects of aerosolized surfactant in patients with stable chronic bronchitis: a prospective randomized controlled trial. JAMA. 1997;278(17):1426–1431. [PubMed] [Google Scholar]
- 86.Griese M, Bufler P, Teller J, Reinhardt D. Nebulization of a bovine surfactant in cystic fibrosis: a pilot study. Eur Respir J. 1997;10(9):1989–1994. [DOI] [PubMed] [Google Scholar]
- 87.Turner JH, Wu J, Dorminy CA, Chandra RK. Safety and tolerability of surfactant nasal irrigation. Int Forum Allergy Rhinol. 2017;7(8):809–812. [DOI] [PubMed] [Google Scholar]
- 88.Yang CL, Chilvers M, Montgomery M, Nolan SJ. Dornase alfa for cystic fibrosis. Paediatr Respir Rev. 2017;21:65–67. [DOI] [PubMed] [Google Scholar]
- 89.Shah PI, Bush A, Canny GJ, et al. Recombinant human DNase I in cystic fibrosis patients with severe pulmonary disease: a short-term, double-blind study followed by six months open-label treatment. Eur Respir J. 1995;8(6):954–958. [PubMed] [Google Scholar]
- 90.Tarrant BJ, Maitre CL, Romero L, et al. Mucoactive agents for adults with acute lung conditions: A systematic review. Heart Lung. 2019;48(2):141–147. [DOI] [PubMed] [Google Scholar]
- 91.Rogers DF. Mucoactive agents for airway mucus hypersecretory diseases. Respir Care. 2007;52(9):1176–1193; discussion 1193-1177. [PubMed] [Google Scholar]
- 92.Tam J, Nash EF, Ratjen F, Tullis E, Stephenson A. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev. 2013(7):CD007168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Gennes PG, Leger L. Dynamics of Entangled Polymer Chains. Annual Review of Physical Chemistry. 1982;33(1):49–61. [Google Scholar]
- 94.Ehre C, Rushton ZL, Wang B, et al. An Improved Inhaled Mucolytic to Treat Airway Muco-obstructive Diseases. Am J Respir Crit Care Med. 2019;199(2):171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Held KD, Biaglow JE. Mechanisms for the Oxygen Radical-Mediated Toxicity of Various Thiol-Containing Compounds in Cultured-Mammalian-Cells. Radiation Research. 1994;139(1):15–23. [PubMed] [Google Scholar]
- 96.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hancock LA, Hennessy CE, Solomon GM, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9(1):5363. [DOI] [PMC free article] [PubMed] [Google Scholar]