

SUMMARY
Single-cell RNA sequencing (scRNA-seq) data are commonly affected by technical artifacts known as “doublets,” which limit cell throughput and lead to spurious biological conclusions. Here, we present a computational doublet detection tool—Doublet-Finder—that identifies doublets using only gene expression data. DoubletFinder predicts doublets according to each real cell’s proximity in gene expression space to artificial doublets created by averaging the transcriptional profile of randomly chosen cell pairs. We first use scRNA-seq datasets where the identity of doublets is known to show that DoubletFinder identifies doublets formed from transcriptionally distinct cells. When these doublets are removed, the identification of differentially expressed genes is enhanced. Second, we provide a method for estimating DoubletFinder input parameters, allowing its application across scRNA-seq datasets with diverse distributions of cell types. Lastly, we present “best practices” for DoubletFinder applications and illustrate that DoubletFinder is insensitive to an experimentally validated kidney cell type with “hybrid” expression features.
Graphical Abstract
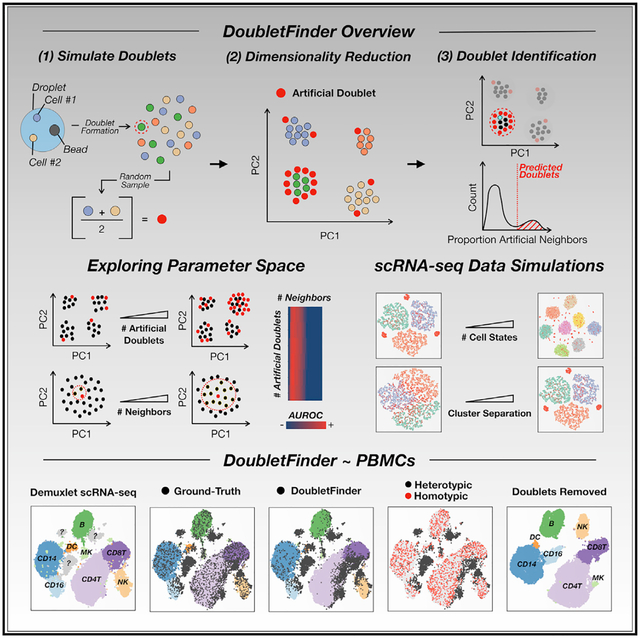
In Brief
scRNA-seq data interpretation is confounded by technical artifacts known as doublets—single-cell transcriptome data representing more than one cell. Moreover, scRNA-seq cellular throughput is purposefully limited to minimize doublet formation rates. By identifying cells sharing expression features with simulated doublets, DoubletFinder detects many real doublets and mitigates these two limitations.
INTRODUCTION
High-throughput single-cell RNA sequencing (scRNA-seq) has evolved into a powerful and scalable assay through the development of combinatorial cell indexing techniques (Cao et al., 2017; Rosenberg et al., 2018) and cellular isolation strategies that utilize nanowells (Gierahn et al., 2017) and droplet microfluidics (Macosko et al., 2015; Klein et al., 2015; Zheng et al., 2017). In droplet microfluidics and nanowell-based scRNA-seq modalities, Poisson loading is used to co-encapsulate individual cells and mRNA capture beads in emulsion oil droplets where the cells are lysed, mRNA is captured on the bead, and transcripts are barcoded by reverse transcription. Since cells are randomly apportioned into droplets, the frequency at which droplets are filled with two cells—forming technical artifacts known as “doublets”—varies according to the input cell concentration with a frequency that follows Poisson statistics (Bloom, 2018). Doublets are known to confound scRNA-seq data analysis (Stegle et al., 2015; Ilicic et al., 2016), and it is common practice to mitigate these effects by sequencing far fewer cells than is theoretically possible in order to minimize doublet formation rates. For this reason, doublet formation fundamentally limits scRNA-seq cell throughput.
Recently developed sample multiplexing approaches can overcome this limitation in some circumstances. For example, genomic (Kang et al., 2018; Guo et al., 2018; Shin et al., 2018) and cellular sample multiplexing techniques (Stoeckius et al., 2018; Gehring et al., 2018; McGinnis et al., 2018; Gaublomme et al., 2018) directly detect most doublets in scRNA-seq data by identifying cells associated with orthogonal sample barcodes or single nucleotide polymorphisms (SNPs). By identifying and removing doublets, these techniques minimize technical artifacts while enabling users to “super-load” droplet microfluidics devices for increased scRNA-seq cell throughput. However, sample multiplexing techniques have limitations in the context of doublet detection. For instance, doublets formed from cells associated with identical sample indices or SNPs cannot be detected. Moreover, sample multiplexing cannot be applied retroactively to existing scRNA-seq datasets.
To address these limitations, we developed DoubletFinder: a computational doublet detection tool that relies solely on gene expression data. DoubletFinder begins by simulating artificial doublets and incorporating these “cells” into existing scRNA-seq data that has been processed using the popular “Seurat” analysis pipeline (Box 1; Satija et al., 2015; Butler et al., 2018). DoubletFinder then distinguishes real doublets from singlets by identifying real cells with high proportions of artificial neighbors in gene expression space. In this study, we describe development and validation of DoubletFinder in three parts. In the first part, we benchmark DoubletFinder against “ground-truth” scRNA-seq datasets where doublets are empirically defined by the sample multiplexing approaches Demuxlet (Kang et al., 2018) and Cell Hashing (Stoeckius et al., 2018). These comparisons reveal that DoubletFinder detects ground-truth false negatives and improves downstream differential gene expression analyses. Moreover, ground-truth comparisons illustrate that DoubletFinder predominantly detects doublets derived from transcriptionally distinct cells—referred to here as “heterotypic” doublets—and is less sensitive to “homotypic” doublets formed from transcriptionally similar cells. In the second part, we leverage scRNA-seq data simulations to demonstrate that DoubletFinder input parameters must be tailored to data with different numbers of cell types and magnitudes of transcriptional heterogeneity. These analyses facilitated the development of a parameter estimation strategy for datasets without ground-truth while also revealing that DoubletFinder is most accurately applied to scRNA-seq data with well-resolved clusters in gene expression space.
Box 1. DoubletFinder “Real-World” Workflow Interfaces with Seurat.
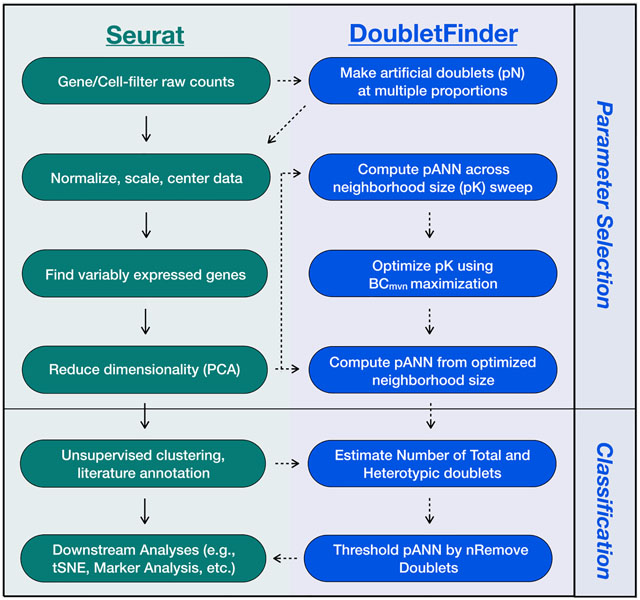
Seurat workflow (green) begins with gene and cell filtering and log2-normalization of filtered raw RNA UMI count matrices. Normalized data are then centered and scaled prior to regression of the undesirable sources of variation. Genes that are abundantly and variably expressed are then defined and used as input for PCA and unsupervised clustering and subsequent literature annotation. These results can then be applied to miscellaneous downstream analyses. DoubletFinder workflow (blue) is split into two stages: parameter selection and doublet classification. During parameter selection, variable numbers of artificial doublets (pN) are generated from filtered raw RNA UMI count matrices. Artificial doublets are then incorporated into existing scRNA-seq data, which are processed using Seurat until after PCA. pANN values are then computed across variable PC space neighborhood sizes (pK). This process is repeated for each pN and pK value, creating a list of pANN values. Optimal pK is then selected using BCmvn maximization, and this pK is applied to the full dataset. Following parameter selection, doublet classification begins by estimating the number of total and heterotypic doublets from the Poisson doublet formation rate with and without homotypic doublet adjustment. Homotypic doublets are estimated as the sum of squared cell-type annotations or unsupervised clustering frequencies. Final doublet classifications are then made by thresholding pANN according to these doublet number predictions, and doublets are then removed prior to subsequent downstream analyses.
In the third part, we apply DoubletFinder to “real-world” data lacking ground-truth doublet labels. Specifically, we test DoubletFinder on an existing mouse kidney scRNA-seq dataset (Park et al., 2018) containing an experimentally validated intermediate cell state that shares gene expression features with two other kidney cell types. We chose this dataset in order to explicitly test whether this strategy for artificial doublet generation (i.e., averaging of expression profiles) leads to DoubletFinder false positives in context with bona fide “hybrid” cell states. DoubletFinder correctly classifies this “hybrid” cell state as singlets, which suggests that DoubletFinder can be broadly applied to scRNA-seq data describing cell-state transitions. This case study also illustrates “best practices” for DoubletFinder application and emphasizes how results should be interpreted with methodological limitations in mind (e.g., undetectable doublets and poor performance on homogeneous data).
RESULTS
Overview of DoubletFinder and Characterization of Its Performance When Ground-Truth Doublet Labels Are Available
DoubletFinder predicts doublets in a fashion that can be split into five distinct steps (Figure 1A). First, DoubletFinder simulates artificial doublets from existing scRNA-seq data by averaging the gene expression profiles of random pairs of cells. Simulating doublets in this fashion preserves cell composition while recapitulating the intermixing of mRNAs from two cells that occurs during doublet formation. Second, DoubletFinder merges and pre-processes real and artificial data using the “Seurat” single-cell analysis pipeline (Satija et al., 2015; Butler et al., 2018). Notably, pre-processing parameters are held constant between the original and merged real-artificial datasets. Third, DoubletFinder performs dimensionality reduction on the merged real-artificial data using principal-component analysis (PCA), producing a low-dimensional space that describes the similarity between real and artificial cells. Fourth, DoubletFinder detects the k nearest neighbors for every real cell in principal component (PC) space, and this information is used to compute each cell’s proportion of artificial nearest neighbors (pANN). Finally, building on the assumption that real and artificial doublets co-localize in PC space, DoubletFinder predicts real doublets as cells with the top n pANN values, where n is set to the total number of expected doublets (see STAR Methods).
Figure 1. DoubletFinder Overview, nUMI Comparison, and Ground-Truth Benchmarking.

(A) Schematic overview of DoubletFinder workflow. Doublet detection is necessary to correctly interpret intermediate cell states (blue, orange) in scRNA-seq data, which could represent developmental intermediates or technical artifacts. Starting with scRNA-seq data pre-processed using Seurat, DoubletFinder integrates artificial doublets (red) into the existing data at a defined proportion (pN). DoubletFinder then defines each cell’s neighborhood in gene expression space (pK, example neighborhood seed in bright blue). The proportion of artificial nearest neighbors (pANN) is then defined, and cells with the top pANN values are predicted as doublets. Doublet removal aids in scRNA-seq data interpretation—e.g., when discerning doublets from legitimate differentiation intermediates.
(B) Schematic describing pN-pK parameter sweep. Increasing pN corresponds with increasing numbers of artificial doublets (red) relative to singlets (black). Increasing pK corresponds with larger neighborhood sizes (red dotted circle, neighbors highlighted in yellow) used during pANN computation.
(C) pN-pK parameter sweep AUC heatmap for Demuxlet and Cell Hashing data.
(D) ROC analysis of logistic regression models trained using DoubletFinder alone (blue), nUMIs alone (red), and both nUMIs and DoubletFinder (orange).
(E) t-SNE visualizations of Demuxlet and DoubletFinder doublets (black) among PBMC cell types. Inset regions exemplify two types of discordance. False-negative DoubletFinder classifications (gold) localize among singlets in gene expression space, while putative false-positive DoubletFinder classifications (red) localize among heterotypic doublets. Mono, monocytes; NK, natural killer cells; MK, megakaryocytes; and DC, dendritic cells.
(F) Density plots describing the proportion of ground-truth doublet neighbors in gene expression space among ground-truth singlets and doublets (left). Singlets (gray) have low doublet neighbor proportions, whereas doublets (red) have widely variable doublet neighbor proportions. Homotypic (Hom.) and heterotypic (Het.) doublets were thresholded at the intersection of single and doublet densities (black dotted line). t-SNE visualization (right) demonstrates that homotypic doublets (red) localize among singlets (gray), unlike heterotypic doublets (black).
(G) RNA UMI boxplots for true-positive doublets (red), putative false-negative doublets (gold), and singlets (black). Data are represented as mean ± SEM.
***statistically significant (p < 2e–16); ns, not significant (p = 0.40).
(H) Marker gene heatmaps for true-positive doublets, false-negative doublets, B cells, and CD14 monocytes.
(I) Bar chart describing the number of additional differentially expressed genes identified following doublet removal.
(J) Heatmap of literature-supported immune cell marker genes identified as differentially expressed genes following doublet removal.
DoubletFinder requires three input parameters expressed as proportions of the merged real-artificial dataset: the number of expected real doublets, the number of artificial doublets (pN) and the neighborhood size (pK) used to compute the number of artificial nearest neighbors. For example, in a dataset with 15,000 real cells, a pN of 0.25 would represent the integration of 5,000 artificial doublets, and a pK of 0.01 would represent a pK of 200 cells. To explore how parameter variation influences DoubletFinder performance, we used existing datasets of peripheral blood mononuclear cells (PBMCs) generated using sample multiplexing techniques (Demuxlet and Cell Hashing). Demuxlet identifies cells belonging to each sample group according to sample-specific SNPs and identifies doublets as cell barcodes associated with mutually exclusive sets of SNPs (Kang et al., 2018). Cell Hashing identifies doublets using a conceptually analogous strategy, except sample-specific SNPs are replaced by sample-specific DNA barcodes that are linked to cells by conjugation to antibodies targeting cell-surface proteins (Stoeckius et al., 2018). Notably, neither method can detect doublets formed from cells associated with the same SNPs or sample barcodes.
We selected these two datasets because they are currently the only publicly available datasets where within-species doublets are directly measured. Moreover, since each dataset was sequenced at variable depths (Demuxlet = 2,438 unique molecular identifiers [UMIs], Cell Hashing = 676 UMIs), we could assess whether sequencing depth influenced DoubletFinder performance. We compared the predictive capacity of DoubletFinder outputs (i.e., a vector of every real cell’s pANN) across a sweep of pN (0.05–0.3) and pK (5e 4–0.3) values using receiver operating characteristic curve (ROC) analysis (Figure 1B). Comparing the relative areas under the curve (AUCs) demonstrates that DoubletFinder performance is largely invariant of pN (Figure 1C). Moreover, optimal parameter regimes are similar for each dataset, which suggests that DoubletFinder performance is insensitive to sequencing depth. These observations demonstrate that pK is the main parameter that must be tuned when applying DoubletFinder to different scRNA-seq data. Therefore, we set pN to 0.25 for all DoubletFinder applications and optimized pK for each dataset.
Using pK values with the highest AUC from Demuxlet and Cell Hashing ROC analysis (pK = 0.01 for both datasets), we next benchmarked DoubletFinder against a commonly used feature for doublet identification in real-world scRNA-seq data—the number of UMIs (nUMIs; Islam et al., 2014; Ziegenhain et al., 2017). UMIs are uniquely associated with individual mRNA transcripts via reverse transcription and enable PCR amplification bias correction as UMIs link each sequenced molecule back to its original mRNA. Since droplets associated with two cells often have more total mRNA molecules than droplets associated with single cells, doublets are commonly removed by setting an upper nUMI threshold. However, this approach has well-established limitations (Kang et al., 2018), as it does not consider technical variability in mRNA capture efficiency or biological variability in cellular RNA content.
To compare the relative predictive capacities of DoubletFinder and nUMIs for doublet detection, we first randomly split the Demuxlet and Cell Hashing datasets into evenly sized test and training sets. Next, we used ROC analysis to compare logistic regression models trained using DoubletFinder alone (i.e., pANN values for every cell), nUMI alone, or a linear combination of both features. DoubletFinder-based models outperformed nUMI-based models for predicting ground-truth doublets in both the Demuxlet (Figure 1D) and Cell Hashing data (Figure S1A). Moreover, models trained with both DoubletFinder and nUMIs performed nearly indistinguishably to DoubletFinder-alone models, demonstrating that the method captures all of the doublet-specific information inherent to nUMIs in this context.
Although DoubletFinder predicts doublets better than nUMIs, it remained unclear whether DoubletFinder results accurately recapitulated the ground-truth doublet labels provided by Demuxlet or Cell Hashing sample classifications. To make these comparisons, we needed to convert the DoubletFinder output (i.e., pANN values for every cell) into a list of singlet and doublet labels. To generate this list, we assigned doublet labels to cells in the Demuxlet and Cell Hashing datasets with the top n pANN values, where n was set to the total number of doublets expected from the empirical sample multiplexing results. For example, since 6,045 doublets were defined by Demuxlet SNP profiling of 8 individuals, and because doublets formed from cells with the same SNPs are classified as singlets by Demuxlet, we estimated that 12.5% of real doublets (864 cells) remained unclassified. To account for both the ground-truth false negatives and the true, Demuxlet-identified doublets, we assigned doublet labels to cells with the top 6,909 pANN values. A similar list was made for the Cell Hashing dataset (see STAR Methods).
Running DoubletFinder on the same data and visualizing doublets on identical t-stochastic neighbor embedding (t-SNE) plots revealed that Demuxlet (Figure 1E), Cell Hashing (Figure S1B), and DoubletFinder doublet classifications were generally concordant, with DoubletFinder identifying few false positives relative to ground-truth (Demuxlet specificity = 0.91, Cell Hashing = 0.91). However, DoubletFinder was insensitive to many ground-truth doublets exhibiting similar gene expression profiles to singlets (Demuxlet sensitivity = 0.73, Figure 1E, orange inset, gold dots; Cell Hashing = 0.64). We hypothesized that these cells represented homotypic doublets—i.e., doublets formed from transcriptionally similar cells that cluster among their composite cell-type singlets in gene expression space. Since DoubletFinder requires putative doublets to cluster separately from singlets in PC space, we did not expect DoubletFinder to robustly detect homotypic doublets. Supporting this hypothesis, DoubletFinder sensitivity was increased when homotypic doublets were identified (Figures 1F, S2A, and S2B; see STAR Methods) and excluded from this analysis (Demuxlet sensitivity = 0.93, Cell Hashing = 0.82), while specificity remained unchanged. Notably, the magnitude of transcriptional divergence necessary for DoubletFinder detection did not always match established cell-type nomenclature, as DoubletFinder identified doublets formed from subsets of CD4+ T cells (Figure S2C). Collectively, these results illustrate that DoubletFinder primarily detects heterotypic doublets—i.e., doublets formed from transcriptionally distinct cells.
DoubletFinder additionally identified a set of doublets left unclassified by Cell Hashing and Demuxlet (Figure 1E, red inset, red dots). As described above, we had estimated that 12.5% of real doublets were formed from cells with the same SNPs. Thus, these doublets would have remained unclassified by Demuxlet but should be detected efficiently by DoubletFinder. If these apparent DoubletFinder false-positive cells (labeled red in Figure 1E) were in fact Demuxlet false negatives, two predictions would follow. First, if putative Demuxlet false negatives were real doublets, then these cells should exhibit enriched nUMIs relative to singlets. Second, Demuxlet false negatives should express marker genes associated with multiple distinct cell states. In line with these predictions, putative false negatives had nUMI levels indistinguishable from true-positive Demuxlet doublets (Wilcoxon rank-sum test, p = 0.4) and were enriched relative to singlets (p < 2e 16, Figure 1G). Equivalent analyses were carried out on the Cell Hashing data with the same result (Figure S1C). Moreover, these ground-truth false negatives expressed marker genes associated with hematopoietic cell types that do not share a common progenitor in peripheral blood (Figure 1H, Demuxlet data; Figure S1D, Cell Hashing data). Collectively, these results suggest that DoubletFinder recapitulates heterotypic doublet classifications made by Demuxlet and Cell Hashing and accurately predicts sample multiplexing false negatives formed from cells associated with identical SNPs or sample barcodes.
A common application of scRNA-seq is to discover genes that are differentially expressed among distinct cell types that are obscured in bulk transcriptomic assays (Satija et al., 2015; Butler et al., 2018; Park et al., 2018). Doublets hinder differential gene expression analyses because doublets often cluster separately in gene expression space while sharing transcriptional features with the cell types from which they are derived. To demonstrate this effect, we compared differential gene expression analysis results between Demuxlet and Cell Hashing datasets before and after removing doublets. Doublet removal results in pronounced increases in the total number of differentially expressed genes (Demuxlet with and without doublets = 2,567 and 4,339; Cell Hashing = 2,185 and 5,598) across nearly every PBMC cell type (Figure 1I). Importantly, many newly identified differentially expressed genes are PBMC cell-type marker genes supported in the literature (Figure 1J; Satija et al., 2015; Butler et al., 2018; Clark et al., 2012; Ancuta et al., 2009; Zhao et al., 2010; Jeevan-Raj et al., 2017; Stoeckle et al., 2009). These results illustrate how doublet detection and removal improves scRNA-seq analysis workflows.
Defining the Relationship between the Parameter pK and the Structure of scRNA-Seq Data
DoubletFinder performance is demonstrably sensitive to changes in the input parameter specifying pK used to compute each cell’s pANN (Figures 1B and 1C). To understand the relationship between scRNA-seq data structure and DoubletFinder performance, we used the “splatter” R package (Zappia et al., 2017) to generate simulated scRNA-seq datasets with 3–8 distinct cell clusters that ranged from being intermixed to completely separated in gene expression space (Figure 2A). Real doublets were simulated by adding the gene expression profiles of randomly selected cells such that 10% of the final data was doublets. We visualized parameter performance by finding the mean AUC for each pK value across all pN since pK selection was previously shown to dominate DoubletFinder performance.
Figure 2. BCMVN Maximization Estimates Ideal DoubletFinder Parameters for Real-World scRNA-Seq Data and Facilitates DoubletFinder Application to Mouse Kidney Data with “Hybrid” Cell States.

(A) Schematic overview of data simulation strategy. scRNA-seq data including doublets (red) with different numbers of cell states (top) and extent of cluster separation in gene expression space (bottom) were simulated. pDE, probability of differential expression.(B) Simulated pN-pK parameter sweep results. Range of pK values coinciding with high mean AUC differ between simulated data with varying numbers of equally separated cell states (pDE, 10.0% for all simulations, top). DoubletFinder performance suffers on the whole when applied to simulated data with variable degrees of cluster separation (number of cell states = 8 for all simulations, bottom).(C) Comparison of BCMVN (teal) and mean AUC distributions (black) enables identification of high AUC pK values for Demuxlet and Cell Hashing data (left). BCMVN distributions for mouse kidney and pancreas data inform pK parameter selection (right). Red dotted lines denote optimal pK values based on peak BCMVN.
(D) t-SNE visualization of DoubletFinder doublet predictions (black) among mouse kidney cell types. DCT, distal convoluted tubule; PT, proximal tubule; Endo, endothelial; and LOH, loop of Henle.
(E) RNA UMI boxplots for doublets (red) and singlets (black). Data are represented as mean ± SEM.
(F) Marker gene heatmaps for doublets, PT cells (beige), and DCT cells (pink).
(G) Bar chart describing the number of additional differentially expressed genes identified following doublet removal.
For most simulations, mean AUC distributions featured an inflection point representing the point at which pKs became too large to enable accurate doublet prediction (Figure 2B). Mean AUC inflection point positions differed for simulations with variable numbers of cell states, suggesting that pK parameter selection is sensitive to the inherent diversity of scRNA-seq data (Figure 2B, top). Moreover, among simulations with the same number of cell-state clusters but varying degrees of cluster separation, mean AUC inflection points were only observed for simulations with well-separated clusters (Figure 2B, bottom). This observation suggests that DoubletFinder performance suffers as a whole when applied to data describing transcriptionally homogeneous cell states. This decrease in performance is common to other computational doublet detection strategies that utilize transcriptomic information alone (Wolock et al., 2018, this issue of Cell Systems) and illustrates a key methodological limitation that should be carefully considered by all prospective users.
Defining “Best Practices” for Real-World DoubletFinder Applications
To identify optimal pK values for real-world scRNA-seq data when ground-truth doublet information is not known (precluding ROC analysis and AUC maximization for pK selection), we suggest that DoubletFinder users calculate the mean-variance-normalized bimodality coefficient (BCMVN; Pfister et al., 2013; Figure S3A; see STAR Methods) of pANN distributions produced during pN-pK parameter sweeps of their data. BCMVN can be used to identify the pK that separates singlets and doublets effectively, without being sensitive to local density differences in gene expression space (Figures S3B and S3C). To demonstrate the utility of BCMVN for DoubletFinder parameter selection, we benchmarked BCMVN against the previously computed ROC results for Demuxlet and Cell Hashing data, as well as two scRNA-seq datasets generated without sample multiplexing (Park et al., 2018; Byrnes et al., 2018). Across all datasets tested, BCMVN distributions featured a single maximum that for the Cell Hashing and Demuxlet datasets, coincided with the pK range maximizing AUC (Figure 2C). Therefore, we propose that BCMVN maximization selects a near-optimal DoubletFinder parameter across a range of scRNA-seq datasets.
We next sought to demonstrate DoubletFinder’s capabilities in a real-world context where ground-truth doublets are not known and BCMVN maximization must be used to determine a reasonable pK. To this end, we applied DoubletFinder to a previously published scRNA-seq dataset describing the mouse kidney. In this study, the authors discover and experimentally validate the existence of a novel cell type—collecting duct transitional cells (CDTCs)—which expresses genes characteristic of two other kidney cell types: collecting duct principal cells (CDPCs) and collecting duct intercalated cells (CDICs) (Park et al., 2018). This dataset represented an intriguing “challenge-case” for DoubletFinder, as we reasoned that legitimate cell types with “hybrid” gene expression profiles may resemble artificial doublets, triggering DoubletFinder false positives.
Beginning with a pre-processed Seurat object (Figure 2D), we first used BCMVN maximization to identify a suitable pK value for these data (pK = 0.09, Figure 2C, top right). We then applied DoubletFinder to the full dataset and classified doublets as cells with the top n pANN values. Initially, n was set according to the Poisson doublet formation rate, as specified for the particular cell-loading density used in the study (Park et al., 2018). This resulted in 913 total doublet predictions, which were highly enriched for nUMIs (Figure 2E) and included a region of heterotypic doublets characterized by the co-expression of proximal tubule and distal convoluted tubule marker genes (Figure 2F). Upon doublet removal, differential gene expression analysis was improved (Figure 2G).
DoubletFinder correctly identified 64% of CDTCs as singlets, despite CDTCs having exceptionally high nUMIs (Figure S4A) and co-expressing both CDPC and CDIC marker genes (Figure S4B). However, these initial results represented an overestimation of the true number of detectable doublets as DoubletFinder was applied without taking homotypic doublets into account. We therefore adjusted the expected doublet number to account for homotypic doublets. Specifically, we grouped cells according to literature-supported cell-type annotations and estimated the proportion of homotypic doublets as the sum of squared cell-type frequencies (Figure S4C; see STAR Methods). This strategy assumes (1) that a cell type’s frequency in the final dataset reflects that cell type’s contribution to the doublet pool, and (2) that user-defined cell-type groups approximate the magnitude of transcriptional divergence necessary to make a detectable heterotypic doublet. In contexts where such annotations are inaccurate or unavailable, unsupervised clustering results should be used instead. This analysis resulted in a revised 473 total heterotypic doublet predictions and allowed us to identify 97% of CDTCs as singlets. This result suggests that DoubletFinder can be insensitive to legitimate cell states with intermediate expression profiles. We suggest that the heterotypic doublet frequency and Poisson doublet formation rates be used as lower and upper bounds for estimating the number of detectable doublets, respectively. We urge DoubletFinder users to interrogate the results of each thresholding strategy against the known biology of the system under study.
DISCUSSION
DoubletFinder is a computational doublet detection method that integrates artificial doublets into existing scRNA-seq data and identifies real doublets as cells enriched for artificial nearest neighbors in gene expression space. DoubletFinder is implemented in the R programming language and is written to interface with the popular Seurat scRNA-seq analysis package (Satija et al., 2015; Butler et al., 2018). However, DoubletFinder is prospectively generalizable to scRNA-seq data analyzed using alternative pipelines as well. In this study, we benchmarked DoubletFinder against ground-truth scRNA-seq data where doublets are directly measured using sample multiplexing techniques such as Demuxlet (Kang et al., 2018) and Cell Hashing (Stoeckius et al., 2018). We leveraged these results to define “best practices” for how DoubletFinder should be applied to real-world scRNA-seq data without ground-truth doublet labels. We then successfully demonstrated these practices on mouse kidney data featuring an experimentally validated cell state that could trigger DoubletFinder false positives (Park et al., 2018).
Ground-truth comparisons revealed a number of DoubletFinder strengths and limitations. For example, DoubletFinder outperforms nUMI thresholding in these data and accurately predicts heterotypic doublets with >90% sensitivity. In contrast, DoubletFinder is insensitive to homotypic doublets, as these cells do not diverge significantly from real singlets in gene expression space. DoubletFinder also identifies false negatives in the Demuxlet and Cell Hashing datasets that are formed from cells associated with identical sample barcodes. For this reason, we view DoubletFinder and sample multiplexing as complementary doublet removal approaches, especially in experimental contexts with relatively low sample numbers. When used in concert, sample multiplexing and computational doublet detection techniques provide an effective solution to the issue of doublets in scRNA-seq data, enabling users to “super-load” droplet microfluidic devices and thereby further increase scRNA-seq cell throughput.
In contexts where sample multiplexing information is unavailable, DoubletFinder detects and removes the preponderance of heterotypic doublets while homotypic doublets remain. The presence of homotypic doublets is unlikely to negatively influence cell-type classification and differential gene expression analysis, as homotypic doublets cluster together with bona fide cell singlets. In fact, simply removing heterotypic doublets improved differential gene expression analysis results in every dataset tested in this study. However, certain scRNA-seq analyses may also benefit from the removal of homotypic doublets. For example, imputation uses the average gene expression profiles of transcriptionally similar cells to infer missing values caused by transcript dropout events (Huang et al., 2018; van Dijk et al., 2018). It is possible that the structure of missing values in singlets and homotypic doublets is distinct, and thus, it remains unclear how the presence of homotypic doublets influences imputation performance.
Beyond exposing DoubletFinder strengths and limitations, ground-truth benchmarking revealed three methodological features that should be considered as users apply DoubletFinder to scRNA-seq data lacking ground-truth doublet labels. First, applying DoubletFinder to simulated scRNA-seq data with poorly resolved clusters demonstrates that DoubletFinder cannot be accurately applied to scRNA-seq data describing transcriptionally similar cells. DoubletFinder users should therefore carefully consider the diversity of their dataset prior to using the method. Second, DoubletFinder input parameters (e.g., pK) must be tuned to datasets with variable numbers of cell states. We predicted pK for ground-truth scRNA-seq data using ROC analysis, which is not possible for real-world data. Instead, we developed a ground-truth-agnostic parameter selection strategy—termed BCMVN maximization—which finds the pK value that optimally separates singlet and doublet pANN distributions. Third, since DoubletFinder is insensitive to homotypic doublets, thresholding DoubletFinder results according to the total number of doublets estimated via Poisson loading statistics will necessarily result in false positives. To account for this issue, we describe how the proportion of homotypic doublets can be estimated from cell-type frequencies described using existing annotations or unsupervised clustering. Using this strategy, one can threshold DoubletFinder results in a fashion that accounts for homotypic doublets and thereby limits false positives.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Zev J. Gartner (zev.gartner@ucsf.edu).
METHOD DETAILS
Seurat Pre-processing Pipeline
DoubletFinder was implemented in the R programming language in a fashion that purposefully interfaces with the Seurat analysis package. The DoubletFinder workflow and how it specifically interfaces with Seurat is outlined in Box 1. DoubletFinder takes as an input a Seurat object that has been pre-processed using the standard Seurat analysis pipeline. Briefly, raw RNA UMI counts are normalized (e.g., log2-transform), centered, and scaled before regression is used to remove undesired sources of variability (e.g., total nUMI). In the standard Seurat workflow, variably expressed genes are then defined via dispersion and mean expression thresholds. In this study, thresholds were chosen that identified ~2000 total genes, as described previously (Satija et al., 2015; Butler et al., 2018). PCA is then performed using this set of variably expressed genes, and statistically-significant PCs are selected (e.g., via inflection point estimation on PC elbow plots). These are the minimum pre-processing requirements prior to running DoubletFinder, although further dimensionality reduction (e.g., t-SNE) and unsupervised clustering were also utilized in this study. Notably, any set of user-defined Seurat pre-processing parameters is compatible with DoubletFinder. Seurat parameters used in this study:
| Seurat Pre-processing Parameters | |||||||
|---|---|---|---|---|---|---|---|
| Data | REF | PCs | Variable gene dispersion threshold |
Variable gene expression threshold |
pN | pK | # of doublet predictions |
| Demuxlet | Kang et al., 2018 | 10 | 0.85 | 0.05 | 0.25 | 0.01 | 6909 cells |
| Cell Hashing | Stoeckius et al., 2018 | 10 | 0.65 | 0.025 | 0.25 | 0.01 | 2687 cells |
| Kidney | Park et al., 2018 | 10 | 0.25 | 0.0125 | 0.25 | 0.09 | 913, 473 cells |
DoubletFinder overview
The DoubletFinder workflow begins with a pre-processed Seurat object, prepared as described above. Artificial doublets are then generated from raw UMI count matrices by averaging the gene expression profiles of cell pairs selected via random sampling with replacement. Sufficient artificial doublets are then generated to comprise 25% of the resulting merged data (pN = 0.25). Next, real and artificial data are merged and pre-processed using the same normalization, scaling, and variable gene definition parameters employed during the original data analysis workflow. Notably, nUMI regression is not performed during merged real-artificial dataset pre-processing in order to preserve differences between singlets and doublets. Using the same number of statistically-significant PCs selected during original data pre-processing, PC cell embeddings are then converted into a Euclidean distance matrix using the ‘rdist’ function from the ‘fields’ R package (Nychka et al., 2017). Each cell’s nearest neighbors are then defined from this distance matrix, and the proportion of artificial nearest neighbors (pANN) is computed for every real cell by dividing its number of artificial neighbors by the neighborhood size (pK). Final doublet classifications are then assigned to the cells with the n highest pANN, where n was set to the total number of expected doublets with or without homotypic doublet adjustment (see ‘Real-World Applications’ below).
Ground-Truth Benchmarking
Optimizing pK Using ROC Analysis
For Cell Hashing and Demuxlet scRNA-seq data, optimal parameters were selected by maximizing the AUC from ROC analysis of pN-pK parameter sweeps. Specifically, Cell Hashing and Demuxlet datasets were first randomly sub-sampled to 10,000 cells in order to maximize computational efficiency during the parameter sweep. Second, artificial doublets were integrated at varying proportions (pN = 0.05–0.30), and merged real-artificial data was pre-processed as described above. Third, the proportion of artificial nearest neighbors was computed for varying neighborhood sizes (pK = 0.0001–0.3) for each real cell. This produced a list of pANN vectors corresponding to each pN-pK combination. Fourth, ground-truth doublet labels and pANN vectors were then evenly split into test and training sets via random sampling without replacement. Fifth, logistic regression models were fit on training cells using the ‘glm’ R function with the ‘family’ and ‘link’ arguments set to ‘binomial’ and ‘logit’, respectively. Logistic regression was used because this technique specifically models the binary nature of singlet/doublet classifications. Sixth, models were applied to test cells and the predictive capacity of each model was compared by computing AUC during ROC analysis, as implemented in the ‘ROCR’ (Sing et al., 2005) and ‘pROC’ (Robin et al., 2011) R packages.
Comparing DoubletFinder and nUMIs
DoubletFinder parameters optimized for the Cell Hashing and Demuxlet datasets using ROC analysis were then used to benchmark the method against nUMI thresholding. Test and training sets were defined as described above, and logistic regression models were fit using DoubletFinder alone, nUMI alone, or a linear combination of both features. Trained models were then applied to test cells, and ROC analysis was used to compare each of the three models.
Classifying Doublets According to Sample Multiplexing Results
For Demuxlet and Cell Hashing data, cells with the n highest pANN values were classified as doublets, where n was defined as the number of ground-truth doublets adjusted according to the expected ground-truth false-negative rate. Notably, we utilized this strategy prior to discovering that DoubletFinder is insensitive to homotypic doublets. Thus, we suggest users interpret DoubletFinder results using the Poisson doublet formation rate with and without adjustment for homotypic doublet proportions (see ‘Real-World Applications’ below).
Defining Homotypic Doublets
To illustrate that DoubletFinder is predominantly sensitive to heterotypic doublets, we sought a strategy to directly distinguish homotypic and heterotypic doublets within ground-truth doublet classifications. Specifically, since homotypic and heterotypic doublets respectively co-localize with singlets and doublets in gene expression space, we reasoned that the two doublet types could be discerned according to the proportion of nearest neighbors that were real doublets. We computed this proportion for each real cell using the pK value optimized by ROC analysis (pK = 0.01). We then visualized the density distributions of doublet neighborhood proportions for real doublets and singlets. We then used the intersection of these distributions as a threshold to split real doublets into homotypic and heterotypic subsets, following the assumption that homotypic doublets and real singlets would have similar doublet neighborhood proportions (Figures 1F and S2A). Homotypic doublets identified using this strategy localize amongst singlets in gene expression space, as expected (Figures 1F and S2B).
Tracking Doublet Composition from Cell Type Annotations
DoubletFinder is insensitive to doublets formed from transcriptionally-similar cells. However, objective criteria describing the magnitude of transcriptional dissimilarity needed to produce a detectable doublet remain unclear. We tested whether literature-supported cell type annotations reflected this magnitude of dissimilarity in the following way. First, we applied DoubletFinder to the Demuxlet dataset while tracking the cell type annotations of every cell pair during artificial doublet generation (Figure S2C, top). Second, for each predicted doublet, we computed the proportion of nearest neighbors that were artificial doublets formed from cells with the same or different cell type annotations. Third, we identified the doublets where > 50% of their neighbors corresponded to artificial doublets formed from cells with the same annotation. We speculated that these doublets were most likely to be real homotypic doublets. Upon visualizing these cells in gene expression space, we observed that many localized amongst the CD4+ T-cell cluster (Figure S2C, bottom left). These doublets also express high levels of CD4+ T-cell markers and do not express marker genes for other PBMCs (Figure S2C, bottom right). Collectively, these results suggest that our analysis successfully distinguished homotypic and heterotypic doublets. Moreover, these results suggest that some cells sharing a single literature-supported cell state annotation may still have sufficient transcriptional heterogeneity to produce doublets that DoubletFinder can detect.
scRNA-Seq Data Simulation
scRNA-seq data was simulated using the ‘splatter’ R package (Zappia et al., 2017), as in (Wolock et al., 2018). Datasets were simulated with 3–8 equally-proportioned cell states, and cluster separation in gene expression space was controlled using the ‘de.prob’ parameter (0.005–0.1) of the ‘splatSimulate’ R function. Simulated doublets were added to these data by adding the UMI counts for random pairs of cells such that 10% of the final data were doublets. Simulated datasets containing doublets were then pre-processed using ‘Seurat’ as described previously, with the number of statistically-significant PCs set to the total number of cell states. Following pre-processing, parameter sweeps, logistic regression modeling, and ROC analysis were performed on each simulated dataset, as described above. Since pK is the main parameter requiring adjustment in different contexts, we visualized our results by finding the mean AUC across all pN values for each pK.
Real-World Applications
Applying DoubletFinder to real-world data lacking ground-truth doublet labels requires two analytical steps that were not performed during benchmarking against Cell Hashing, Demuxlet, and simulated data. First, when ground-truth doublets labels are unavailable, DoubletFinder parameters must be selected using a ground-truth-agnostic strategy called mean-variance-normalized bimodality coefficient (BCMVN) maximization. Second, since DoubletFinder is insensitive to homotypic doublets, thresholding results based on the Poisson doublet formation rate will necessarily result in false-positives. Thus, DoubletFinder results can be interpreted after adjusting the number of expected doublets to account for the estimated proportion of homotypic doublets in the data. These two processes are described below.
Optimizing pK with BCMVN Maximization
The bimodality coefficient (BC) measures deviations from unimodality in data distributions (Pfister et al., 2013). For DoubletFinder parameter fitting, we reasoned that parameter sets that produced non-unimodal pANN distributions would optimally separate singlets from doublets and, as a result, would perform the best. Thus, for the Demuxlet (Kang et al., 2018), Cell Hashing (Stoeckius et al., 2018), mouse kidney (Park et al., 2018), and mouse pancreas (Byrnes et al., 2018) scRNA-seq datasets, we tested every pANN distribution generated during pN-pK parameter sweeps to find those with elevated BC values. Specifically, we computed BC as is implemented in the ‘bimodality_coefficient’ function in the ‘modes’ R package (Deevi, 2016), which is formalized as:
Where γ is the pANN distribution skewness (i.e., peak width), k is the kurtosis (i.e., peak sharpness), and n is the sample size. We then measured the BC mean and variance for each pK across all pN values tested, as it was previously shown that DoubletFinder performance is not influenced by the number of generated artificial doublets.
We documented the results of this workflow when applied to the Cell Hashing data as a representative example (Figure S3). When pK values are too high (e.g., pK > 0.1), singlets and doublets have similar proportions of artificial nearest neighbors, and the resulting pANN distributions are associated with low BC and AUC (Figure S3B). In contrast, when pK values are too low (e.g., pK = 5e-4), DoubletFinder performance suffers because neighborhoods in gene expression space are dominated by local effects that result in multimodal pANN distributions (Figure S3A, top left). Since these distributions are not unimodal, they are associated with high BC. However, local effects are sensitive to the number of artificial doublets integrated into the dataset (pN), resulting in elevated BC variance for the associated pK values (Figure S3B, pink). Finally, ideal pK values (e.g., pK = 0.01) generate long-tailed pANN distributions (Figure S3A, mid left) that are characterized by high AUC and high BC with low variance (Figure S3B, red). Since high BC values with low-variance predicted high AUC parameter sets in the Cell Hashing data, we leveraged these observations to devise a new metric for pK parameter selection – BCMVN – formalized as:
Where μBC and are the BC mean and variance, respectively, for each pK across pN values. BCMVN distributions feature a single, visually-discernible maximum for the four datasets tested in this study (Figure 2D). For ground-truth datasets, this maximum corresponds with the ideal pK value identified via ROC analysis.
Estimating Homotypic Doublet Proportions
In this study, homotypic doublet proportions were modeled as the sum of squared cell state frequencies. For example, consider a scRNA-seq dataset with five unique cell states present at the following proportions:
Where pCi is the proportion of cell state i. The proportion of homotypic doublets present in this data is then estimated as:
The final number of detectable (i.e., heterotypic) doublets is then defined by adjusting the total number of doublets (i.e., as determined by the Poisson doublet formation rate) by the homotypic doublet proportion:
Where DDR and TDR are the detectable and total doublet rates, respectively. This strategy follows the assumption that, during droplet microfluidics-based cell capture, the probability that an emulsion oil droplet is filled with a cell from state i matches the proportion of cell state i in the final scRNA-seq dataset. This assumption does not consider differential doublet formation propensities between cell types (e.g., due to adhesive properties, cell size, etc.).
Notably, the accuracy of this strategy depends on whether cell state annotations accurately group cells with transcriptional profiles that are sufficiently similar to preclude formation of a heterotypic doublet. This magnitude of transcriptional similarity is difficult to define and is likely dataset-dependent. For example, since DoubletFinder detected doublets formed from subsets of CD4+ T-cells, this specific annotation would not be ideal for homotypic doublet estimation. However, unsupervised clustering results and/or literature-supported cell state annotations from existing scRNA-seq data are the best approximations, and represent a lower bound for the total number of doublets.
For the mouse kidney data with which this strategy was implemented, we assigned doublet labels to cells with the top n pANN values, where n was set to the Poisson doublet formation rate with and without homotypic doublet adjustment. This strategy results in two sets of doublet predictions associated with varying stringencies. With the unadjusted Poisson threshold, DoubletFinder users can be confident that all heterotypic doublets were removed, albeit along with a subset of real singlets. In contrast, the homotypic-adjusted threshold preserves the most existing data while potentially leaving real doublets remaining.
QUANTIFICATION AND STATISTICAL ANALYSIS
nUMI Statistical Analysis
Statistically-significant differences between nUMIs in Demuxlet and Cell Hashing singlets and doublets were defined using the Wilcoxon rank sum test implemented with the ‘pairwisewilcox.test’ R function. Multiple comparison correction was performed using the Benjamini-Hochberg procedure. In this context, n represents the total nUMIs associated with one cell (Demuxlet n = 33,328; Cell Hashing n = 15,178).
Sensitivity and Specificity
Sensitivity and specificity were computed for ground-truth scRNA-seq data before and after homotypic doublet definition, as described above. Sensitivity and specific calculations were performed using the ‘caret’ R package (Kuhn, 2008).
Differential Gene Expression Analysis
Differential gene expression analysis comparisons between scRNA-seq datasets before and after doublet removal was performed with the ‘FindMarkers’ function in ‘Seurat’. Statistical significance was tested using the likelihood-ratio test for single-cell gene expression (McDavid et al., 2013), and marker genes were defined as statistically-significant genes with 3-fold expression enrichment. For the Cell Hashing and Demuxlet datasets, doublet removal included all doublets classified either by sample-multiplexing or DoubletFinder. For mouse kidney scRNA-seq data, only DoubletFinder-defined doublets were removed.
DATA AND SOFTWARE AVAILABILITY
Cell Hashing (GEO: GSE108313), Demuxlet (GEO: GSE96583), mouse kidney (GEO: GSE107585), and mouse pancreas (GEO: GSE101099) UMI count matrices were downloaded from the Gene Expression Omnibus. DoubletFinder is implemented as a fast, easy-to-use R package that interfaces with Seurat version 2.0 and higher. DoubletFinder can be downloaded from GitHub (https://github.com/chris-mcginnis-ucsf/DoubletFinder) and is available as an executable Compute Capsule on Code Ocean (DOI: https://doi.org/10.24433/CO.4902498.v1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| soRNA-seq-UMI counts of PBMCs demultiplexed using Demuxlet | Kang et al., 2018 | GEO: GSE96583 |
| scRNA-seq-UMI counts of PBMCs demultiplexed using Cell Hashing | Stoeckius et al., 2018 | GEO:GSE108313 |
| scRNA-seq-UMI counts of mouse kidney cells | Park et al., 2018 | GEO: GSE107585 |
| scRNA-seq-UMI counts of mouse pancreas cells | Byrnes et al., 2018 | GEO:GSE101099 |
| Software and Algorithms | ||
| Seurat |
Satija et al., 2015 Butler et al., 2018 |
https://github.com/satijalab/seurat |
| Splatter | Zappia et al., 2017 | https://github.com/Oshlack/splatter |
| ROCR | Sing et al., 2005 | https://github.com/ipa-tys/ROCR |
| pROC | Robin et al., 2011 | https://github.com/xrobin/pROC |
| Caret | Kuhn, 2008 | https://cran.r-project.org/web/packages/caret/caret.pdf |
| Modes | Deevi, 2016 | https://cran.r-project.org/web/packages/modes/modes.pdf |
| Fields | Nychka et al., 2017 | https://cran.r-project.org/web/packages/fields/fields.pdf |
| DoubletFinder | This paper | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
Highlights.
DoubletFinder uses gene expression features to predict doublets in scRNA-seq data
DoubletFinder identifies doublets derived from transcriptionally distinct cells
Doublet removal improves differential gene expression analysis performance
DoubletFinder is insensitive to bona fide cells with “hybrid” expression profiles
ACKNOWLEDGMENTS
We thank Matt Thomson (California Institute of Technology) and Jimmie Ye (UCSF) for helpful discussion, as well as Lauren Byrnes (UCSF), Marlon Stoeckius (New York Genome Center), and Shiwei Zheng (New York University) for providing data access. This research was supported in part by grants from the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221); the NIH Common Fund (DP2 HD080351–01); the NSF (MCB-1330864); and the UCSF Center for Cellular Construction (DBI-1548297), an NSF Science and Technology Center. Z.J.G. is a Chan-Zuckerberg Biohub Investigator. L.M.M. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2239–15).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2019.03.003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, and Gabuzda D (2009). Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16- monocyte subsets. BMC Genomics 10, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD (2018). Estimating the frequency of multiplets in single-cell RNA sequencing from cell-mixing experiments. PeerJ 6, e5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes LE, Wong DM, Subramaniam M, Meyer NP, Gilchrist CL, Knox SM, Tward AD, Ye CJ, and Sneddon JB (2018). Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun 9, 3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HL, Banks R, Jones L, Hornick TR, Higgins PA, Burant CJ, and Canaday DH (2012). Characterization of MHC-II antigen presentation by B cells and monocytes from older individuals. Clin. Immunol 144, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deevi S (2016). Modes: Find the Modes and Assess the Modality of Complex and Mixture Distributions, Especially with Big Datasets. R package, version0.7.0. https://rdrr.io/cran/modes/. [Google Scholar]
- Gaublomme JT, Li B, McCabe C, Knecht A, Drokhlyansky E, van Wittenberghe N, Waldman J, Dionne D, Nguyen L, De Jager P, et al. (2018). Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. bioRxiv 10.1101/476036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring J, Park JH, Chen S, Thomson M, and Pachter L (2018). Highly multiplexed single-cell RNA-seq for defining cell population and transcriptional spaces. bioRxiv 10.1101/315333. [DOI] [Google Scholar]
- Gierahn TM, Wadsworth MH, 2nd, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, and Shalek AK (2017). Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Biddy BA, Kamimoto K, Kong W, and Morris SA (2018). CellTag Indexing: a genetic barcode-based multiplexing tool for single- cell technologies. bioRxiv 10.1101/335547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray JI, Raj A, Li M, and Zhang NR (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, and Teichmann SA (2016). Classification of low quality cells from single-cell RNA-seq data. Genome Biol 17, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, and Linnarsson S (2014). Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 11, 163–166. [DOI] [PubMed] [Google Scholar]
- Jeevan-Raj B, Gehrig J, Charmoy M, Chennupati V, Grandclément C, Angelino P, Delorenzi M, and Held W (2017). The transcription factor Tcf1 contributes to normal NK cell development and function by limiting the expression of granzymes. Cell Rep 20, 613–626. [DOI] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M (2008). Building predictive models in R using the caret package. J. Stat. Softw 28, 1–26.27774042 [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDavid A, Finak G, Chattopadyay PK, Dominguez M, Lamoreaux L, Ma SS, Roederer M, and Gottardo R (2013). Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 29, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Patterson DM, Winkler J, Hein MY, Srivastava V, Conrad DN, Murrow LM, Weissman JS, Werb Z, Chow ED, et al. (2018). Multi-seq: scalable sample multiplexing for single-cell RNA sequencing using lipid tagged indices. bioRxiv 10.1101/387241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nychka D, Furrer R, Paige J, and Sain S (2017). Fields: tools for spatial data. R package, version 9.6. https://cran.r-project.org/web/packages/fields/index.html. [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Suszták K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister R, Schwarz KA, Janczyk M, Dale R, and Freeman JB (2013). Good things peak in pairs: a note on the bimodality coefficient. Front. Psychol 4, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, and Müller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Lee W, Lee JH, and Bang D (2018). Multiplexed single-cell RNA-seq via transient barcoding for drug screening. bioRxiv 10.1101/359851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, and Lengauer T (2005). ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Stegle O, Teichmann SA, and Marioni JC (2015). Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet 16, 133–145. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckle C, Gouttefangeas C, Hammer M, Weber E, Melms A, and Tolosa E (2009). Cathepsin W expressed exclusively in CD8+ T cells and NK cells, is secreted during target cell killing but is not essential for cytotoxicity in human CTLs. Exp. Hematol 37, 266–275. [DOI] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2018). Scrublet: computational identification of cell doublets in single-cell transcriptome data. bioRxiv 10.1101/357358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia L, Phipson B, and Oshlack A (2017). Splatter: simulation of single-cell RNA sequencing data. Genome Biol 18, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Tan Y, Wong W, Sem X, Zhang H, Han H, Ong SM, Wong KL, Yeap WH, Sze SK, et al. (2010). The CD14+/lowCD16+ monocyte subset is more susceptible to spontaneous and oxidant-induced apoptosis than the CD14+CD16 subset. Cell Death Dis 1, e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, Leonhardt H, Heyn H, Hellmann I, and Enard W (2017). Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643.e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cell Hashing (GEO: GSE108313), Demuxlet (GEO: GSE96583), mouse kidney (GEO: GSE107585), and mouse pancreas (GEO: GSE101099) UMI count matrices were downloaded from the Gene Expression Omnibus. DoubletFinder is implemented as a fast, easy-to-use R package that interfaces with Seurat version 2.0 and higher. DoubletFinder can be downloaded from GitHub (https://github.com/chris-mcginnis-ucsf/DoubletFinder) and is available as an executable Compute Capsule on Code Ocean (DOI: https://doi.org/10.24433/CO.4902498.v1).