

Epidermal growth factor receptor signaling regulates sleep homeostasis in zebrafish and is implicated in regulating human sleep.
Abstract
The genetic bases for most human sleep disorders and for variation in human sleep quantity and quality are largely unknown. Using the zebrafish, a diurnal vertebrate, to investigate the genetic regulation of sleep, we found that epidermal growth factor receptor (EGFR) signaling is necessary and sufficient for normal sleep levels and is required for the normal homeostatic response to sleep deprivation. We observed that EGFR signaling promotes sleep via mitogen-activated protein kinase/extracellular signal–regulated kinase and RFamide neuropeptide signaling and that it regulates RFamide neuropeptide expression and neuronal activity. Consistent with these findings, analysis of a large cohort of human genetic data from participants of European ancestry revealed that common variants in genes within the EGFR signaling pathway are associated with variation in human sleep quantity and quality. These results indicate that EGFR signaling and its downstream pathways play a central and ancient role in regulating sleep and provide new therapeutic targets for sleep disorders.
INTRODUCTION
Identifying how sleep is regulated is a critical health priority. Sleep loss and sleep disorders are among the most common, yet frequently overlooked, human health problems. An estimated 50 to 70 million Americans suffer from a chronic sleep disorder (1), but the genetic and neural mechanisms that regulate human sleep remain poorly understood. Large-scale human genome-wide association studies (GWAS) have recently identified genomic regions associated with variation in human sleep and several sleep disorders (2–8), but in most cases, the causative gene(s) remains unclear, and most genes identified in these studies have not been validated in vivo. With the exception of narcolepsy, which results from loss of hypocretin (Hcrt) neurons (1), and circadian disorders that affect sleep timing (1), the genetic basis for variation in human sleep and sleep disorders is largely unknown.
Sleep is an evolutionarily conserved behavioral state defined as a period of rapidly reversible immobility and increased arousal threshold that is under homeostatic control, such that prolonged wakefulness leads to a compensatory increase in sleep duration and intensity (9). This definition is valid for complex organisms such as humans, for simpler vertebrate animal models such as rodents and zebrafish, and for invertebrates such as Caenorhabditis elegans and Drosophila melanogaster (9, 10). However, while recent studies have begun to reveal mechanisms that regulate sleep in either invertebrate or vertebrate model organisms, there is relatively little evidence for conserved genetic or neuronal mechanisms that regulate sleep in both vertebrates and invertebrates. While neuronal circuits that regulate sleep in Drosophila and C. elegans have been described in detail (11), there are no clear vertebrate homologs for these invertebrate neuronal systems. Similarly, the few signaling pathways implicated in regulating vertebrate sleep, such as Hcrt- and melanin-concentrating hormone signaling, lack unambiguous invertebrate orthologs (10). As a result, it is unclear whether mechanisms that regulate sleep evolved independently in invertebrate and vertebrate species and whether mechanisms identified in invertebrates are relevant to sleep in humans. Identifying conserved mechanisms, if they exist, would be valuable because they would likely have central roles in regulating sleep and may provide novel therapeutic targets for sleep disorders.
Among the peptidergic signaling pathways known to regulate invertebrate sleep, the pathway with perhaps the clearest homology in vertebrates is that of the epidermal growth factor (EGF) receptor (EGFR) and its ligands. EGFR signaling has primarily been studied for its roles in development and physiology, but more recently it has also been implicated in regulating several behaviors (12–20). Genetic gain- and loss-of-function studies in Drosophila and C. elegans have demonstrated that EGFR signaling is both necessary and sufficient for normal sleep levels in these invertebrate animals (12, 13, 16, 19, 21, 22). Furthermore, a GWAS using a panel of inbred wild-caught Drosophila lines identified an association between common variants in several EGFR pathway genes and sleep duration (17), as did an artificial selection screen for long and short sleep duration in an outbred Drosophila population (18). These GWAS observations suggest that EGFR signaling plays a role in natural sleep variation in Drosophila. However, mechanisms that link EGFR signaling to downstream sleep regulators are poorly understood. It is also unclear whether EGFR signaling regulates sleep in vertebrate model organisms and in humans.
Here, we address the role of EGFR signaling and downstream mechanisms in regulating sleep using the zebrafish, a vertebrate with established, behaviorally defined diurnal sleep (23). This model organism has several advantages over more commonly used rodent species for modeling aspects of human sleep, including a simpler, yet anatomically and molecularly conserved, vertebrate brain; rapid development with complex behaviors such as sleep present in 5-day-old animals; amenability to high-throughput behavioral assays; and amenability to genetic and pharmacological manipulations (10). Furthermore, zebrafish sleep has genetic, pharmacological, and neuroanatomical similarities to mammalian sleep (10), suggesting that findings in zebrafish will translate to mammals.
Using genetics and pharmacology, we show that EGFR signaling is both necessary and sufficient for normal sleep duration, promotes consolidation and maintenance of sleep, and is required for the normal sleep rebound response to sleep deprivation (SD) in zebrafish. In addition, we show that EGFR signaling promotes sleep in zebrafish via mitogen-activated protein kinase (MAPK)/extracellular signal–regulated kinase (ERK) and RFamide neuropeptide signaling, and describe a mechanism linking the EGFR and RFamide neuropeptide systems. Last, by analyzing a large cohort of human genetic data (24), we identify associations between sleep duration and quality measures with common variants in genes that participate in EGFR signaling. These results establish EGFR signaling as an evolutionarily conserved sleep-promoting system with implications for human sleep disorders.
RESULTS
Transforming growth factor, alpha overexpression increases sleep
To explore the role of EGFR signaling in vertebrate sleep, we first characterized the expression pattern of each zebrafish ortholog of EGFR and its ligands in the brain at 5 days postfertilization (dpf) using in situ hybridization (ISH). We found that one EGFR paralog, egfra, is expressed in juxtaventricular cells that colocalize with sox2 (Fig. 1, A, A″, and B), a marker of glial and ependymal cells (25), as well as in the hypothalamus, hindbrain, tectum, and cerebellum (Fig. 1, A′ and A″). The EGFR ligands transforming growth factor, alpha (TGFa) and EGF are implicated in regulating mammalian behavior (14, 20, 26), and each gene has a single zebrafish ortholog. Zebrafish egf is expressed just dorsal to egfra-expressing cells in the diencephalon (fig. S1A). Similar to mammals and zebrafish egf (27), zebrafish tgfa is also expressed in juxtaventricular cells in the diencephalon, just dorsal to egfra-expressing cells (Fig. 1, C and C′), that also express sox2 (Fig. 1, D and D′). We observed a significant day/night oscillation in tgfa mRNA level by reverse transcription quantitative polymerase chain reaction (qPCR) [P < 0.05, one-way analysis of variance (ANOVA)], with peak tgfa expression at 12 a.m. 150% higher than the trough at 12 p.m. in animals entrained under 14-hour light/10-hour dark conditions until 6 dpf (P < 0.05, one-way ANOVA, Holm-Sidak test; fig. S1, B and C), consistent with a potential role for tgfa in regulating sleep. On the basis of these results, we tested the roles of tgfa and egfra in zebrafish sleep.
Fig. 1. TGFa overexpression increases sleep.
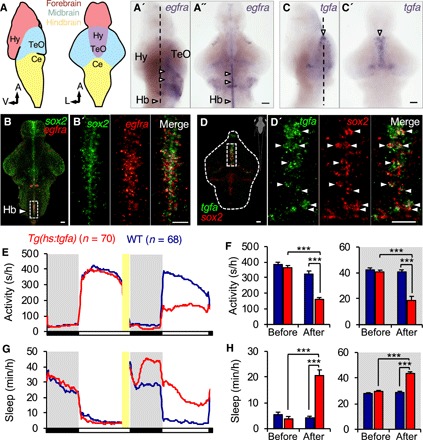
(A to D) ISH of egfra and tgfa in the 5-dpf zebrafish brain (schematic) (A). A, anterior; L, lateral; V, ventral; Ce, cerebellum; Hy, hypothalamus; TeO, tectum. (A′ and A″) Sagittal (A′) and dorsal (A″) views of egfra expression in juxtaventricular cells (white arrowheads). (B and B′) egfra coexpression with sox2 in these cells. (C and C′) Sagittal (C) and dorsal (C′) views of tgfa expression in cells just dorsal to juxtaventricular cells in the diencephalon (white arrowheads). (D and D′) tgfa coexpression with sox2 in these cells (white arrowheads). Dashed lines in (A′) and (C) indicate the horizontal planes shown in (A″) and (C′). Boxed regions in (B) and (D) are magnified in (B′) and (D′). Dashed line in (D) shows outline of brain. Scale bars, 30 μm (B, B′, D, and D′) and 50 μm (A″ and C′). (E to H) Following a 1-hour HS (yellow bars), Tg(hs:tgfa) animals were less active (E and F) and slept more (G and H) than their WT siblings. Pre-HS and post-HS are calculated for the day or night before, and the day or night after, HS, respectively. White and black bars indicate day (14 hours) and night (10 hours). Data are obtained from two pooled experiments. Bar graphs show mean ± SEM. n = number of animals. m/h, minutes/hour; s/h, second/hour. ***P < 0.0001 by two-way ANOVA with Holm-Sidak test.
To test whether EGFR signaling is sufficient to promote sleep, we used a genetic strategy to conditionally overexpress TGFa while monitoring behavior using a video tracking assay (28). As previously established using locomotor activity and arousal threshold assays, a period of 1min (or longer) of inactivity corresponds to a sleep bout in zebrafish larvae (28). We generated Tg(hs:tgfa) zebrafish, in which a heat shock (HS)–inducible promoter regulates expression of zebrafish tgfa. Before HS, we observed no difference in locomotor activity or sleep between transgenic animals and their wild-type (WT) siblings (Fig. 1, E to H). In contrast, after HS-induced TGFa overexpression, transgenic animals were less active and slept more than their WT siblings during both the night and the following day (night activity, −53%; day activity, −52%; night sleep, +54%; day sleep, +400%; P < 0.005, two-way ANOVA, Holm-Sidak test) (Fig. 1, E to H). The increase in sleep was most pronounced during the day, when zebrafish are normally most awake. This phenotype was due to an increase in sleep bout length during both the night and day, an increase in the number of sleep bouts during the day, and a decrease in wake bout length during the day (fig. S1, D to F). TGFa-overexpressing animals also exhibited a decrease in sleep latency (time to first sleep bout following lights on or off) during the day and a decrease in daytime and nighttime waking activity (locomotor activity while awake) (fig. S1, G and H). Thus, activation of EGFR signaling results in increased sleep due to consolidation of the sleep state and decreased sleep latency.
Genetic loss of EGFR signaling decreases sleep
We next tested the hypothesis that EGFR signaling is required for normal sleep levels by creating zebrafish with mutations in egfra and its ligands. Mice containing a null mutation in Tgfa are viable and fertile (29), but sleep studies using this mutant have not been reported. Using the transcription activator–like effector nuclease (TALEN) method (30), we generated a zebrafish tgfa mutant that is predicted to lack all known functional domains and should thus be a null mutation (fig. S2A). tgfa−/− zebrafish are viable and fertile, with no apparent morphological abnormalities. Consistent with the TGFa overexpression phenotype, tgfa−/− animals were 15% more active and slept 28% less than their tgfa+/+ siblings during the day (activity, P < 0.005; sleep, P < 0.01; one-way ANOVA, Holm-Sidak test; fig. S1, I to L). These effects were primarily due to fewer sleep bouts and higher levels of waking activity (fig. S1, M to Q).
To ask whether EGF also contributes to sleep regulation, we used CRISPR-Cas9 (31) to isolate an egf mutant that is predicted to lack all known functional domains after the initial three EGF-like domains and should thus be a null mutation (fig. S2B). Similar to tgfa−/− animals, egf−/− animals were 26% more active and showed a trend of less sleep than their egf+/+ siblings during the day (activity, P < 0.01; sleep, P = 0.10; one-way ANOVA, Holm-Sidak test; fig. S1, R to U), with sleep architecture phenotypes more modest than those observed in tgfa mutants (fig. S1, V to Z). To ask whether egf and tgfa act redundantly, we compared the behavior of egf−/−; tgfa−/− animals to their egf+/−; tgfa−/− and egf+/+; tgfa−/− siblings. We found that tgfa−/−; egf−/− animals are viable and fertile, with no apparent morphological abnormalities. egf−/−; tgfa−/− animals were more active and slept less during the day (activity, +20%; sleep, −45%; P < 0.005, one-way ANOVA, Holm-Sidak test) and slept less at night (−13%; P < 0.05, one-way ANOVA, Holm-Sidak test) compared to their egf+/+; tgfa−/− siblings (Fig. 2, A to D). The decrease in sleep was due to fewer sleep bouts and longer wake bouts during the day and shorter sleep bouts at night (fig. S1, AA to AE), suggesting that loss of both egf and tgfa results in consolidation of wakefulness during the day and sleep fragmentation at night. These results suggest that tgfa and egf have partially redundant roles in EGFR-mediated sleep since loss of both genes resulted in a stronger sleep defect (day sleep, −45%; P < 0.005; night sleep, −13%; P < 0.05; one-way ANOVA, Holm-Sidak test; Fig. 2, C and D) than loss of tgfa alone (day sleep, −28%; P < 0.01; night sleep, −4.7%, P > 0.05; one-way ANOVA, Holm-Sidak test; fig. S1, I to Q).
Fig. 2. Loss of EGFR signaling decreases sleep.
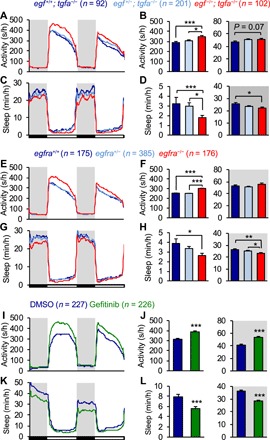
(A to D) egf−/−; tgfa−/− animals were more active during the day and slept less during the day and night than egf+/+; tgfa−/− siblings. Larvae were generated by mating egf+/−; tgfa+/− to egf+/−; tgfa−/− animals. For clarity, data for other genotypes are not shown. (E to H) egfra−/− animals were more active during the day and slept less during the day and night than egfra+/+ siblings. (I to L) Gefitinib-treated WT animals were more active and slept less during the day and night than their dimethyl sulfoxide (DMSO)–treated siblings. Pooled data from nine (A to D), eight (E to H), and six (I to L) experiments are shown. Bar graphs show mean ± SEM. n = number of animals. *P ≤ 0.05, **P < 0.01, and ***P < 0.005 for indicated comparisons by one-way ANOVA and Holm-Sidak test (A to H) or Student’s t test (I to L).
We next tested whether EGFR is required for normal sleep levels in zebrafish. Egfr knockout mice are embryonic lethal, likely because of defects in placental development (32), thus preventing genetic loss-of-function sleep studies. However, since zebrafish develop ex utero without a placenta and contain at least two egfr paralogs (egfra and egfrb) (33), we reasoned that we could analyze the requirement for EGFR in zebrafish sleep. Since we found that egfra is expressed in juxtaventricular cells adjacent to tgfa- and egf-expressing cells (Fig. 1, A′, A″, C, and C′, and fig. S1A), we used CRISPR-Cas9 to generate a zebrafish egfra mutant that is predicted to lack the transmembrane and intracellular domains (fig. S2C). egfra−/− animals are viable and lack obvious morphological abnormalities. Similar to EGFR ligand double mutants, egfra−/− animals were more active and showed a trend of less sleep during the day (activity, +17%; P < 0.005; sleep, −22%; P = 0.08; one-way ANOVA, Holm-Sidak test) and also slept less at night (−11%; P < 0.005, one-way ANOVA, Holm-Sidak test) compared to their egfra+/+ siblings (Fig. 2, E to H). Similar to EGFR ligand double mutants, the egfra mutant phenotype was primarily due to fewer sleep bouts during the day and shorter sleep bouts at night (fig. S1, AF to AJ), suggesting that loss of egfra results in sleep fragmentation at night. Together, these results suggest that tgfa, egf, and egfra are required for normal sleep levels in zebrafish. More broadly, the zebrafish genome contains several additional EGFR ligands, a second egfr paralog (33), and several ERBB family co-receptors that can form heterodimers with EGFR that can be activated by EGFR ligands; some of these proteins may also play roles in EGFR signaling–mediated sleep.
Pharmacological inhibition of EGFR signaling decreases sleep
To address the possibility that EGFR family members, in addition to egfra, promote sleep, we acutely and selectively inhibited the function of EGFRs by treating WT animals with the EGFR small-molecule inhibitors gefitinib and erlotinib (34). These compounds inhibit receptor function by binding to the adenosine triphosphate–binding pocket of the tyrosine kinase domain (34), which has >90% amino acid similarity between humans and zebrafish (33). We found that gefitinib-treated WT animals were more active (day, +23%; night, +30%) and slept less (day, −29%; night, −22%) than vehicle-treated siblings during both the day and night (P < 0.0001, Student’s t test; Fig. 2, I to L). This phenotype was due to fewer sleep bouts during the day, shorter sleep bouts at night, and longer wake bouts during the day and night (fig. S3, C to E), suggesting that inhibition of EGFR signaling results in fragmented sleep at night and consolidated wakefulness during the day and night. Gefitinib treatment also increased waking activity and increased sleep latency during the day and night (fig. S3, F and G), suggesting that EGFR signaling is required for the normal initiation of sleep. Erlotinib treatment induced a similar behavioral phenotype (fig. S3, H to P). Gefitinib treatment did not further decrease sleep in egfra−/− animals (fig. S3, A and B), suggesting that pharmacological inhibitors of EGFR promote sleep largely by inhibiting egfra, that gefitinib-induced sleep is not due to nonspecific effects on other proteins, and that EGFR ligands promote sleep primarily via egfra. Together, these pharmacological and genetic results demonstrate that EGFR signaling is required for normal sleep levels.
EGFR signaling modulates arousal threshold
Sleep is distinguished from quiet wakefulness by an increased arousal threshold, which we assayed by monitoring the behavioral response of zebrafish to a mechano-acoustic stimulus (35). We quantified the fraction of animals that responded to the stimulus at 14 different intensities and used these data to construct stimulus-response curves. We observed that the tapping intensity at which the half-maximal response occurred [effective tap power 50 (ETP50)] for TGFa-overexpressing larvae was 252% higher than that of their WT siblings (F1,834 = 20.95, P < 0.0001 by extra sum-of-squares F test) (Fig. 3A). Conversely, the ETP50 for gefitinib-treated animals was 37% lower than that of their vehicle-treated siblings (F1,834 = 18.16, P < 0.0001 by extra sum-of-squares F test) (Fig. 3B). Similarly, the ETP50 for erlotinib-treated animals was 30% lower than that of their vehicle-treated siblings (F1,834 = 4.9, P < 0.05 by extra sum-of-squares F test) (Fig. 3C). These results demonstrate that TGFa overexpression increases arousal threshold, whereas inhibition of EGFR signaling decreases arousal threshold, consistent with modulation of a sleep state.
Fig. 3. EGFR signaling regulates arousal threshold and sleep homeostasis.
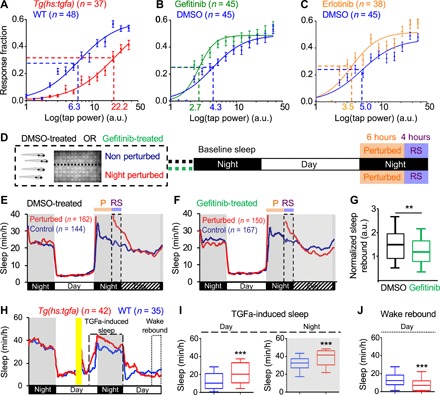
Representative stimulus response curves for Tg(hs:tgfa) animals and their WT siblings following HS (A), gefitinib and DMSO vehicle-treated WT siblings (B) and erlotinib and DMSO vehicle-treated WT siblings (C). Data points indicate mean ± SEM fraction of animals that responded to the stimulus. Dashed lines mark ETP50 values. (A) Tg(hs:tgfa) animals had an ETP50 value of 22.2 versus 6.3 for WT siblings (252% increase; F1,834 = 20.95, P < 0.0001 by extra sum-of-squares F test). (B) Gefitinib-treated animals had an ETP50 of 2.7 versus 4.3 for DMSO-treated siblings (37% decrease; F1,834 = 18.16, P < 0.0001 by extra sum-of-squares F test). (C) Erlotinib-treated animals had an ETP50 of 3.5 versus 5.0 for DMSO-treated siblings (30% decrease; F1,834 = 4.9, P < 0.05 by extra sum-of-squares F test). a.u., arbitrary units. n = number of animals. (D) SD paradigm. (E and F) Sleep behavioral traces for animals treated with DMSO (E) or gefitinib (F) starting at 5 dpf, subjected to SD during the first 6 hours of the night at 7 dpf (P; orange), and monitored thereafter in the dark. The 4-hour periods of RS (purple) are indicated with dashed boxes. (G) Normalized sleep rebound in DMSO- or gefitinib-treated siblings following SD. Normalized sleep rebound = amount of sleep for each perturbed animal during the first 4 hours of RS divided by the average amount of sleep of all nonperturbed controls during this time period. (H to J) After a 1-hour HS in the middle of the day (yellow bar), Tg(hs:tgfa) animals showed increased daytime and nighttime sleep for ~24 hours (H and I), followed by decreased sleep (H and J). Sleep in (I) and (J) is quantified for the boxed regions in (H), which includes the entire night and the last 3 hours of each day. Pooled data from five (D to G) and two (H to J) experiments are shown. Black, white, and hatched bars under behavioral traces indicate night (10 hours), day (14 hours), and subjective day (14 hours), respectively. n = number of animals. **P < 0.01 and ***P < 0.001 by Mann-Whitney test.
EGFR signaling is required for homeostatic regulation of sleep
Sleep is postulated to be controlled by two processes: a homeostatic process that increases during wakefulness and dissipates during sleep and a circadian process that controls when sleep occurs during the 24-hour day/night cycle (9). While invertebrate studies have demonstrated a role for EGFR signaling in promoting sleep (12, 13), it is unknown whether it does so as part of the homeostatic or circadian process. To distinguish between these possibilities, we first tested whether EGFR signaling is required for circadian regulation of sleep by entraining WT animals in a 14-hour light/10-hour dark cycle until 5 dpf and then treating animals with either gefitinib or dimethyl sulfoxide (DMSO) vehicle control while monitoring their behavior in either constant light or constant dark (fig. S4, A to D). While gefitinib treatment decreased sleep compared to controls, the circadian pattern of sleep was maintained with no obvious effect on circadian period length or phase, suggesting that EGFR signaling is not necessary for circadian regulation of sleep.
To explore the role of EGFR signaling in homeostatic regulation of sleep, we developed a large-scale sleep deprivation (SD) assay. We monitored the behavior of at least 60 animals from 5 to 8 dpf in a 96-well plate that was cut in half. On the beginning of the third night of the assay (7 dpf), we disrupted the sleep of half the animals using acoustic stimuli (see Materials and Methods) in the dark for 6 hours while leaving the other half of the 96-well plate in the video tracker as a nonperturbed control. Light was kept off during the remaining 4 hours of night and the following day to avoid masking effects of light on any recovery sleep (RS) response. We observed a significant increase in sleep during the remaining 4 hours of night for perturbed animals compared to nonperturbed sibling controls (fig. S5, A to C), similar to the RS observed in other species in response to SD. This increase in sleep persisted during the following subjective day (fig. S5A) but returned to normal levels by the following night (fig. S5D). To verify that the increased RS resulted from SD and not simply fatigue or some other response to the stimulus, we applied the same acoustic stimulation to developmentally identical siblings during the day, when the animals spend most of their time awake (fig. S5E). We observed no increase in sleep in perturbed animals compared to nonperturbed sibling controls following daytime perturbation (fig. S5, E to H), consistent with the increased sleep observed following perturbation at night being due to loss of sleep. We calculated a normalized sleep rebound value for each animal as the amount of sleep during the first 4 hours of RS divided by the average amount of sleep of all nonperturbed controls during this time period. Normalized sleep rebound was significantly higher in animals perturbed during the night, when zebrafish spend most of the time asleep, compared to those perturbed during the day, when zebrafish spend most of the time awake (fig. S5I), as expected for a perturbation that specifically disrupts sleep. These results indicate that this SD assay is both specific and robust in its ability to detect a homeostatic response to SD.
We next used this assay to test whether EGFR signaling is required for homeostatic regulation of sleep. To do so, we sleep-deprived WT siblings treated with either the EGFR inhibitor gefitinib or DMSO vehicle control in neighboring video trackers for 6 hours at night and quantified sleep rebound during the remaining 4 hours of night (Fig. 3, D to G). We observed a 28% reduction in normalized sleep rebound for gefitinib-treated animals compared to DMSO-treated controls (median response = 1.15 for gefitinib versus 1.59 for DMSO, P < 0.01, Mann-Whitney test; Fig. 3G and fig. S5, J to O). This result indicates that the amount of rebound sleep following SD is reduced in gefitinib-treated animals compared to DMSO-treated controls and, thus, that EGFR signaling participates in the homeostatic regulation of sleep.
If EGFR signaling regulates sleep homeostasis, then sleep induced by activation of EGFR signaling might be followed by a period of decreased sleep. When TGFa overexpression was induced in the middle of the day, we observed an initial increase in sleep that persisted for ~24 hours (day, P < 0.001; night, P < 0.001, two-way ANOVA with Holm-Sidak test; Fig. 3, H and I), followed by a decrease in sleep (P < 0.001, two-way ANOVA with Holm-Sidak test; Fig. 3, H and J) compared to sibling controls. These data are consistent with the hypothesis that EGFR signaling regulates sleep homeostasis.
TGFa overexpression–induced sleep is primarily mediated by EGFRa
To investigate the cellular mechanism through which TGFa promotes sleep, we first asked which cells in the brain are activated by TGFa overexpression by performing ISH using a probe for c-fos, a marker of neural activity (36). We used Tg(hs:tgfa) animals and their WT siblings fixed 4 hours after a midday HS, when Tg(hs:tgfa) animals sleep ~100% more than their WT siblings (Fig. 4A). We observed that tgfa overexpression resulted in strong c-fos expression in juxtaventricular cells (Fig. 4C), similar to the expression pattern of egfra (Fig. 1A″). In contrast, only low levels of c-fos expression were present in identically treated WT siblings (Fig. 4B), indicating that the strong c-fos expression in Tg(hs:tgfa) animals was not due to the HS. Double fluorescence ISH (FISH) confirmed that TGFa overexpression induced c-fos expression in egfra-expressing juxtaventricular cells (Fig. 4, D and D′). This result is consistent with the hypothesis that TGFa overexpression–induced sleep is mediated, at least in part, by egfra-expressing juxtaventricular cells in the brain.
Fig. 4. TGFa overexpression–induced sleep requires EGFRa.
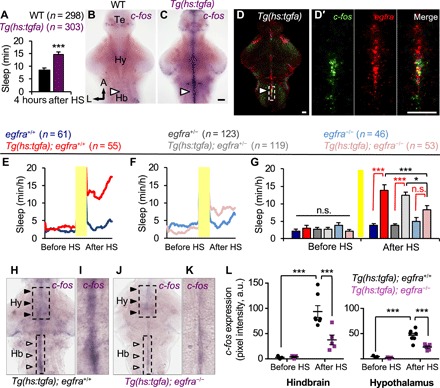
(A) Total sleep induced following TGFa overexpression quantified during the fourth hour after HS. (B and C) c-fos expression in a Tg(hs:tgfa) brain at 4 hours after HS shows activation of juxtaventricular cells in the telencephalon, hypothalamus, and hindbrain (arrowhead) (C), but not in the brain of an identically treated WT sibling (B). (D and D′) Double FISH of a Tg(hs:tgfa) brain fixed at 4 hours after HS shows coexpression of egfra and c-fos in juxtaventricular cells. Boxed region in (D) is magnified in (D′). Images are 2.9-μm-thick (D) and 0.7-μm-thick (D′) confocal sections. (E to G) TGFa overexpression increased sleep in transgenic animals compared to nontransgenic siblings in egfra+/+, but not in egfra−/− animals (red comparisons) (G). TGFa overexpression–induced sleep was significantly decreased in egfra−/− animals compared to egfra+/− and egfra+/+ (black comparisons) (G). Yellow bars indicate HS. Pre- and post-HS data are calculated for the day of HS. Data are obtained from six pooled experiments. Bar graphs show mean ± SEM. n.s., not significant. (H to L) Juxtaventricular c-fos expression that is induced by TGFa overexpression (white arrowheads) is suppressed in egfra−/− animals (J and K) compared to egfra+/+ animals (H and I) at 4 hours after HS. Hindbrain (Hb)–boxed regions in (H) and (J) are magnified in (I) and (K). (L) Quantification of c-fos expression in hindbrain and hypothalamus egfra-expressing cells [boxed regions in (H) and (J)]. Data points represent single brains. n = number of animals. *P < 0.05 and ***P < 0.005 by two-way ANOVA with Holm-Sidak test. Te, telencephalon; Hb, hindbrain. Scale bars, 30 μm.
To test this hypothesis, we compared the behavioral effects of TGFa overexpression on sleep in egfra−/− and egfra+/+ siblings. We found that Tg(hs:tgfa); egfra+/+ animals slept 270% more than their egfra+/+ siblings after HS-induced TGFa overexpression (P < 0.0001, two-way ANOVA, Holm-Sidak test; Fig. 4, E and G). These effects were strongly suppressed in Tg(hs:tgfa); egfra−/− animals compared to their egfra−/− siblings (P = 0.42, two-way ANOVA with Holm-Sidak test; Fig. 4, F and G). This result indicates that TGFa overexpression–induced sleep is largely mediated by EGFRa. Consistent with this result, TGFa overexpression–induced c-fos expression was significantly reduced in egfra−/− animals (Fig. 4, H to L). Together, these data suggest that EGFR ligands promote sleep primarily by activating egfra-expressing juxtaventricular cells in the brain.
Inhibition of MAPK/ERK signaling suppresses EGFR-induced sleep
EGFR can act via multiple signal transduction pathways, but genetic (13) and pharmacological (37) studies in Drosophila suggest that EGFR signaling promotes sleep, at least in part, via MAPK/ERK signaling. To test whether the MAPK/ERK pathway is required for EGFR-mediated sleep in zebrafish, we treated Tg(hs:tgfa) larvae immediately after HS with either of two MEK1/2 (MAPK kinase 1/2) antagonists previously demonstrated to be effective in zebrafish (38). While these antagonists had no effect on sleep or activity in WT animals at the concentrations tested, Tg(hs:tgfa) animals treated with either 3 μM SL327 (fig. S6, A to F) or 15 μM U0126 (fig. S6, G to L) were significantly more active and slept less after HS than vehicle-treated Tg(hs:tgfa) siblings (P < 0.001, two-way ANOVA, Holm-Sidak test), although they were still less active and slept more than their drug-treated WT siblings (P < 0.001, two-way ANOVA, Holm-Sidak test). SL327 treatment reduced both the number and length of sleep bouts in Tg(hs:tgfa) animals, while U0126 treatment only reduced the number of sleep bouts (fig. S6, C, F, I, and L), possibly because SL327 had a more potent effect on behavior than U0126. Thus, both MEK1/2 antagonists suppressed TGFa overexpression–induced sleep, suggesting that EGFR signaling regulates sleep, in part, via the MAPK/ERK pathway in zebrafish. These results link previous independent observations describing changes in ERK signaling following EGFR-induced sleep in Drosophila (13) and a role for ERK in regulating invertebrate and vertebrate sleep (37, 39).
TGFa overexpression–induced sleep is suppressed by mutation of npvf
In C. elegans, EGF overexpression and cellular stress induce sleep by activating EGFR in the ALA interneuron (12). Several RFamide family neuropeptides are expressed at high levels in the ALA neuron, including flp-13 and flp-24, and overexpression of these peptides is sufficient to induce sleep (21). It has been reported that EGF-induced sleep is attenuated by ~50% in flp-13 mutants (22) or in flp-13; flp-24 double mutants (21) and that heat stress–induced sleep in Drosophila is suppressed in mutants that lack the RFamide family neuropeptide fmrfamide or its receptor (40). These observations suggest that RFamide neuropeptides mediate the sedating effects of EGFR signaling in these invertebrate species. We recently showed that the vertebrate RFamide neuropeptide VF (NPVF), a possible homolog of flp-13 and fmrfamide, is necessary and sufficient to promote sleep in zebrafish (35), similar to our present findings for EGFR signaling. On the basis of these observations, we hypothesized that EGFR signaling promotes sleep, in part, via NPVF signaling in zebrafish. We tested this hypothesis by overexpressing TGFa in npvf-mutant animals (35). We found that TGFa-overexpressing animals slept 450% more than their nontransgenic siblings in control npvf+/− animals (P < 0.0001, two-way ANOVA, Holm-Sidak test; Fig. 5E and fig. S7K), similar to the effect of TGFa overexpression in WT animals (Fig. 1, E to H). However, TGFa overexpression–induced sleep was suppressed by 50% in npvf−/− animals compared to TGFa overexpression–induced sleep in their npvf+/− siblings (P < 0.0001, two-way ANOVA, Tukey’s post hoc test; Fig. 5E and fig. S7K). This result suggests that NPVF signaling plays a key role in mediating EGFR signaling–induced sleep in zebrafish, consistent with observations in Drosophila (40) and C. elegans (21, 22).
Fig. 5. EGFR signaling promotes sleep via NPVF.
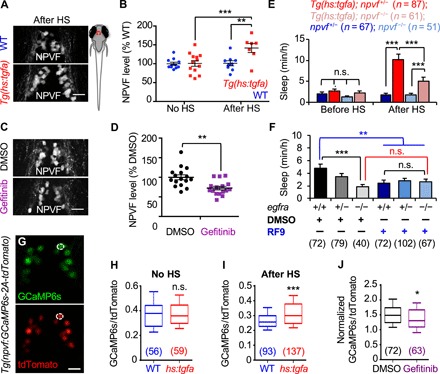
(A to D) Larval zebrafish schematic. Red box indicates hypothalamic region shown in (A), (C), and (G). Immunostaining with an NPVF-specific antibody reveals higher NPVF protein levels at 2 hours after HS for Tg(hs:tgfa) animals compared to WT siblings (A and B) and lower NPVF protein levels in animals fixed at 3 hours after treatment with gefitinib compared to DMSO-treated controls (C and D). Representative images are shown in (A) and (C), and average pixel intensity of NPVF labeling is quantified in (B) and (D); each dot represents one animal. (E) TGFa-induced sleep was partially suppressed in npvf−/− animals compared to npvf+/− siblings. Animals were heat-shocked at 3 p.m. Pre- and post-HS quantify sleep for the day of HS. (F) egfra−/− animals slept less than their egfra+/+ siblings (black comparison), and RF9 suppressed sleep in egfra+/+ animals compared to DMSO-treated egfra+/+ siblings (blue comparison), but RF9 treatment did not further decrease sleep in egfra−/− animals compared to DMSO-treated egfra−/− siblings (red comparison). RF9 was added on the afternoon of 5 dpf, and sleep was quantified during the day at 6 and 7 dpf. (G) Representative images from a Tg(npvf:GCaMP6s-p2A-tdTomato) animal. An individual NPVF neuron is indicated with a white circle. (H) No difference in GCaMP6s/tdTomato values was observed between non–heat-shocked Tg(hs:tgfa) and WT siblings. (I) GCaMP6s/tdTomato values were significantly increased in Tg(hs:tgfa) animals compared to WT siblings at 2 hours after HS. (J) Normalized GCaMP6s/tdTomato values were significantly decreased in WT gefitinib-treated animals compared to DMSO-treated siblings. Normalized GCaMP6s/tdTomato = GCaMP6s/tdTomato value in each neuron at 2 hours after the addition of either gefitinib or DMSO divided by the value in the same neuron before treatment is shown. Bar graphs (E and F) and lines (B and D) indicate mean ± SEM. Numbers in parentheses indicate number of animals (E and F) or neurons (H to J). *P < 0.05, **P < 0.01, and ***P < 0.001 by two-way ANOVA with Holm-Sidak test (E and F) or by two-tailed Wilcoxon matched-pairs signed-rank test (H to J). Scale bars, 20 μm.
Because loss of npvf suppressed EGFR-induced sleep, we hypothesized that NPVF signaling acts downstream of EGFR to regulate normal levels of sleep. To test this hypothesis, we assayed sleep in egfra mutants in the presence or absence of RF9, a small-molecule inhibitor of NPFFR1 and NPFFR2, receptors for the mature RFRP1 and RFRP3 peptides derived from the NPVF prepropeptide (41). If EGFR signaling and NPVF signaling act independently to promote sleep, then inhibition of both pathways should result in an additive effect on sleep. Alternatively, if NPVF signaling acts downstream of EGFR, inhibiting both pathways should not have an additive effect. We found that egfra−/− animals treated with DMSO vehicle control slept significantly less than their egfra+/+ and egfra+/− siblings during the day (Fig. 5F), as expected. RF9 treatment also decreased sleep in egfra+/+ and egfra+/− animals (Fig. 5F), as previously reported (35). However, RF9 treatment did not further decrease sleep in egfra−/− animals compared to egfra−/− animals treated with DMSO vehicle control (Fig. 5F). These results are consistent with the hypothesis that NPVF signaling acts downstream of EGFR signaling to promote normal sleep levels.
EGFR signaling regulates NPVF expression
While genetic experiments in C. elegans (21, 22) and Drosophila (40) and our experiments in zebrafish suggest that EGFR signaling promotes sleep via RFamide neuropeptides, it is unknown how the EGFR and RFamide neuropeptide systems interact. One possibility is that EGFR signaling regulates expression of RFamide neuropeptides. We tested this hypothesis by assaying NPVF mRNA and protein levels in response to gain or loss of EGFR signaling. Using ISH, we found that overexpression of TGFa increased npvf mRNA level in endogenous npvf-expressing neurons by 68% compared to WT siblings (P < 0.05, two-way ANOVA, Holm-Sidak test; fig. S7, A and B). Conversely, treatment with the EGFR antagonist gefitinib decreased npvf mRNA level in these neurons by 19% (P < 0.05, Student’s t test; fig. S7, C and D). Similarly, using an NPVF-specific antibody (42), we found that TGFa overexpression increased NPVF protein level in the hypothalamus by 57% compared to WT siblings (P < 0.005, two-way ANOVA, Holm-Sidak test; Fig. 5, A and B), while gefitinib treatment decreased NPVF protein level by 17% compared to DMSO-treated siblings (P < 0.01, Student’s t test; Fig. 5, C and D). In contrast, overexpression of the sleep-promoting neuropeptide Y (NPY) (43) or the wake-promoting neuropeptide Hcrt (28) did not affect NPVF protein levels (fig. S7, E to H). In addition, TGFa overexpression did not affect Hcrt protein levels (fig. S7, I and J). These results suggest that EGFR signaling regulates sleep by specifically regulating expression of NPVF.
EGFR signaling modulates the activity of npvf-expressing neurons
We previously showed that optogenetic and chemogenetic stimulation of npvf-expressing neurons is sufficient to induce sleep (35), suggesting that, in addition to affecting NPVF expression, EGFR signaling may also promote sleep by stimulating npvf-expressing neurons. To test this hypothesis, we generated Tg(npvf:GCaMP6s-T2A-tdTomato) animals, in which npvf-expressing neurons express stoichiometric levels of GCaMP6s and tdTomato (44). We used GCaMP6s to monitor calcium levels, a proxy for neuronal activity, in npvf-expressing neurons and normalized GCaMP6s fluorescence intensity values to tdTomato fluorescence to correct for any changes in transgene expression or movement artifacts during live imaging. To test the hypothesis that EGFR signaling is sufficient to stimulate npvf-expressing neurons, we mated Tg(npvf:GCaMP6s-T2A-tdTomato) to Tg(hs:tgfa)/+ fish, imaged GCaMP6s and tdTomato in 5-dpf animals (Fig. 5G), and then used PCR genotyping to identify Tg(hs:tgfa) and WT siblings. Control animals not subjected to a HS displayed no difference in GCaMP6s/tdTomato fluorescence between Tg(hs:tgfa) and WT siblings (Fig. 5H). However, at 2 hours after HS, Tg(hs:tgfa) animals displayed significantly higher levels of GCaMP6s/tdTomato fluorescence compared to WT siblings (P < 0.005, two-tailed Wilcoxon matched-pairs signed-rank test; Fig. 5I), suggesting that TGFa overexpression stimulates npvf-expressing neurons.
We next tested whether EGFR signaling is necessary for normal activity levels of npvf-expressing neurons by inhibiting EGFR signaling. We first quantified baseline GCaMP6s/tdTomato fluorescence levels in Tg(npvf:GCaMP6s-T2A-tdTomato) animals. We then treated animals with either gefitinib or DMSO vehicle control and reimaged the same neurons 90 min later. Last, we calculated normalized GCaMP6s/tdTomato fluorescence values by dividing posttreatment values by baseline values. Consistent with the TGFa overexpression result, we found that inhibition of EGFR signaling using gefitinib treatment decreased normalized GCaMP6s/tdTomato fluorescence values compared to DMSO-treated siblings (P < 0.05, two-tailed Wilcoxon matched-pairs signed-rank test; Fig. 5J). This result suggests that EGFR signaling is required to maintain normal activity levels of npvf-expressing neurons. Together, these observations suggest that EGFR signaling promotes sleep, at least in part, by stimulating npvf-expressing neurons. More broadly, our results indicate that EGFR signaling promotes sleep via RFamide neuropeptides in both invertebrates and vertebrates.
Common genetic variants at EGFR signaling–associated genes affect human sleep
Having shown that regulation of sleep by EGFR signaling is conserved between invertebrate and vertebrate animal models, we next asked whether this conservation extends to humans. We tested for an association between self-reported sleep traits and genetic variation in EGFR signaling–associated gene regions in participants of European ancestry from the U.K. Biobank (tables S1 and S2) (24). ERBB4 is a member of the ERBB family of receptor tyrosine kinases that can heterodimerize with ERBB1/EGFR and can be activated by EGF family ligands (45) and thus participates in EGFR signaling. We found that genetic variation (rs7607363 G allele) at the ERBB4 genomic locus is associated with increased frequency of excessive daytime sleepiness (EDS) (P = 8.00 × 10−9, linear regression analysis; Fig. 6, A and B, and table S1). We also found that genetic variation (rs1846644 C allele) at the genomic locus of kinase suppressor of ras 2 (KSR2), a scaffolding protein with established roles in EGFR/MAPK signaling (46), is associated with increased sleep duration (P = 5.30 × 10−9, linear regression analysis), increased frequency of daytime napping (P = 2.00 × 10−41, linear regression analysis), and increased frequency of EDS (P = 2.50 × 10−27, linear regression analysis) (Fig. 6, A and C to E, and table S1). In addition, pathway analysis for EDS revealed associations at EGFR signaling pathways enriched in the sleep propensity subtype of daytime sleepiness (4). Together, these results suggest that the identified genetic variants in the ERBB4 and KSR2 loci associate with measures of increased sleep.
Fig. 6. Genetic variants at KSR2 and ERBB4 associate with human sleep quality and quantity.
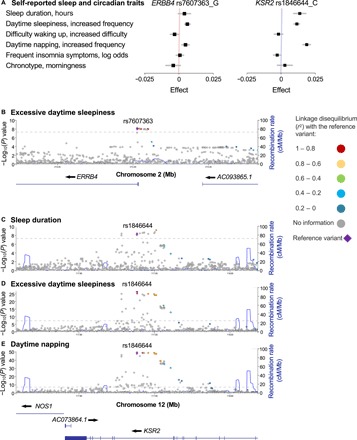
(A) Forest plot of associations between genetic variant rs7607363 in ERBB4 and rs1846644 in KSR2 with sleep quality and quantity measures. Black boxes indicate effect estimate, and gray lines represent 95% confidence interval. Data are based on 453,964 human subjects of European ancestry in the U.K. Biobank. Full results for each single-nucleotide polymorphism (SNP) are shown in table S1. (B) Regional association plot for genome-wide significant association between ERBB4 rs7607363 and EDS. (C to E). Regional association plots for genome-wide significant association between KSR2 rs1846644 and sleep duration (C), EDS (D), and daytime napping (E). Genes within each region are shown in the lower panels. Blue lines indicate recombination rate. Filled circles show the −log10 P value for each SNP, with the named SNP shown in purple. Additional SNPs in the locus are colored according to correlation (r2) with the lead SNP, estimated by LocusZoom based on the CEU HapMap haplotypes. Dashed lines represent the genome-wide significance threshold (P < 5 × 10−8).
Expression quantitative trait loci (eQTL) analysis of the ERBB4 variant rs7607363 showed increased ERBB4 expression associated with the minor allele that is associated with increased sleep (fig. S8A) and no association with expression levels of neighboring genes, suggesting a gain of function associated with this single-nucleotide polymorphism (SNP). The lead SNPs in the ERBB4 and KSR2 loci, as well as several SNPs highly linked to the lead SNPs, are aligned within each gene, with recombination hotspots separating them from neighboring genes (Fig. 6, B to E). These observations provide supporting evidence that the SNPs are linked to ERBB4 and KSR2. However, since SNPs may influence distant genes, we cannot be absolutely certain that ERBB4 or KSR2 is the causal gene at each locus. Despite this caveat, these results suggest that common variants in EGFR signaling pathway components underlie some of the natural variation observed in human sleep.
To validate the association of KSR2 and ERBB4 with sleep traits in vivo, we treated zebrafish with small-molecule inhibitors of these proteins (fig. S8). First, we treated WT zebrafish with APS-2-79 (fig. S8, J to Q), a small molecule that stabilizes KSR2 in an inactive state (46). Treatment with this drug resulted in decreased sleep during both the day (−38%) and night (−14%) compared to DMSO vehicle-treated controls (day, P < 0.05; night, P < 0.005, Student’s t test; fig. S8, M and N). These effects were primarily due to shorter sleep bouts at night and a trend of higher activity and waking activity during the day (fig. S8, J to L and P). This phenotype is consistent with the finding that Ksr2 knockout mice exhibit increased locomotor activity (47). Next, we treated WT zebrafish with spironolactone (fig. S8, B to I), an ERBB4 small-molecule inhibitor (48). Treatment with this drug resulted in a 7% increase in locomotor activity and a 34% decrease in sleep during the day (activity, P < 0.05; sleep, P < 0.01, Student’s t test), with no significant effect at night, compared to DMSO vehicle-treated siblings (fig. S8, B, C, E, and F). These phenotypes were associated with higher waking activity and fewer sleep bouts during the day (fig. S8, D and G). Thus, inhibition of KSR2 or ERBB4 phenocopies loss of EGFR signaling, although the spironolactone result should be interpreted with caution because this drug has been shown to also target other proteins (48). These results validate a genetic pathway that underlies some of the observed variation in human sleep quality and quantity.
DISCUSSION
There is abundant evidence that sleep is behaviorally conserved throughout evolution (9), and genetic mechanisms that regulate sleep have been described in both vertebrate and invertebrate species (9). However, while it has been shown that genes that regulate circadian rhythms and thus affect sleep timing are conserved in invertebrates and humans (1, 9), there is relatively little clear and unambiguous evidence for conserved mechanisms that regulate sleep in both vertebrates and invertebrates, including humans. As a result, the extent to which mechanisms that underlie sleep evolved independently in vertebrates and invertebrates remains unclear. Identifying such mechanisms is important not only from an evolutionary standpoint but also because these mechanisms would likely play a central role in regulating sleep in all organisms. Here, we describe a genetic mechanism that regulates sleep in both invertebrates and vertebrates and provide evidence that these findings may extend to humans.
We report that EGFR signaling regulates sleep in the zebrafish, a diurnal vertebrate. We found that TGFa overexpression increased sleep, whereas mutation of EGFR or its ligands, or pharmacological inhibition of EGFR signaling, decreased sleep. The reduced sleep resulting from inhibition of EGFR signaling was due to defects in both initiation and maintenance of sleep, consistent with the human sleep disorder insomnia. While gain- and loss-of-function studies have shown that EGFR signaling is both necessary and sufficient for normal sleep levels in C. elegans and Drosophila (12, 13, 16), our results extend these invertebrate studies by demonstrating that this mechanism is conserved in a vertebrate and identify a previously undescribed role of EGFR signaling in regulating sleep homeostasis. In both zebrafish and Drosophila (13), animals with elevated EGFR signaling had more and longer sleep bouts, whereas pharmacological inhibition of EGFR signaling resulted in shorter sleep bouts, indicating that EGFR signaling promotes consolidation of the sleep state. Combined with our discovery that common variants in two genes that participate in EGFR signaling are associated with variation in human sleep, this study provides one of the first clear examples of a mechanism that regulates sleep in both invertebrate and vertebrate species and suggests that these findings may extend to humans. These results reveal a clear evolutionary link between invertebrate and vertebrate sleep, suggest that EGFR signaling participates in an ancient and central sleep regulatory mechanism, and provide a basis to explore central and ancient mechanisms that regulate vertebrate sleep.
In contrast to our findings using zebrafish and previous invertebrate studies (12, 13, 16, 19), the role of EGFR signaling in mammalian sleep is unclear. While EGFR signaling has been shown to regulate various rodent behaviors (14, 15), attempts to explore its requirement for sleep have been confounded by the embryonic lethality of murine Egfr-null mutants (32). Consistent with a role for EGFR signaling in rodent sleep, one study using mice homozygous for the Egfrwa2 hypomorphic allele, which produces a receptor with reduced tyrosine kinase activity, observed increased daytime locomotor activity (14). However, this allele contains a linked hypomorphic mutation in Wnt3a (14) that may confound sleep studies. Wnt signaling is required for brain development (49) and has been implicated in regulating sleep duration in Drosophila (18). Mice lacking TGFa or EGF are viable and fertile, but sleep studies using these mutants have not been described. Reports of EGFR gain-of-function manipulations on mammalian sleep are also inconclusive. Acute intracerebroventricular injection of EGF into rabbits was sufficient to increase sleep (20), although the effect required the use of high EGF levels that are unlikely to be physiologically relevant. In contrast, infusion of TGFa had no effect on sleep duration in hamsters, although it did suppress wheel running (14, 15). These conflicting gain-of-function results could be due to the use of small numbers of animals, nonphysiological peptide concentrations resulting in nonspecific behavioral effects, or the use of in vitro synthesized peptides that may lack posttranslational modifications required for function. Other indirect evidence supporting a role for EGFR signaling in mammalian sleep was provided by the recent finding that inhibition of ERK results in reduced sleep in rodents (39). However, ERK participates in many signal transduction pathways, so it is unclear whether this effect is specific to EGFR signaling. Thus, before our study, it remained unclear whether EGFR signaling plays a role in vertebrate sleep.
Consistent with a conserved role for EGFR in sleep is its expression in the hindbrain and hypothalamus in both rodents and zebrafish. These brain structures are anatomically and molecularly conserved in mammals and zebrafish (10) and play prominent roles in sleep regulation (50). In rodents, Egfr is expressed in juxtaventricular radial-glial–like cells named tanycytes (51), which are responsive to photoperiod and regulate energy balance (52), and Tgfa is expressed in nearby ependymal cells (27, 53). Similarly, in zebrafish, egfra is expressed in juxtaventricular cells that express the glial cell markers sox2 and glial fibrillary acidic protein, and tgfa is expressed in an adjacent sox2-expressing glial cell population. These observations suggest a conserved neuroanatomical basis for Tgfa/Egfr-regulated sleep. This possibility is supported by the observations that TGFa overexpression in zebrafish results in c-fos expression in egfra-expressing juxtaventricular cells, while intracerebroventricular injection of either TGFa or EGF in hamsters similarly results in ERK activation in Egfr-expressing juxtaventricular tanycytes (15, 51). Studies in mammals have shown that these tanycytes can sense and transport molecules from the cerebrospinal fluid (CSF) to brain regions implicated in regulating sleep (54) and that CSF contains sleep-inducing factors (55). Together, these observations suggest that Tgfa/Egfr signaling may regulate sleep through interactions between CSF, specialized glial cells, and neurons. This possibility contrasts with most previously described neuropeptidergic and neuromodulatory mechanisms that regulate sleep, which act via communication between neurons, but is consistent with the model that tanycytes act as gatekeepers that monitor homeostatic signals in CSF (52), including those that regulate sleep (55).
Before this study, the molecular and cellular mechanisms through which EGFR regulates invertebrate sleep were unclear. It has been shown that EGF-induced sleep in C. elegans is suppressed by mutation of the RFamide neuropeptides flp-13 and flp-24 (21, 22). Likewise, in Drosophila, cellular stress–induced sleep, which is thought to be mediated by EGFR signaling, is suppressed by mutation of the RFamide neuropeptide fmrfamide (40). However, these studies only demonstrated genetic interactions, and the mechanism linking the EGFR and RFamide systems was unknown. We previously showed that NPVF, a member of the vertebrate RFamide family of neuropeptides, as well as npvf-expressing neurons, are necessary and sufficient for normal sleep amount in zebrafish (35), similar to our findings for EGFR signaling in this study. Our demonstration that EGFR signaling promotes sleep in part via the NPVF system in zebrafish suggests that this aspect of EGFR-regulated sleep is conserved, although it is unclear whether the vertebrate and invertebrate RFamide proteins are true orthologs owing to low sequence conservation (35). In addition to showing a genetic interaction, we found that EGFR signaling regulates both NPVF expression and the activity of npvf-expressing neurons, providing a molecular and cellular link between the two pathways. However, we did not observe egfra expression in npvf-expressing neurons, so it remains unclear how EGFR signaling affects NPVF expression and the activity of npvf-expressing neurons. One possibility is that egfra is expressed in npvf-expressing neurons at levels too low to detect using ISH. Alternatively, presently unknown genes and/or neurons may act to link the EGFR and NPVF systems. Further studies are also needed to determine whether EGFR signaling regulates the expression of invertebrate RFamide neuropeptides and the activity of neurons that express them, and whether mechanisms that act upstream of EGFR signaling to promote invertebrate sleep, such as cellular stress, are conserved in vertebrates.
Are these experimental findings in model systems relevant to humans? Our analysis of several self-reported sleep traits (N = 450,000) in humans of European ancestry from the U.K. Biobank (4–8, 24) identified genome-wide significant associations of several sleep traits with common variants in the KSR2 and ERBB4 genomic loci. KSR2 is a scaffolding protein that enables interaction of the EGFR signaling components RAF, MEK, and ERK, and thus is crucial for EGFR signaling (46). ERBB4 is a member of the ERBB1/EGFR receptor tyrosine kinase subfamily that binds several EGFR ligands and forms heterodimers with other ERBB family members, including EGFR (45). Our results suggest that common variants in EGFR signaling pathway genes underlie some of the variation observed in human sleep, including traits such as sleep duration, daytime napping, and EDS, suggesting gain-of-function effects leading to an increased sleep propensity. We found that genetic loss of egfra, egf, or tgfa, as well as pharmacological inhibition of EGFR, KSR2, or ERBB4, results in reduced sleep in zebrafish, consistent with the human data. Our results are consistent with a report that elevated TGFa serum levels are associated with fatigue in humans (56). Furthermore, our results suggest that the disrupted sleep commonly observed in RASopathies (57), a group of human developmental syndromes caused by mutations in genes involved in Ras-MAPK signaling, may be directly caused by abnormal EGFR signaling, rather than occurring as a secondary consequence of abnormal development. Last, we note that except for narcolepsy, which is due to loss of Hcrt neurons (1), and circadian disorders that affect sleep timing (1), the basis for human sleep disorders is largely unknown, and our study provides one of the first examples of a genetic mechanism that likely affects human sleep. Further studies using animal models, such as zebrafish, will provide a deeper mechanistic understanding of the role of EGFR signaling in sleep and how abnormal EGFR signaling results in sleep disorders.
MATERIALS AND METHODS
Experimental model
Zebrafish experiments and husbandry followed standard protocols (58) in accordance with Caltech Institutional Animal Care and Use Committee guidelines. Larval zebrafish were studied before the onset of sexual differentiation, and all behavioral experiments were performed using siblings with the same genetic background, differing only in the presence of a transgene, mutation of a specific gene, or treatment with drugs and appropriate vehicle control. The age of animals used in each experiment is described in the manuscript, in each figure legend, or in this section. All experiments used animals from a TLAB strain. Unless otherwise noted, larvae were raised and behaviorally tested in a 14-hour light/10-hour dark cycle, with white lights on from 9 a.m. to 11 p.m.
Transgenic and mutant zebrafish
Tg(hs:tgfa)
Zebrafish tgfa complementary DNA (cDNA) was isolated using 5′ and 3′ RACE (FirstChoice RLM-RACE, AM1700, Thermo Fisher Scientific), and the open reading frame (ORF) was cloned downstream of the zebrafish hsp70c promoter (59) in a vector containing flanking I–SceI endonuclease recognition sites. We generated stable transgenic animals by injecting plasmids with I–SceI (New England Biolabs Inc.) into the cell of embryos at the one-cell stage (60). To identify transgenic founders, we outcrossed potential founders, heat-shocked their progeny at 5 dpf, fixed animals 30 min later, and performed tgfa ISH. At least two lines that produced strong and ubiquitous overexpression and lacked pre-HS expression were selected and tested for behavioral phenotypes. The two independent Tg(hs:tgfa) transgenic lines produced similar behavioral phenotypes, but all data in the paper are from the line that produced stronger phenotypes. Transgenic animals were identified by PCR using the primers 5′-CGGGCCACCATGATGTAT-3′ and 5′-GGTTTGTCCAAACTCATCAATGT-3′ with a product size of 647 base pairs (bp).
Tg(npvf:GCaMP6s-2A-tdTomato)
Using Gibson assembly, the 3.9-kb npvf promoter (35) was cloned upstream of GCaMP6s (61), a T2A sequence (62), and tdTomato. Transgenic lines were generated using the I–SceI method (60).
tgfa mutant
Using TALENs (30), we isolated a zebrafish tgfa mutant that contains a 7-bp deletion (ORF nucleotides 25 to 31: 5′-ACAATAT-3′) that shifts the translational reading frame after the eighth amino acid and introduces a premature stop codon that should truncate the protein from 189 to 61 amino acids (fig. S2A). The predicted mutant protein lacks the signal peptide, protease cleavage sites, and epidermal growth factor–like domain and should thus be nonfunctional. Tgfa−/− zebrafish are viable, and homozygous mutant males and females are fertile, with no gross morphological abnormalities. Mutant animals were genotyped using the primers 5′-GACGACGCTGAGAATCTTTCATC-3′ and 5′-CAATCGTTTTGGTCTTTGCAT-3′, which produce a 188- or 181-bp band for the WT or mutant allele, respectively.
egf mutant
Using CRISPR-Cas9 (31), we isolated a zebrafish egf mutant that contains a 19-bp insertion and a 2-bp deletion after ORF nucleotide 426 that shifts the translational reading frame and introduces a premature stop codon that should truncate the protein from 1177 to 457 amino acids (fig. S2B). The predicted mutant protein lacks five EGF-like domains and the transmembrane domain and should thus be nonfunctional. Egf−/− zebrafish are viable, and homozygous mutant males and females are fertile, with no gross morphological abnormalities. Mutant animals were genotyped using the primers 5′-GCTGTGGATTGTGATCATGC-3′ and 5′-TGCAGAAAAACAGCCTGAAA-3′, which produce a 204- or 221-bp band for the WT or mutant allele, respectively.
egfra mutant
Using CRISPR-Cas9 (31), we generated a zebrafish egfra mutant that contains an 11-bp deletion and a 27-bp insertion after nucleotide position 792 that shifts the translational reading frame and introduces a premature stop codon upstream of the transmembrane and intracellular domains, and should thus produce a non-functional protein (fig. S2C). egfra−/− animals were viable and lacked obvious morphological abnormalities. egfra−/− males were fertile, but egfra−/− females were seemingly infertile. Mutant animals were genotyped using the primers 5′-ACTTCCAGGATGAAGGGACA-3′ and 5′-CTTACGTGGGCATGTCTTGA-3′, which produced a 137- or 153-bp band for the WT or mutant allele, respectively.
Tg(hs:hcrt) zf12Tg (28), Tg(hs:npy) ct853Tg (43), and npvf ct845 mutant (35) zebrafish have been previously described.
Zebrafish behavioral assays
Locomotor activity assay
Individual larvae were placed into each well of a 96-well plate (7701-1651, Whatman) containing 650 μl of E3 embryo medium [5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4 (pH 7.4)] at 4 dpf and acclimated overnight in the video tracker before behavioral analysis beginning at 5 dpf. Plates were sealed with an optical adhesive film (4311971, Applied Biosystems) to prevent evaporation, except for drug experiments. Animals in unsealed plates can appear to have higher amounts of sleep compared to those in sealed plates because as water evaporates from each well, it forms a concave meniscus at the top of the well, which creates a slight visual distortion that causes the tracking system to miss some animal movements. To mitigate this issue, each well was filled to the top with water each morning during unsealed plate experiments. The sealing process introduces air bubbles in some wells, which are discarded from analysis. Animals were blindly assigned a position in the plate and were genotyped after the behavioral experiment was complete. Locomotor activity was monitored using a video tracking system (Viewpoint Life Sciences) with a Dinion one-third inch monochrome camera (Dragonfly 2, Point Grey) fitted with a fixed-angle megapixel lens (M5018-MP, Computar) and infrared filter. For HS-induced overexpression experiments, larvae were heat-shocked at 37°C for 1 hour starting at 3 p.m., 4 p.m., or 9:45 p.m. at 5 dpf. The movement of each larva was captured at 15 Hz and recorded using the quantization mode with 1-min time bins. The 96-well plate and camera were housed inside a custom-modified ZebraBox (Viewpoint Life Sciences) that was continuously illuminated with infrared LEDs and illuminated with white LEDs from 9 a.m. to 11 p.m., except as noted in experiments where animals were entrained in a 14-hour light/10-hour dark cycle and then assayed in constant light or constant dark. The 96-well plate was housed in a chamber filled with recirculating water to maintain a constant temperature of 28.5°C. The parameters used for detection were as follows: detection threshold, 15; burst, 29; freeze, 3, which were determined empirically. Data were processed using custom PERL and MATLAB (MathWorks Inc.) scripts, and statistical tests were performed using Prism (GraphPad) for ANOVA analysis and Excel (Microsoft) for Student’s t test.
A movement was defined as a pixel displacement between adjacent video frames preceded and followed by a period of inactivity of at least 67 ms (the limit of temporal resolution). Any 1-min period with no movement was defined as 1 min of sleep based on arousal threshold changes (28). A sleep bout was defined as a continuous string of sleep minutes. Sleep latency was defined as the length of time from lights on or off to the start of the first sleep bout. Average activity was defined as the average amount of locomotor activity in seconds per hour, including sleep bouts. Average waking activity was defined as the average amount of activity in seconds per hour, excluding sleep bouts.
Arousal threshold assay
We used an Arduino-based automated driver to control two solenoids (28P-I-12, Guardian Electric) that delivered a tap over an intensity range of 4095 settings (from 0.01 to 40.95) to a 96-well plate in the video tracker (35). We used taps ranging from a power setting of 1 to 36.31. Taps of 14 different intensities were applied in a random order with an intertrial interval of 1 min during the day or night for Tg(hs:tgfa) or gefitinib and erlotinib experiments, respectively. Previous studies showed that a 15-s intertrial interval is sufficient to prevent behavioral habituation (63). The background probability of movement was calculated by identifying for each genotype the fraction of animals that moved 5 s before all stimuli delivered during an experiment (14 different tap powers × 30 trials at each intensity = 420 data points per animal). This value was subtracted from the average response fraction value for each tap event. The response of larvae to the stimuli was monitored using the video tracking software and was analyzed using MATLAB and Excel. Statistical analysis was performed using the Variable Slope log(dose) response curve fitting module of Prism.
SD assay
At 4 dpf, up to 84 animals were placed in a 96-well plate that had been cut into two separable halves that can be rejoined to fit inside the video tracker. Animals were monitored for 48 hours (5 to 6 dpf) to measure baseline sleep. Experiments in which significant differences were observed between the two halves of the plate during the 24 hours before perturbation were excluded from analyses. Immediately before the start of the third night of analysis (7 dpf), a half-plate from each of two video trackers was transferred and fastened to the center of a 245-mm2 square dish (431301, Corning) containing 20 glass beads, 6 plastic marbles, and 8 fixed pegs that prevent the marbles and beads from only circling the perimeter of the plate during nutation. For night-perturbed animals, the plates were wrapped in aluminum foil to shield the animals from light, fastened to a nutator (B3D1020, Benchmark Mini BioMixer), and nutated in a dark 28°C incubator for 6 hours. For day-perturbed experiments, animals of the same developmental stage were treated the same, except the plates were exposed to light during perturbation during the day at 7 dpf. In addition to mechanical stimulation, we simultaneously subjected the animals to auditory stimuli by playing songs from a percussive recording in random order (Death Magic, Health) at maximum volume using a portable Bluetooth speaker (DKnight MagicBox II) that was housed in the incubator. For each SD half-plate, the corresponding half remained inside the video tracker to serve as a nonperturbed control. At the end of the 6-hour perturbation, the SD half-plates were rejoined with their nonperturbed control halves in their respective video tracker. Night-perturbed samples were kept in the dark when transferred from the SD assay into video trackers, and both night- and day-perturbed samples were maintained in dark for the rest of the experiment to avoid masking effects of light on any sleep recovery response.
Behavioral data analysis
sort_fish_sttime_192.pl. is a Perl script (28) that converts data acquired by the Viewpoint system to a format that is useful for MATLAB analysis. perl_batch_192well.m is a MATLAB script that allows the sort_fish_sttime_192.pl. script to run using MATLAB. TapAnalysis.m is a MATLAB script that analyzes tapping assay data and generates a table that lists the number of animals that moved following each tap. VT_analysis.m is a MATLAB script [modified from (28)] that performs sleep/wake analyses. These scripts are posted online (35), and detailed instructions on their use will be provided upon request.
Histology
In situ hybridization
Samples were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 16 hours at room temperature. ISH was performed using digoxigenin (DIG)–labeled antisense riboprobes (DIG RNA Labeling Kit, Roche) (35). Double FISH was performed using DIG- and fluorescein-labeled riboprobes and the TSA Plus Fluorescein and Cyanine 3 Systems kit (PerkinElmer). Probes specific for npvf (35) and c-fos (35) have been described. The tgfa probe was transcribed using a PCR product amplified from zebrafish cDNA using the primers 5′-CGCGTGCCTTCATCTTTATT-3′ (forward) and 5′-TCCCACTGCCCATATTGAAC-3′ (reverse) and then serially amplified with the same forward primer and a T7 sequence (lowercase) added to the primer 5′-ggatcctaatacgactcactatagggTCCCACTGCCCATATTGAAC-3′ (reverse). The egfra probe was transcribed using a PCR product amplified from zebrafish cDNA using the primers 5′-TCTGCTTGACCTCCACAGTG-3′ (forward) and 5′-CAAGGGCAAAAGTACCTGATCG-3′ (reverse) and then serially amplified with the same forward primer and a T7 sequence (lowercase) added to a reverse primer located within that amplicon: 5′-ggatcctaatacgactcactatagggTGTAACAAGCTGAAGTAACGTGC-3′. The sox2 probe was transcribed using a PCR product amplified from zebrafish cDNA using the primers 5′-GGTCAACCAGAGGATGGACA-3′ (forward) and 5′-AACTGCGTACAGGCCACATT-3′ (reverse) and then serially amplified with the inner primer 5′-GGCCGACAGCTACGCGCATATGAA-3′ (forward) and a T7 sequence (lowercase) added to a primer located within that amplicon: 5′-taatacgactcactatagggGCTGGAGAGTGCCTCTGTTC-3′.
Immunohistochemistry
Samples were fixed in 2% PFA in PBS overnight at 4°C and then washed with 0.25% Triton X-100/PBS (PBTx). Brains were dissected and blocked in 2% goat serum/2% DMSO/PBTx overnight at 4°C. Antibody incubations were performed in blocking solution overnight at 4°C using rabbit anti-NPVF (1:200; ab122738, Abcam) or rabbit anti-Hcrt (1:500; AB3704, MilliporeSigma) primary antibodies and Alexa Fluor 568 secondary antibody (1:500; Life Technologies). Samples were mounted in Vectashield (H-1000, Vector Labs) and imaged using a Zeiss LSM 780 confocal microscope.
Pharmacology
A dose-response experiment was first performed for each drug to determine its optimal concentration. Each drug was dissolved in DMSO and then added to E3 medium. The following drug concentrations were tested: DMSO vehicle control, 0.1, 0.5, 1, 5, 10, 50, and 100 μM. Gefitinib (13166, Cayman Chemical) and erlotinib (10483, Cayman Chemical) were not soluble at 50 or 100 μM, and treatment of zebrafish with 10 μM of either drug produced developmental defects in a small number of animals. Treatment with 5 μM gefitinib or erlotinib did not cause developmental defects but did result in increased locomotor activity and decreased sleep compared to DMSO vehicle-treated controls. Optimal concentrations for other drugs were determined in a similar fashion and were used in experiments at the following concentrations: 15 μM U0126 (1144, Tocris), 3 μM SL327 (1969, Tocris), 10 μM RF9 (3672, Tocris), 0.2 μM spironolactone (2968, Tocris), and 5 μM APS-2-79 (S8355, Selleck Chemicals). Drug solutions were freshly prepared before each experiment. Controls were exposed to DMSO at concentrations identical to experimental conditions, at no more than 0.1% DMSO. Drugs and DMSO vehicle were loaded into alternating rows of a 96-well plate or loaded on opposite sides of a 96-well plate. WT larvae from the same clutch were then added to the plate. For RF9 experiments and gefitinib constant dark and constant light experiments, drug and DMSO vehicle were loaded into separate sides of a 96-well plate with an empty row in between. WT larvae from the same clutch were added to the plate, and plates were sealed with an optical adhesive film (4311971, Applied Biosystems) to prevent evaporation.
Quantitative polymerase chain reaction
Zebrafish were raised on a 14-hour light/10-hour dark cycle at 28.5°C with lights on at 9 a.m. and lights off at 11 p.m. At 5 dpf, total RNA was collected using TRIzol reagent (15596-026, Life Technologies) from 20 pooled larvae per biological replicate every 6 hours for 36 hours, with three independent biological replicates at each time point. cDNA was synthesized from 5 μg of total RNA using SuperScript III Reverse Transcriptase (18080-051, Invitrogen), and qPCR was carried out using SYBR Green Master Mix (4364346, Life Technologies) using an ABI PRISM 7900HT (Life Technologies) instrument using the primers 5′-GTGTGTGGTGGGCAGTGTC-3′ and 5′-CCAACAGGAGAGGGTGTGAC-3′ for tgfa and 5′-ATCCAGACCCCAATACAAC-3′ and 5′-GGGAGACTCTGCTCCTTCT-3′ for per1b. ef1a was used as a reference gene using the primers 5′-CAGCTGATCGTTGGAGTCAA-3′ and 5′-TGTATGCGCTGACTTCCTTG-3′. Relative expression levels were determined using the ΔΔCt method (64), normalized to the highest cycle threshold (Ct) value for each gene.
Imaging
Dissected brains were coverslip-mounted in Vectashield (H-1000, Vector Labs) or 80% glycerol in PBS and imaged using a compound microscope [AxioImager with EC Plan-Neofluar 10×/0.30–numerical aperture (NA) air objective or Plan-Apochromat 20×/0.8-NA air objective, Zeiss] for chromogenic ISH samples or using a confocal microscope (LSM 780 with Plan-Apochromat 10×/0.45-NA air objective, LD LCI Plan-Apochromat 25×/0.8-NA Imm Corr objective, or LD C-Apochromat 40×/1.1-NA water objective; Zeiss) for FISH samples. Fluorescein and cyanine 3 were imaged in separate channels with 488- and 561-nm lasers, respectively. Confocal images were displayed as an optical section of approximately 1–airy unit thickness.
For c-fos and npvf mRNA quantification, chromogenic ISH was performed using c-fos and npvf riboprobes on 5-dpf dissected brains, with experimental and control samples developed in the same tube blinded to genotype (Fig. 4, H to L, and fig. S7, A and B) or with experimental and control samples developed in different tubes in parallel and treated identically (fig. S7, C and D). Samples were imaged using a Zeiss compound microscope and then genotyped by PCR. Regions of interest (ROIs) were drawn over hypothalamic or hindbrain regions containing ISH signal, along with a nonlabeled area of the brain that was selected for background correction. The ISH signal was quantified using ImageJ (65), with background subtraction correction. Average pixel intensity within the ROI was quantified blind to genotype or treatment.
For NPVF (Fig. 5, A and B, and fig. S7, E to H) and Hcrt (fig. S7, I and J) protein quantification, experimental and control brains were immunostained in the same tube, imaged using a Zeiss confocal microscope, and then genotyped by PCR. For NPVF protein quantification on drug-treated animals (Fig. 5, C and D), experimental and control brains were immunostained in different tubes in parallel and treated identically. An ROI containing all NPVF soma was drawn in each brain hemisphere to calculate mean fluorescence intensity, and an unlabeled area in the image was selected for background correction using ImageJ (65). ROIs were kept the same size and shape but moved to contain as many NPVF soma as possible across images. Average pixel fluorescence intensity was quantified blind to genotype or treatment.
For GCaMP6s experiments, 5-dpf Tg(npvf:GCaMP6s-2A-tdTomato) animals were mounted in low-melting agarose and then imaged once per minute for 5 min. The average pixel intensity of GCaMP6s and tdTomato fluorescence in each neuron was averaged for the five time points. tdTomato fluorescence was used to normalize GCaMP6s fluorescence to control for movement artifacts or changes in transgene expression. For Tg(hs:tgfa) experiments, these animals and their WT siblings were either not heat-shocked or heat-shocked for 30 min at 37°C and then embedded in agarose 30 min later. Animals were imaged 2 hours after heat shock. For gefitinib experiments, animals were head-fixed in agarose and imaged, then treated with either DMSO or 10 μM gefitinib in E3 medium, and then imaged again 30 min later. A rolling ball algorithm (ImageJ) was used for background correction. ROIs were drawn over each cell in the red channel, and the average pixel intensity of the green channel (GCaMP6s) was divided by the average pixel intensity of the red channel (tdTomato). NPVF soma with saturated fluorescence were excluded from quantification. Images were quantified blind to treatment or condition.
Human study population and phenotypes
This study was conducted on participants of European ancestry from the U.K. Biobank study of more than 500,000 people aged 40 to 69 living in the United Kingdom (24). We identified two genome-wide significant signals that localize within genes broadly implicated in EGFR/MAPK signaling using summary statistics of GWAS available at the Sleep Disorder Knowledge Portal: http://sleepdisordergenetics.org/ (4–8). Self-reported heath questionnaire, anthropometric assessments, and demographic information were collected at baseline. To assess chronotype, subjects were asked, “Do you consider yourself to be…” with response options “Definitely a ‘morning’ person,” “More a ‘morning’ than ‘evening person,” “More an ‘evening’ than a ‘morning’ person,” “Definitely an ‘evening’ person,” “Do not know,” and “Prefer not to answer,” which were coded as 2, 1, −1, −2, 0, and missing, respectively. To assess difficulty waking up, participants were asked, “On an average day, how easy do you find getting up in the morning?” with responses “Not at all easy,” “Not very easy,” “Fairly easy,” “Very easy,” “Do not know,” and “Prefer not to say.” To assess self-reported sleep duration, participants were asked, “About how many hours sleep do you get in every 24 hours? (please include naps),” with responses in hour increments. To assess frequent insomnia symptoms, subjects were asked, “Do you have trouble falling asleep at night or do you wake up in the middle of the night?” with responses “never/rarely,” “sometimes,” “usually,” and “prefer not to answer.” Subjects who responded “Prefer not to answer” (n = 637) were set to missing. Insomnia symptoms were dichotomized into controls (“never/rarely”) and cases with frequent insomnia symptoms (“usually”), with those reporting “sometimes” excluded. To assess daytime napping, subjects were asked, “Do you have a nap during the day?” with response options “Never/rarely,” “Sometimes,” “Usually,” and “Prefer not to answer.” Self-reported EDS was ascertained using the question “How likely are you to dose off or fall asleep during the daytime when you don’t mean to? (e.g., when working, reading, or driving)” with response options “Never/rarely,” “Sometimes,” “Often,” “All of the time,” “Do not know,” and “Prefer not to answer.” Subjects reporting “Do not know” and “Prefer not to answer” were set to missing. Other responses were coded continuously as 1 to 4 corresponding to the severity of EDS. Subjective overall health was reported in response to the question “In general, how would you rate your overall health?” Those who self-reported “Excellent” or “Good” overall health were classified as “healthy,” and those who self-reported “Fair” or “Poor” overall health were classified as “unhealthy.”
Genetic association analysis
Genome-wide genotyping was performed by the U.K. Biobank, and genotyping, quality control, and imputation procedures have been previously described in detail (66). We further clustered subjects into four ancestry clusters using K-means clustering on the principal components, identifying 453,964 subjects of European ancestry. Genetic association analysis was performed, as previously described (7), in related subjects of European ancestry using BOLT (67) linear mixed models and an additive genetic model adjusted for age, sex, 10 principal components, genotyping array, and genetic correlation matrix with a maximum per-SNP missingness of 10% and per-sample missingness of 40%. We tested for an association between SNPs in the genomic intervals encompassing ERBB4 and KSR2. Genomic intervals were defined using recombination hotspots upstream and downstream of the gene of interest and correspond to the National Center for Biotechnology Information Build 37/hg19 positions 211.94 to 213.49 Mb for ERBB4 and 117.81 to 118.47 Mb for KSR2. Linear/logistic regression was performed adjusting for age, sex, 10 principal components of ancestry, and genotyping array. Additional adjustment for body mass index was performed in the regression model for difficulty waking up and EDS. Association testing for daytime napping excluded those with self-reported poor overall health (“unhealthy”; n = 60,712). Reported SNPs were significant at the stringent genome-wide significant level used in GWAS (P < 5 × 10−8). Locus zoom plots were generated using LocusZoom using the HG19 1000 genomes European genetic panel for genetic linkage data (68).
ERBB4 and KSR2 SNP eQTL analysis in Genome-Tissue Expression
The Genome-Tissue Expression (GTEx) Project (69) data portal (accessed 1 July 2019) was used to investigate genetic effects of the genome-wide significant association signals at ERBB4 and KSR2 on gene expression across 48 tissues (data source: GTEx data version V7; n = 620 individuals). GTEx version V7 eQTL analysis (described in the data portal) used gene expression measured using RNA sequencing (Illumina TruSeq) with inclusion of samples with an RNA integrity number (as measured by Agilent Bioanalyzer) of 6.0 or higher, and quality control measures included genes >0.1 transcripts per kilobase million in at least 20% of samples and ≥6 reads in at least 20% of samples. Expression values were quantile-normalized between samples, and for each gene, expression values were normalized across samples using an inverse normal transform. SNPs were genotyped in GTEx using whole-genome sequencing from blood samples. SNPs with a minor allele frequency ≥1% were used. Association analyses were linear regression analyses that were adjusted for sex, genotyping platform, and the top three genotyping-based principal components to adjust for potential population stratification by race/ethnicity and sample size–dependent complex nongenetic PEER (probabilistic estimation of expression residuals) factors (70). The normalized effect size of the eQTLs was defined as the slope of the linear regression based on the human genome reference GRCh37/hg19. The normalized effect sizes were computed in a normalized space where magnitude had no direct biological interpretation. A false discovery rate < 0.05 was used to correct for multiple hypothesis testing.
Statistical analysis
Behavioral data were processed using MATLAB (MathWorks), graphs were generated using Excel (Microsoft), and statistical analyses were performed using Prism 6 (GraphPad). The number of humans, animals, and statistical tests used are stated in each figure or figure legend. Line graphs in Figs. 1 to 4 and figs. S1 and S3 to S8 were generated from raw data and averaged over 1-hour bins in 10-min intervals to show underlying behavioral trends. Bar graphs show mean ± SEM. Box plots extend from the 25th to the 75th percentile, with the median marked by a horizontal line in the box, and whiskers extend from the 10th to the 90th percentile of data points. In all zebrafish statistical tests, the significance threshold was set to P < 0.05, and P values were adjusted for multiple comparisons where appropriate. Parametric analyses were applied because the data followed an approximately normal distribution in nearly all cases. Unpaired two-tailed Student’s t test was performed using Excel. One-way and two-way ANOVA and post hoc tests to correct for multiple comparisons were performed using Prism. The Holm-Sidak post hoc test was used to correct for multiple comparisons and to allow pairwise comparison of means for all samples. Asterisks in figures denote statistics for pairwise or multiple comparisons as indicated. The GCaMP6s/tdTomato fluorescence data and sleep rebound data were not normally distributed, so a Wilcoxon rank-sum nonparametric test was used to test for significant differences.
Supplementary Material
Acknowledgments
We thank members of the Prober Lab, P. Sternberg, R. Nath, H. Wang, C. Oikonomou, and J. Bedont for feedback. We also thank H. S. Dashti, C. Singh, D. Chilin, V. Sapin, T. Cammidge, H. Hurley, and C. Cook for technical assistance. Funding: This work was supported by grants from the National Institutes of Health (NIH) (D.A.L.: K99NS097683 and F32NS084769; G.O.: F32NS082010; D.A.P.: R01NS070911 and R01NS101158; J.M.L.: T32HL007567; R.S.: R01DK107859, R01HL113338, R01DK102696, and R01DK105072) and the Mallinckrodt (to D.A.P.), Rita Allen (to D.A.P.), and Brain and Behavior Research (to D.A.P. and D.A.L.) foundations. The GTEx Project was supported by the Common Fund of the Office of the Director of the NIH and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Author contributions: D.A.L., J.L., and D.A.P. conceived the project and designed the experiments. D.A.L. performed most of the experiments and analyzed the data, with assistance from J.L., Y.H., A.J.H., S.L.H., G.O., U.P., J.E. and D.A.P. J.M.L., H.W., and R.S. performed the sleep genetic association analysis. D.A.L. and D.A.P. wrote the manuscript with input from J.M.L. and R.S. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Custom PERL and MATLAB code used for zebrafish behavioral analysis is available upon request and posted online (35). Human sleep genetic association analysis was conducted using the U.K. Biobank Resource under application number 6818.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax4249/DC1
Fig. S1. Effects of gain and loss of EGFR signaling on sleep architecture.
Fig. S2. Amino acid alignment of human and zebrafish TGFa, EGF, and EGFR.
Fig. S3. Gefitinib does not enhance egfra−/− phenotype and effects of EGFR inhibitors on sleep architecture.
Fig. S4. EGFR signaling is not required for behavioral circadian rhythms.
Fig. S5. Validation of an SD assay, and EGFR signaling is required for normal homeostatic regulation of sleep.
Fig. S6. Inhibition of MAPK/ERK signaling suppresses TGFa overexpression–induced sleep.
Fig. S7. EGFR signaling regulates npvf expression, and TGFa overexpression–induced sleep is suppressed in npvf mutant animals.
Fig. S8. Association of ERBB4 sleepiness allele with increased ERBB4 expression in humans and pharmacological inhibition of KSR2 or ERBB4 decrease sleep in zebrafish.
Table S1. Variants at ERBB4 and KSR2 associate with self-reported measures of sleep quality and quantity in U.K. Biobank subjects.
Table S2. Descriptive characteristics of U.K. Biobank subjects of European ancestry used for sleep trait analysis.
REFERENCES AND NOTES
- 1.H. R. Colten, Institute of Medicine, Board on Health Sciences Policy, Committee on Sleep Medicine and Research, in Sleep Disorders and Sleep Deprivation: An Unmet Public Health Problem, B. M. Altevogt, H. R. Colten, Eds. (National Academies Press, 2006). [PubMed] [Google Scholar]
- 2.Jones S. E., Tyrrell J., Wood A. R., Beaumont R. N., Ruth K. S., Tuke M. A., Yaghootkar H., Hu Y., Teder-Laving M., Hayward C., Roenneberg T., Wilson J. F., del Greco F., Hicks A. A., Shin C., Yun C. H., Lee S. K., Metspalu A., Byrne E. M., Gehrman P. R., Tiemeier H., Allebrandt K. V., Freathy R. M., Murray A., Hinds D. A., Frayling T. M., Weedon M. N., Genome-wide association analyses in 128,266 individuals identifies new morningness and sleep duration loci. PLOS Genet. 12, e1006125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Y., Shmygelska A., Tran D., Eriksson N., Tung J. Y., Hinds D. A., GWAS of 89,283 individuals identifies genetic variants associated with self-reporting of being a morning person. Nat. Commun. 7, 10448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H., Lane J. M., Jones S. E., Dashti H. S., Ollila H. M., Wood A. R., van Hees V. T., Brumpton B., Winsvold B. S., Kantojärvi K., Palviainen T., Cade B. E., Sofer T., Song Y., Patel K., Anderson S. G., Bechtold D. A., Bowden J., Emsley R., Kyle S. D., Little M. A., Loudon A. S., Scheer F. A. J. L., Purcell S. M., Richmond R. C., Spiegelhalder K., Tyrrell J., Zhu X., Hublin C., Kaprio J. A., Kristiansson K., Sulkava S., Paunio T., Hveem K., Nielsen J. B., Willer C. J., Zwart J.-A., Strand L. B., Frayling T. M., Ray D., Lawlor D. A., Rutter M. K., Weedon M. N., Redline S., Saxena R., Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat. Commun. 10, 3503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dashti H. S., Jones S. E., Wood A. R., Lane J. M., van Hees V. T., Wang H., Rhodes J. A., Song Y., Patel K., Anderson S. G., Beaumont R. N., Bechtold D. A., Bowden J., Cade B. E., Garaulet M., Kyle S. D., Little M. A., Loudon A. S., Luik A. I., Scheer F. A. J. L., Spiegelhalder K., Tyrrell J., Gottlieb D. J., Tiemeier H., Ray D. W., Purcell S. M., Frayling T. M., Redline S., Lawlor D. A., Rutter M. K., Weedon M. N., Saxena R., Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat. Commun. 10, 1100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S. E., van Hees V. T., Mazzotti D. R., Marques-Vidal P., Sabia S., van der Spek A., Dashti H. S., Engmann J., Kocevska D., Tyrrell J., Beaumont R. N., Hillsdon M., Ruth K. S., Tuke M. A., Yaghootkar H., Sharp S. A., Ji Y., Harrison J. W., Freathy R. M., Murray A., Luik A. I., Amin N., Lane J. M., Saxena R., Rutter M. K., Tiemeier H., Kutalik Z., Kumari M., Frayling T. M., Weedon M. N., Gehrman P. R., Wood A. R., Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat. Commun. 10, 1585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane J. M., Jones S. E., Dashti H. S., Wood A. R., Aragam K. G., van Hees V. T., Strand L. B., Winsvold B. S., Wang H., Bowden J., Song Y., Patel K., Anderson S. G., Beaumont R. N., Bechtold D. A., Cade B. E., Haas M., Kathiresan S., Little M. A., Luik A. I., Loudon A. S., Purcell S., Richmond R. C., Scheer F. A. J. L., Schormair B., Tyrrell J., Winkelman J. W., Winkelmann J.; HUNT All In Sleep, Hveem K., Zhao C., Nielsen J. B., Willer C. J., Redline S., Spiegelhalder K., Kyle S. D., Ray D. W., Zwart J. A., Brumpton B., Frayling T. M., Lawlor D. A., Rutter M. K., Weedon M. N., Saxena R., Biological and clinical insights from genetics of insomnia symptoms. Nat. Genet. 51, 387–393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones S. E., Lane J. M., Wood A. R., van Hees V. T., Tyrrell J., Beaumont R. N., Jeffries A. R., Dashti H. S., Hillsdon M., Ruth K. S., Tuke M. A., Yaghootkar H., Sharp S. A., Jie Y., Thompson W. D., Harrison J. W., Dawes A., Byrne E. M., Tiemeier H., Allebrandt K. V., Bowden J., Ray D. W., Freathy R. M., Murray A., Mazzotti D. R., Gehrman P. R., Lawlor D. A., Frayling T. M., Rutter M. K., Hinds D. A., Saxena R., Weedon M. N., Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 10, 343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joiner W. J., Unraveling the evolutionary determinants of sleep. Curr. Biol. 26, R1073–R1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oikonomou G., Prober D. A., Attacking sleep from a new angle: Contributions from zebrafish. Curr. Opin. Neurobiol. 44, 80–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bringmann H., Sleep-active neurons: Conserved motors of sleep. Genetics 208, 1279–1289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Buskirk C., Sternberg P. W., Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nat. Neurosci. 10, 1300–1307 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Foltenyi K., Greenspan R. J., Newport J. W., Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila. Nat. Neurosci. 10, 1160–1167 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Kramer A., Yang F.-C., Snodgrass P., Li X., Scammell T. E., Davis F. C., Weitz C. J., Regulation of daily locomotor activity and sleep by hypothalamic EGF receptor signaling. Science 294, 2511–2515 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Gilbert J., Davis F. C., Behavioral effects of systemic transforming growth factor-alpha in Syrian hamsters. Behav. Brain Res. 198, 440–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donlea J. M., Ramanan N., Shaw P. J., Use-dependent plasticity in clock neurons regulates sleep need in Drosophila. Science 324, 105–108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harbison S. T., McCoy L. J., Mackay T. F., Genome-wide association study of sleep in Drosophila melanogaster. BMC Genomics 14, 281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harbison S. T., Serrano Negron Y. L., Hansen N. F., Lobell A. S., Selection for long and short sleep duration in Drosophila melanogaster reveals the complex genetic network underlying natural variation in sleep. PLOS Genet. 13, e1007098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill A. J., Mansfield R., Lopez J. M., Raizen D. M., Van Buskirk C., Cellular stress induces a protective sleep-like state in C. elegans. Curr. Biol. 24, 2399–2405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kushikata T., Fang J., Chen Z., Wang Y., Krueger J. M., Epidermal growth factor enhances spontaneous sleep in rabbits. Am. J. Physiol. 275, R509–R514 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Nath R. D., Chow E. S., Wang H., Schwarz E. M., Sternberg P. W., C. elegans stress-induced sleep emerges from the collective action of multiple neuropeptides. Curr. Biol. 26, 2446–2455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson M. D., Lee K. H., Churgin M. A., Hill A. J., van Buskirk C., Fang-Yen C., Raizen D. M., FMRFamide-like FLP-13 neuropeptides promote quiescence following heat stress in Caenorhabditis elegans. Curr. Biol. 24, 2406–2410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhdanova I. V., Wang S. Y., Leclair O. U., Danilova N. P., Melatonin promotes sleep-like state in zebrafish. Brain Res. 903, 263–268 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M., Liu B., Matthews P., Ong G., Pell J., Silman A., Young A., Sprosen T., Peakman T., Collins R., UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salvatierra J., Lee D. A., Zibetti C., Duran-Moreno M., Yoo S., Newman E. A., Wang H., Bedont J. L., de Melo J., Miranda-Angulo A. L., Gil-Perotin S., Garcia-Verdugo J. M., Blackshaw S., The LIM homeodomain factor Lhx2 is required for hypothalamic tanycyte specification and differentiation. J. Neurosci. 34, 16809–16820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snodgrass-Belt P., Gilbert J. L., Davis F. C., Central administration of transforming growth factor-alpha and neuregulin-1 suppress active behaviors and cause weight loss in hamsters. Brain Res. 1038, 171–182 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Ma Y. J., Junier M. P., Costa M. E., Ojeda S. R., Transforming growth factor-alpha gene expression in the hypothalamus is developmentally regulated and linked to sexual maturation. Neuron 9, 657–670 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Prober D. A., Rihel J., Onah A. A., Sung R. J., Schier A. F., Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J. Neurosci. 26, 13400–13410 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mann G. B., Fowler K. J., Gabriel A., Nice E. C., Williams R. L., Dunn A. R., Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell 73, 249–261 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Chen S., Oikonomou G., Chiu C. N., Niles B. J., Liu J., Lee D. A., Antoshechkin I., Prober D. A., A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 41, 2769–2778 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J. R., Joung J. K., Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sibilia M., Wagner E. F., Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269, 234–238 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Laisney J. A., Braasch I., Walter R. B., Meierjohann S., Schartl M., Lineage-specific co-evolution of the Egf receptor/ligand signaling system. BMC Evol. Biol. 10, 27 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hubbard S. R., Miller W. T., Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 19, 117–123 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee D. A., Andreev A., Truong T. V., Chen A., Hill A. J., Oikonomou G., Pham U., Hong Y. K., Tran S., Glass L., Sapin V., Engle J., Fraser S. E., Prober D. A., Genetic and neuronal regulation of sleep by neuropeptide VF. eLife 6, e25727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S., Chiu C. N., McArthur K. L., Fetcho J. R., Prober D. A., TRP channel mediated neuronal activation and ablation in freely behaving zebrafish. Nat. Methods 13, 147–150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanderheyden W. M., Gerstner J. R., Tanenhaus A., Yin J. C., Shaw P. J., ERK phosphorylation regulates sleep and plasticity in Drosophila. PLOS ONE 8, e81554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong C. C., Peterson Q. P., Hong J. Y., Peterson R. T., Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and MAP kinase/ERK signaling. Curr. Biol. 16, 1366–1372 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikhail C., Vaucher A., Jimenez S., Tafti M., ERK signaling pathway regulates sleep duration through activity-induced gene expression during wakefulness. Sci. Signal. 10, eaai9219 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Lenz O., Xiong J., Nelson M. D., Raizen D. M., Williams J. A., FMRFamide signaling promotes stress-induced sleep in Drosophila. Brain Behav. Immun. 47, 141–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simonin F., Schmitt M., Laulin J. P., Laboureyras E., Jhamandas J. H., MacTavish D., Matifas A., Mollereau C., Laurent P., Parmentier M., Kieffer B. L., Bourguignon J. J., Simonnet G., RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 103, 466–471 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madelaine R., Lovett-Barron M., Halluin C., Andalman A. S., Liang J., Skariah G. M., Leung L. C., Burns V. M., Mourrain P., The hypothalamic NPVF circuit modulates ventral raphe activity during nociception. Sci. Rep. 7, 41528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh C., Rihel J., Prober D. A., Neuropeptide Y regulates sleep by modulating noradrenergic signaling. Curr. Biol. 27, 3796–3811.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daniels R. W., Rossano A. J., Macleod G. T., Ganetzky B., Expression of multiple transgenes from a single construct using viral 2A peptides in Drosophila. PLOS ONE 9, e100637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen B. D., Green J. M., Foy L., Fell H. P., HER4-mediated biological and biochemical properties in NIH 3T3 cells. Evidence for HER1-HER4 heterodimers. J. Biol. Chem. 271, 4813–4818 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Dhawan N. S., Scopton A. P., Dar A. C., Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature 537, 112–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costanzo-Garvey D. L., Pfluger P. T., Dougherty M. K., Stock J. L., Boehm M., Chaika O., Fernandez M. R., Fisher K., Kortum R. L., Hong E. G., Jun J. Y., Ko H. J., Schreiner A., Volle D. J., Treece T., Swift A. L., Winer M., Chen D., Wu M., Leon L. R., Shaw A. S., McNeish J., Kim J. K., Morrison D. K., Tschöp M. H., Lewis R. E., KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity, and insulin sensitivity. Cell Metab. 10, 366–378 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wehr M. C., Hinrichs W., Brzózka M. M., Unterbarnscheidt T., Herholt A., Wintgens J. P., Papiol S., Soto-Bernardini M. C., Kravchenko M., Zhang M., Nave K. A., Wichert S. P., Falkai P., Zhang W., Schwab M. H., Rossner M. J., Spironolactone is an antagonist of NRG1-ERBB4 signaling and schizophrenia-relevant endophenotypes in mice. EMBO Mol. Med. 9, 1448–1462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee S. M., Tole S., Grove E., McMahon A. P., A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127, 457–467 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Weber F., Dan Y., Circuit-based interrogation of sleep control. Nature 538, 51–59 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Ma Y. J., Hill D. F., Junier M. P., Costa M. E., Felder S. E., Ojeda S. R., Expression of epidermal growth factor receptor changes in the hypothalamus during the onset of female puberty. Mol. Cell. Neurosci. 5, 246–262 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Lee D. A., Blackshaw S., Feed your head: Neurodevelopmental control of feeding and metabolism. Annu. Rev. Physiol. 76, 197–223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li X., Sankrithi N., Davis F. C., Transforming growth factor-alpha is expressed in astrocytes of the suprachiasmatic nucleus in hamster: Role of glial cells in circadian clocks. Neuroreport 13, 2143–2147 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Feng C. Y., Wiggins L. M., von Bartheld C. S., The locus ceruleus responds to signaling molecules obtained from the CSF by transfer through tanycytes. J. Neurosci. 31, 9147–9158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krueger J. M., Rector D. M., Churchill L., Sleep and cytokines. Sleep Med. Clin. 2, 161–169 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rich T., Innominato P. F., Boerner J., Mormont M. C., Iacobelli S., Baron B., Jasmin C., Lévi F., Elevated serum cytokines correlated with altered behavior, serum cortisol rhythm, and dampened 24-hour rest-activity patterns in patients with metastatic colorectal cancer. Clin. Cancer Res. 11, 1757–1764 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Rauen K. A., Huson S. M., Burkitt-Wright E., Evans D. G., Farschtschi S., Ferner R. E., Gutmann D. H., Hanemann C. O., Kerr B., Legius E., Parada L. F., Patton M., Peltonen J., Ratner N., Riccardi V. M., van der Vaart T., Vikkula M., Viskochil D. H., Zenker M., Upadhyaya M., Recent developments in neurofibromatoses and RASopathies: Management, diagnosis and current and future therapeutic avenues. Am. J. Med. Genet. A 167A, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.M. Westerfield, The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio), M. Westerfield, O. R. Eugene, Eds. (University of Oregon Press, 1993). [Google Scholar]
- 59.Halloran M. C., Sato-Maeda M., Warren J. T., Su F., Lele Z., Krone P. H., Kuwada J. Y., Shoji W., Laser-induced gene expression in specific cells of transgenic zebrafish. Development 127, 1953–1960 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Thermes V., Grabher C., Ristoratore F., Bourrat F., Choulika A., Wittbrodt J., Joly J. S., I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech. Dev. 118, 91–98 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Chen T. W., Wardill T. J., Sun Y., Pulver S. R., Renninger S. L., Baohan A., Schreiter E. R., Kerr R. A., Orger M. B., Jayaraman V., Looger L. L., Svoboda K., Kim D. S., Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Provost E., Rhee J., Leach S. D., Viral 2A peptides allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis 45, 625–629 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Woods I. G., Schoppik D., Shi V. J., Zimmerman S., Coleman H. A., Greenwood J., Soucy E. R., Schier A. F., Neuropeptidergic signaling partitions arousal behaviors in zebrafish. J. Neurosci. 34, 3142–3160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bycroft C., Freeman C., Petkova D., Band G., Elliott L. T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., Cortes A., Welsh S., Young A., Effingham M., McVean G., Leslie S., Allen N., Donnelly P., Marchini J., The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loh P. R., Tucker G., Bulik-Sullivan B. K., Vilhjálmsson B. J., Finucane H. K., Salem R. M., Chasman D. I., Ridker P. M., Neale B. M., Berger B., Patterson N., Price A. L., Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pruim R. J., Welch R. P., Sanna S., Teslovich T. M., Chines P. S., Gliedt T. P., Boehnke M., Abecasis G. R., Willer C. J., LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.GTEx Consortium , The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stegle O., Parts L., Durbin R., Winn J., A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLOS Comput. Biol. 6, e1000770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax4249/DC1
Fig. S1. Effects of gain and loss of EGFR signaling on sleep architecture.
Fig. S2. Amino acid alignment of human and zebrafish TGFa, EGF, and EGFR.
Fig. S3. Gefitinib does not enhance egfra−/− phenotype and effects of EGFR inhibitors on sleep architecture.
Fig. S4. EGFR signaling is not required for behavioral circadian rhythms.
Fig. S5. Validation of an SD assay, and EGFR signaling is required for normal homeostatic regulation of sleep.
Fig. S6. Inhibition of MAPK/ERK signaling suppresses TGFa overexpression–induced sleep.
Fig. S7. EGFR signaling regulates npvf expression, and TGFa overexpression–induced sleep is suppressed in npvf mutant animals.
Fig. S8. Association of ERBB4 sleepiness allele with increased ERBB4 expression in humans and pharmacological inhibition of KSR2 or ERBB4 decrease sleep in zebrafish.
Table S1. Variants at ERBB4 and KSR2 associate with self-reported measures of sleep quality and quantity in U.K. Biobank subjects.
Table S2. Descriptive characteristics of U.K. Biobank subjects of European ancestry used for sleep trait analysis.