

Notable differences exist between synthetic and native apatite in their crystal size and state of existence in bone tissue.
Abstract
Hydroxyapatite (HA) plays an important role in clinical bone repair. However, it remains a challenge to accurately determine its changes during bone reconstruction and to identify its differences from native bone apatite. Here, terbium (Tb) doped uniform HA nanocrystals were implanted into bone tissue and compared with native bone apatite. These comparisons demonstrated the occurrence of compositional and structural alteration of the implanted HA nanocrystals, and their gradual degradation, during bone reconstruction. They also revealed notable differences between HA nanocrystals and bone apatite crystals in crystal size, distribution pattern, and state of existence in bone tissue. Although synthetic HA nanocrystals could osteointegrate with bone tissue, they still seemed to be treated as foreign material in this tissue and thus were gradually degraded. These findings can help to identify and rethink the function of synthetic apatite and bone apatite, which will benefit future design and application of biomimetic bone repair materials.
INTRODUCTION
Bone is an important organ supporting human movement and has a hierarchical structure in three dimensions (1, 2); however, the number of bone injuries caused by disease, trauma, and osteoporosis is increasing yearly. The repair of bone fractures and defects is an eternal subject in clinical medicine. Calcium phosphate materials, represented here by hydroxyapatite (HA), include block or granulate ceramics (3), coatings (4, 5), bone cements (6), and scaffolds (7, 8) and have shown advantages in bone tissue repair (9). Nanohydroxyapatite (nHA), in particular, has attracted increasing attention from researchers and clinicians because of its high bioactivity and close similarity to native bone mineral (10, 11). In addition, calcium ions in the HA crystal structure can be substituted by other cations (3) and by lanthanide ions; the latter can endow HA material with traceable downconversion or upconversion fluorescence without influencing its biocompatibility (12–14).
Bone mineral is composed primarily of apatite crystals (15, 16), which result from deposition and mineralization of calcium and phosphorus ions on the bone matrix secreted by osteoblasts (17, 18). Osteoclasts lie in resorption craters on bone surfaces (19), secrete acidic substances to dissolve apatite crystals, secrete proteases to digest the bone matrix, and form bone resorption pits (20). Subsequently, osteoblasts migrate to the absorbed site and secrete the bone matrix, which then mineralizes to form new bone. Within mature bone tissue, mineralized collagen fibrils are insoluble, and the bone apatite crystals are stable and not easily dissociated; but the crystal surface can exchange ions with other ions in body fluid (Na, K, Mg, etc.) to maintain a stable electrolyte concentration in the living body (21, 22).
After synthetic HA materials or nHA crystals are implanted into bone defects, new bone tissue will form on or bond with the surface of the HA material (3) to realize reconstruction of the damaged bone tissue. However, there are still scientific challenges to be resolved. For example, once the synthetic nHA crystals are implanted into bone defects to participate in the bone reconstruction process, will they perform in the bone matrix in the same manner as the native bone apatite crystals? Will they degrade, and how can researchers determine whether they have degraded? How can implanted nHA crystals be effectively distinguished from bone apatite crystals in the reconstructed bone tissue? The resolution of these problems has important value for clinical application of synthetic HA nanocrystals or HA materials and will provide a reference for future creation of new biomimetic bone repair materials.
For this purpose, we carefully designed an experimental strategy to solve the above challenges. First, uniform Tb-doped HA nanocrystals were synthesized using a hydrothermal method and then implanted into bone defects. Microcomputed tomography (micro-CT), laser scanning confocal microscopy (LSCM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), and energy-dispersive x-ray spectrometry (EDX) were used to determine the in vivo distribution of these nanocrystals and to track whether their uniform shape, composition, and crystal structure were altered during new bone reconstruction as well as to reveal differences between the implanted HA nanocrystals and native bone apatite crystals by investigating their state in the bone matrix. The results may help to identify and rethink the function of synthetic apatite and bone apatite, which will benefit the design and clinical application of biomimetic HA-based biomaterials in the future.
RESULTS AND DISCUSSION
The hydrothermally synthesized HA nanocrystals displayed a uniform rod-shape morphology with a length of approximately 99.9 ± 10.8 nm (Fig. 1A) and had a crystal structure similar to that of pure HA (Fig. 1B). Terbium (Tb) ions were also incorporated into the HA crystals to distinguish the synthetic HA nanocrystals from the bone apatite crystals by tracing the Tb during analysis of the animal samples. The results showed that the incorporation of Tb did not obviously change the rod-shape morphology (Fig. 1D) or crystal structure (Fig. 1, B and C) of the HA nanocrystals, and the average crystal length was approximately 100 ± 8.7 nm (inset in Fig. 1D). The characteristic x-ray diffraction (XRD) peaks at 25.9°, 32°, 32.3°, 40°, 47°, and 50° shown in Fig. 1B correspond to the (002), (211), (112), (310), (222), and (213) crystal planes of the HA structure, respectively. The infrared (IR) spectra of HA and HA-Tb in Fig. 1C show PO43− bands at 1043 to 960 cm−1 (stretching vibration) and at 606 to 575 cm−1 (bending vibration) and an ─OH peak at 3570 cm−1. The XPS spectra in Fig. 1E demonstrate the presence of the basic Ca, P, and O elements and the successful incorporation of Tb in the HA crystal. Both the HA nanocrystals and the HA-Tb nanocrystals exhibited good cytocompatibility based on their similar effects on cell proliferation compared with the control (Fig. 1F), which was also confirmed previously (23). The results indicate that the hydrothermal process is suitable for synthesis of uniform HA or uniform ion-doped HA-Tb nanocrystals, and the latter was selected for the animal experiments because of its distinguishable uniform shape and the presence of Tb, both of which are traceable in new bone tissue.
Fig. 1. Synthesis and characterization of HA and HA-Tb nanocrystals.
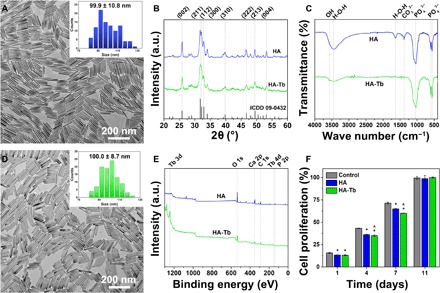
TEM images of crystal morphologies, with insets showing size distribution (A and D), XRD patterns (B), IR spectra (C), XPS spectra (E), and cell proliferation effects (F) of HA and HA-Tb nanocrystals. (*versus control, ^versus HA for P < 0.05). ICDD, the International Centre for Diffraction Data; a.u., arbitrary units.
After implantation of the HA-Tb powder into bone defects in rabbit femoral condyles for 1, 3, and 6 months, bone samples were harvested, and micro-CT and TEM analyses were performed. The micro-CT images in Fig. 2 show that the HA-Tb powder was still accumulated in the cylindrical bone defect for the first month (Fig. 2, A and D), but in the third month, the HA-Tb powder was separated by the ingrowing new bone tissue (Fig. 2, B and E), and some of the powder particles had also diffused into the surrounding cancellous bone tissue. By the sixth month, the reconstruction of new bone tissue has basically completed, and less HA-Tb powder remained in the bone tissue (Fig. 2, C and F). The results demonstrate that the synthetic HA-Tb nanocrystal powder was partially degraded during bone reconstruction or in the process of bone defect repair, and the defect space occupied by the HA-Tb powder was gradually replaced or filled by the new bone tissue. The results also show that micro-CT is a very useful tool for observation of implanted HA-Tb material and changes in the material distribution region with implantation time.
Fig. 2. Micro-CT images of HA-Tb powder in bone tissue.
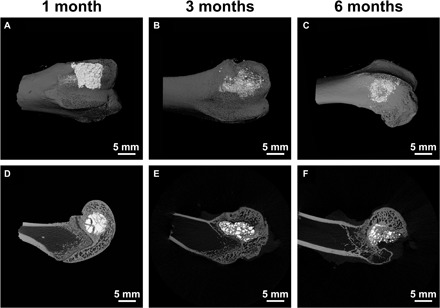
After implantation for 1, 3, and 6 months, the overall distribution and degradation change of the implanted HA-Tb material and bone remodeling at the macro level. (A to C) Reconstructed images. (D to F) Scanned single-layer images.
Notably, the bone tissue is displayed as a gray image, and the implanted HA-Tb powder is bright white in the image. However, the native bone apatite minerals in bone tissue cannot be clearly observed and distinguished via micro-CT. This might be because the bone apatite crystals are well dispersed in the bone matrix and too small to detect under the current micro-CT resolution. Another reason is the lack of Tb or a similar ion in the bone apatite crystals, because the lanthanide Tb ions have a large K-edge value (52.0) and high x-ray absorbance efficiency (24), thus providing clearer micro-CT imaging of the HA-Tb material.
Figure 3 shows LSCM images of bone tissue, the HA-Tb particles with green fluorescence (excitation, 232 nm; emission, 552 nm), and their correlating overlapping images after implantation for 1, 3, and 6 months. As seen from Fig. 3 (A to C), in contrast with Fig. 3 (G to I), without the participation of the green fluorescence provided by the HA-Tb particles (Fig. 3, D to F), it is difficult to clearly determine the implanted HA material and its distribution in or interrelation with the bone tissue or the distribution and amount change in the implanted particles during bone reconstruction. However, with the aid of the green HA-Tb fluorescence, we can clearly observe the changes in the implanted HA-Tb particles, which were in an agglomerated distribution at 1 month (Fig. 3G), and gradually degraded into smaller particles and separated by the new bone tissue at 3 months (Fig. 3H). At 6 months, the HA-Tb particles became finer, were fewer in number, and were dispersed in the interspace of the bone tissue (Fig. 3I). Meanwhile, Fig. 3 (A to C) shows that new bone or osteoid tissue appeared in the bone defect at 1 month, a large amount of trabecular bone formed at 3 months, and a mature woven bone structure formed at 6 months. The LSCM imaging results are consistent with the micro-CT imaging observations (Fig. 2), both indicating that the implanted HA particles underwent degradation during the process of new bone reconstruction. However, the micro-CT imaging shows the overall distribution of the implanted HA material and bone remodeling at the macro level, while the LSCM images exhibit the detailed interrelation between the HA material and bone tissue at the micro level.
Fig. 3. LSCM images of bone tissue and HA-Tb particles.
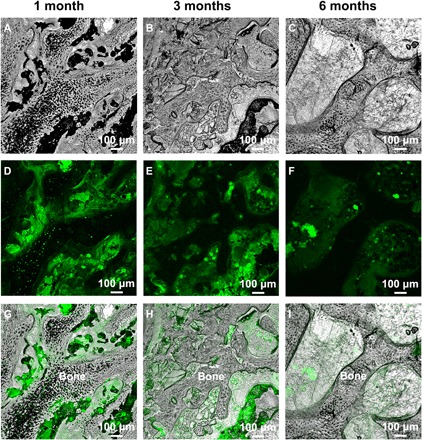
LSCM images of bone tissue (A to C), HA-Tb particles with green fluorescence (D to F), and their overlapping images (G to I) after implantation for 1, 3, and 6 months. The images reveal the detailed interrelation between the HA-Tb material and bone tissue, and the distribution and amount change of the implanted HA-Tb material during bone reconstruction at the micro level.
The TEM micrographs in Fig. 4 exhibit the details of the HA-Tb nanocrystals implanted in new bone tissue and the change in their uniform morphology and size with implantation time. At 1 month after implantation, the HA-Tb nanocrystals are still agglomerated (Fig. 4A) and, on the whole, maintain their uniform rod-shape morphology (Fig. 4D) and their HA crystal structure (Fig. 4G) but has undergone a certain reduction in the nanocrystal size (average, 78.4 ± 24.5 nm) compared with the original size shown in Fig. 1D (average, 100.0 ± 8.7 nm). At 3 months after implantation, the bone tissue has grown between and osteointegrated with the HA-Tb nanocrystals (Fig. 4B), and they appear glued together. At this point, the nanocrystals have already experienced obvious degradation in the bone tissue, becoming rougher, irregular, and smaller (white arrow, Fig. 4E). However, the nanocrystals still have an HA crystalline structure (Fig. 4H). At 6 months, the implanted HA-Tb nanocrystals have been fused into the new bone (Fig. 4C); there are fewer HA-Tb nanocrystals in the bone matrix, and the nanocrystals have become finer and more irregular (Fig. 4F). The crystalline structure or crystal symmetry of the degraded HA-Tb particles has been greatly destroyed (Fig. 4I).
Fig. 4. TEM analysis of the HA-Tb nanocrystals in new bone tissue.

TEM micrographs (A to F) and SAED patterns (G to I) of the implanted HA-Tb nanocrystals in new bone tissue at 1, 3, and 6 months, demonstrating the change in their uniform morphology, crystal size, and crystal structure with implantation time.
To compare and reveal differences between the implanted HA nanocrystals and the native bone apatite crystals, TEM micrographs, SAED patterns, and EDX mapping of natural bone tissue from the rabbit femoral condyle are shown in Fig. 5. The mature bone tissue displays a reticular fibrous structure in which even the periodic banding structure (~ 67-nm axial pitch) of the collagen fibers is clearly visible (Fig. 5A). Collagen is a crystalline fibrinogen embedded in the bone matrix containing apatite minerals (25). The SAED pattern (inset in Fig. 5A) and the EDX mapping (c1) demonstrate that the black area [indicated by the white arrow in Fig. 5A or marked with a + (c1) in the magnified image of the bone tissue in Fig. 5C] is the mineralized region, which shows an HA diffraction ring structure and contains the main Ca, P, and O elements of the HA. The gray area is the nonmineralized region of bone tissue, which has no HA crystal diffraction ring structure (inset in Fig. 5B) and exhibits almost no calcium and phosphorus peaks in the EDX mapping analysis (c2). The results demonstrate that the mineralized apatite of bone itself has an HA phase diffraction ring structure but does not show an observable precise or defined shape and size; the size of the bone apatite crystals should be very small and in the subnanometer range. It is difficult to clearly observe the morphology of a single bone apatite crystal, even in the high-resolution image (like Fig. 5C), which only shows a black mineralized region. In their study of natural bone, Tertuliano and Greer revealed the composite composition and hierarchical structure of bone tissue (16); however, the precise shape and size of the mineralized bone apatite crystals cannot be clearly determined in their TEM images, although few reports have suggested a mean thickness of the apatite mineral platelets of approximately 2 to 3 nm (25, 26). More attention should be paid to the investigation and development of smaller (less than 3 nm) or subnanometer HA crystals, which may show more important biological characteristics and application prospects.
Fig. 5. TEM analysis of the native apatite in bone tissue.
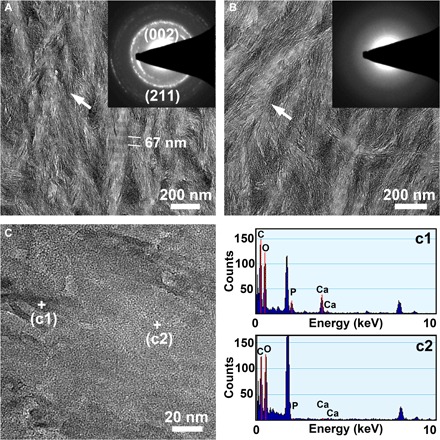
TEM micrographs (A to C), SAED patterns [insets in (A) and (B)], and EDX mapping (c1, c2) of the natural bone tissue from the rabbit femoral condyle demonstrating that the native mineralized bone apatite has a diffraction structure with an HA phase but does not show an observable precise or defined shape and size; the size of bone apatite crystals should be in the subnanometer range.
Figure 6 shows TEM micrographs and the corresponding EDX mapping of the degraded HA-Tb particles after 6 months in the bone tissue. The degraded particles exhibit either a regional (Fig. 6, A and B) or a sporadic (Fig. 6C) distribution in the bone matrix, with irregular morphology and a very small particle size. However, the tiny particles still contain the Ca, P, and O elements and the doping Tb element, except for a slight difference in their spectral peak strength (Fig. 6, D to F). In addition, the degradation of HA-Tb nanocrystals was not completely synchronous and consistent; some degraded into fine needles and some into small particles. These differences are likely closely related to their surrounding microenvironment in the bone matrix and need further investigation in the future.
Fig. 6. TEM analysis of the degraded HA-Tb particles.
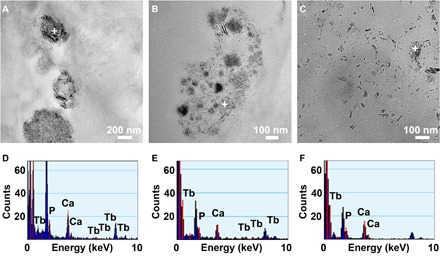
TEM micrographs (A to C) and corresponding EDX mapping (D to F) of the degraded HA-Tb particles after 6 months in the bone tissue, suggesting that the differences in distribution form (regional or sporadic) and morphology (acicular or granular) of the degraded particles should be closely related to their microenvironment in the bone matrix.
The results shown in Figs. 4 to 6 indicate that native bone apatite has a crystal structure similar to that of the implanted synthetic HA (HA-Tb) crystals, but the two materials have obvious differences in several aspects: (i) the native bone apatite crystals formed by mineralization of calcium and phosphorus ions on bone organic matrix are much smaller but more stable than the synthetic HA nanocrystals, and the latter will undergo obvious degradation and structural change in the bone matrix, while the bone apatite within mature bone tissue is in a stable state and not easily dissociated (21, 22); (ii) although the implanted HA nanocrystals can form osteointegration with the bone tissue during bone reconstruction, they are not directly involved in the mineralization of the bone matrix, which is obviously different from the osteogenic process of bone apatite mineralization; and (iii) the state of existence and distribution pattern in the reconstructed bone tissue are distinctly different between the synthetic HA nanocrystals and the native bone apatite crystals.
The reason for the differences may be related to osteonectin, osteocalcin, or acid phospholipids. For example, in the process of new bone reconstruction, osteonectin (a phosphoprotein) attached to collagen can combine with free calcium ions to form a nucleation center for bone apatite deposition and combine the deposited bone apatite crystals with bone collagen (27). Moreover, osteocalcin (an acidic protein) has a high affinity with bone apatite and Ca2+ ions and participates in calcium transport and bone calcification (28, 29). These factors determine and promote the mineralization of apatite crystals on the bone matrix. This indicates that there should be a mutually identifiable relation or synergistic effect among the bone matrix, related proteins, and bone apatite crystals. As to the synthetic HA nanocrystals, although they can bond or osteointegrate with bone tissue, they seem to be “isolated” by the bone matrix (as in Fig. 6A) or are still regarded as “foreign body” to the bone tissue and may lack the innate mutual recognition or synergistic effects of the mineralized bone apatite; thus, the synthetic HA nanocrystals are gradually degraded in the bone tissue. A quantitative evaluation is necessary in the future to focus on the immunohistochemical analysis of the osteonectin, osteocalcin, or acid phospholipids in both the harvested samples and the control group, to examine and quantify the synergistic relationship of natural apatite with related proteins and the difference in such synergistic effect between natural and synthetic apatite.
From the XRD patterns and IR spectra shown in fig. S1, we can see other differences between the bone apatite crystals and the synthetic HA nanocrystals. The bone apatite crystals from rabbit femoral condyles exhibit an envelope curve of HA diffraction with weak crystallization (measured crystallinity is 51.48%), while the hydrothermal synthetic HA (HA-Tb) nanocrystals have a higher crystallinity of 69.25%, and the characteristic peaks corresponding to different crystal planes are more distinct (fig. S1A). The lattice parameters of bone apatite are a = b = 0.946 nm and c = 0.688 nm, while the lattice parameters of synthetic HA-Tb nanocrystals are a = b = 0.943 nm and c = 0.688 nm; the a (= b) parameter between them shows a slight difference. In the IR spectra (fig. S1B), the bone apatite shows a higher carbonate (CO32−) (at approximately 1500 cm−1) and water (H2O) (at approximately 3500 and 1600 cm−1) content and does not exhibit the spectral ─OH peak of HA at 3570 cm−1. Why bone apatite does not display the ─OH peak may not be caused by its carbonate groups occupying the hydroxyl position of the HA structure, because in synthetic apatite, the carbonate groups can also occupy the hydroxyl position but still show a spectral peak of ─OH groups. Therefore, there are two possible explanations for such a difference. One is that the water molecules may occupy the position of hydroxyl groups in bone apatite, thus blocking the appearance of the hydroxyl spectral peak. Figure S2 shows the partial lattice models along the hydroxyl channel for HA, HA-Tb, and the H2O-substituted HA (with a water molecule replacing an OH position and a Ca position). When the trivalent Tb3+ ion replaces the bivalent Ca2+ ion in the HA lattice, there will be a Ca vacancy in the lattice due to the need for charge balance. When a water molecule occupies a hydroxyl position, one H atom of the water molecule will also occupy an adjacent calcium position (fig. S2, Bone) to maintain the stability of the HA crystal structure (30). Another key factor that has been neglected is that organic small molecules composed of C, H, and O may embed into the bone apatite lattice during the bone mineralization process, which is also an important aspect worthy of further research in the future.
Although clinicians and researchers have typically utilized HA materials for bone tissue repair, they still pay insufficient attention to and lack detailed investigations of in vivo changes over time and of the differences between the synthetic HA crystals and native bone apatite crystals. By using Tb-doped HA uniform nanocrystals and via combined analyses of micro-CT, LSCM, TEM, SAED, and EDX, this study has successfully demonstrated the degradation changes of HA material in bone tissue and the details of its morphology, composition, and structure alterations, as well as the differences between the synthetic HA nanocrystals and the native bone apatite crystals. The differences in their state of existence and mechanism of action in bone tissue, which are related to their formation process (in vitro preprepared or in vivo biological synthesis), crystal size (nanometer or subnanometer), crystal composition (─OH or organic groups), and possible recognition/synergistic interaction with related proteins, will have a direct and crucial effect on bone regeneration and reconstruction. The degradation process represented by the uniform HA nanocrystals indicates that, although it can carry out bone bonding or osteointegration, the synthetic nanoapatite is still a foreign body in bone tissue and cannot participate directly in bone mineralization in the same way as native bone apatite. The comparative study of synthetic HA crystals and bone-native HA crystals requires researchers to develop a new generation of apatite crystals that are more similar to bone apatite and may be directly involved in bone mineralization. This achievement will bring a brighter prospect for future application of synthetic apatite in bone reconstruction and will pave a way for the creation of new biomimetic bone repair materials.
CONCLUSION
The advantage of using uniform HA nanocrystals is that they can better reveal the in vivo detailed changes of implanted HA material during bone reconstruction through observation of their morphological, compositional, and structural alterations, while it is difficult to assess changes with conventional irregular HA particles or HA nanocrystals. The addition of a certain number of Tb ions in an HA lattice can provide elemental tracking, micro-CT imaging, and fluorescence imaging capabilities and will not change the crystal structure, morphology, or biocompatibility of the HA crystals. By investigating the bone reconstruction process with uniform HA-Tb nanocrystals, we demonstrated the gradual degradation, composition, and crystal structure changes in HA nanocrystals in bone tissue and revealed differences between the implanted HA nanocrystals and bone apatite crystals with regard to crystal size, distribution pattern, and state of existence in the bone tissue. These results will facilitate an in-depth understanding of the respective characteristics of synthetic HA crystals and bone apatite crystals and guide future design of new biomimetic bone repair materials. This research also suggests the need for further investigations and the development of smaller subnanometer HA crystals, which might have important scientific value and clinical significance in exploiting the biological characteristics and application of HA materials.
MATERIALS AND METHODS
Materials and reagents
Analytical-grade Tb(NO3)3, octadecylamine, and oleic acid were purchased from Shanghai Aladdin Co. Ltd., China. Analytical reagent (AR)–grade Ca(NO3)2, Na3PO4, and ethanol were purchased from Chengdu Kelong Chemical Co. Ltd., China. Other chemical reagents that were obtained commercially were of AR grade and were used without further purification.
Preparation of uniform HA and HA-Tb nanocrystals
HA and HA-Tb nanocrystals were synthesized by referring to the literature (23). A reaction formula, (10 − x)Ca(NO3)2 + xTb(NO3)3 + Na3PO4 → (Ca10−xTbx)(PO4)6OH2, was adopted, in which x = 0 to 1. Briefly, octadecylamine (1 g) was dissolved in oleic acid (8 ml) and ethanol (32 ml) under magnetic stirring in a Teflon-lined autoclave (100 ml). Next, aqueous solutions of Ca(NO3)2 (3.6 to 4 mmol, 14 ml) and Tb(NO3)3 (0 to 0.4 mmol, 4 ml) were added and stirred for 5 min, followed by the addition of Na3PO4 (2.4 mmol, 14 ml) solution. Afterward, the resulting solution was agitated for 10 min and hydrothermally treated at 160°C for 6 hours. After cooling to room temperature, the white precipitate was collected through centrifugation (5 min at 2700g), fully washed using cyclohexane, ethanol, and deionized water, and then freeze dried.
Characterization
The morphology of the HA-Tb nanocrystals was observed using TEM on an FEI Tecnai G2 F20 instrument at 200 kV. The XRD patterns were acquired using a PANalytical Empyrean instrument in the 2θ range from 20° to 60° with Cu Kα radiation (λ = 1.5406 Å). The Fourier transform IR spectra were recorded in a transmission mode at a wave number range of 500 to 4000 cm−1 (PerkinElmer 6000). The elements were measured via both EDX mapping on TEM and XPS analyses (AXIS Ultra DLD, Kratos, UK). The photoluminescence was recorded using a Hitachi F-7000 fluorescence spectrophotometer. The HA-Tb nanocrystal powder implanted in the bone defects was scanned using micro-CT (vivaCT 80, SCANCO Medical AG, Switzerland) and a two-photon LSCM (LSM 780 NLO, ZEISS, Germany).
Cell culture and proliferation
Osteoblastic MG63 cells were used for cell culture. The cells were cultured in F12 medium (GIBCO, USA) supplemented with 10% calf serum (GIBCO, USA) and 1% penicillin and streptomycin (100 U/ml, GIBCO, USA) and maintained at 37°C with 5% CO2 and 90% humidity. The medium was replaced every 2 to 3 days, and uniform subculturing was carried out with 0.25% trypsin. Then, the MG63 cells were cultured with HA or HA-Tb solution (100 μg/ml). The cells were seeded into 96-well tissue culture plates (Corning, USA) at a density of 2 × 104 cells per well. Subsequently, the seeded samples were cultured in a humidified incubator (37°C, 5% CO2), and the medium was changed every 2 days. MG63 cells cultured in F12 medium served as the control. After incubation for 1, 4, 7, and 11 days, the samples were evaluated using a cell counting kit-8 assay, and the optical density value of the solution was recorded using a microplate reader (PerkinElmer Wallac 1420) at 490 nm to determine cell proliferation. The results were expressed as means ± SD based on measurements from triplicate wells.
Animal experiments
All animal protocols were approved and performed in compliance with the guidelines from the Research Ethics Committee of the State Key Laboratory of Oral Diseases and West China Hospital of Stomatology, Sichuan University (permit number: WCHSIRB-D-2016-134) according to the Regulations on the Administration of Experimental Animals of the People’s Republic of China. The freeze-dried HA-Tb powder was first sterilized with an autoclave at 120°C, then mixed into a paste with sterile distilled water, and implanted into femoral bone defects created in six adult New Zealand white rabbits, which were randomly divided into three groups (n = 2 in each group). After shaving and disinfection of the hindlimbs, a cylindrical bone defect (φ6 mm × 5 mm) was drilled in the distal femoral condyle of each hindlimb. The HA-Tb powder was implanted into the defects, and samples were harvested along with surrounding tissue at 1, 3, and 6 months after implantation. The harvested samples were fixed in 4% buffered paraformaldehyde and scanned via micro-CT. On the basis of the original CT scans of each sample, three-dimensional images were reconstructed using a finite element analysis software to display the new bone tissue and determine the material in the new bone tissue. The fixed samples were also embedded in polymethyl methacrylate and cut into thin sections after being dehydrated through gradient ethanol and cleaned in xylene. The unstained histological sections were observed via LSCM (LSM 780 NLO, ZEISS, Germany) to detect the HA-Tb material in the new bone tissue. TEM was also used to observe the implanted HA-Tb nanocrystals and the mineralized apatite in the bone tissue. The fixed samples were cut into ultrathin sections using an Ultracut ultramicrotome (Leica uc6/fc6) and were lightly stained with 1% uranyl acetate and lead citrate for TEM observation.
Statistical analysis
All results were expressed as means ± SD. Statistical comparisons between groups were analyzed using one-way analysis of variance (ANOVA). A value of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Special thanks to L. Chen for the micro-CT analysis, as well as G. Yuan and S. Wang for the TEM observation. Funding: We acknowledge the support of funds from the National Natural Science Foundation of China (grant no. 31700828), the Postdoctoral Science Foundation of China (grant no. 2018 M633381), and the Postdoctoral Science Foundation of Sichuan University (grant no. 2017SCU12055). Author contributions: X.L. designed and conducted most of the experiments. Q.Z. helped conduct the animal experiments. W.L. and H.C. directed the experiments and supervised the project. The manuscript was written with contributions from all the authors. All authors have approved the final version of this manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay6484/DC1
Fig. S1. Crystallization and composition differences of bone apatite and HA-Tb nanocrystals.
Fig. S2. Partial lattice models of HA, HA-Tb, and H2O-substituted HA.
REFERENCES AND NOTES
- 1.Reznikov N., Shahar R., Weiner S., Bone hierarchical structure in three dimensions. Acta Biomater. 10, 3815–3826 (2014). [DOI] [PubMed] [Google Scholar]
- 2.K. L. Moore, A. F. Dalley, A. M. R. Agur, in Clinically Oriented Anatomy, C. Taylor, Ed. (Wolters Kluwer, 2017), pp. 18–27. [Google Scholar]
- 3.L. L. Hench, An Introduction to Bioceramics. An Introduction to Bioceramics, C. Taylor, Ed. (World Scientific, ed. 2, 2013). [Google Scholar]
- 4.Goodman S. B., Yao Z., Keeney M., Yang F., The future of biologic coatings for orthopaedic implants. Biomaterials 34, 3174–3183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pillai R. S., Frasnelli M., Sglavo V. M., HA/β-TCP plasma sprayed coatings on Ti substrate for biomedical applications. Ceram. Int. 44, 1328–1333 (2018). [Google Scholar]
- 6.Barinov S. M., Komlev V. S., Calcium phosphate bone cements. Inorg. Mater. 47, 1470–1485 (2011). [PubMed] [Google Scholar]
- 7.Maji S., Agarwal T., Das J., Maiti T. K., Development of gelatin/carboxymethyl chitosan/nano-hydroxyapatite composite 3D macroporous scaffold for bone tissue engineering applications. Carbohydr. Polym. 189, 115–125 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Denry I., Kuhn L. T., Design and characterization of calcium phosphate ceramic scaffolds for bone tissue engineering. Dent. Mater. 32, 43–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eliaz N., Metoki N., Calcium phosphate bioceramics: A review of their history, structure, properties, coating technologies and biomedical applications. Materials 10, 334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J., Xu Y., Li S., Seifert G. V., Becker M. L., Three-dimensional printing of nano hydroxyapatite/poly(ester urea) composite scaffolds with enhanced bioactivity. Biomacromolecules 18, 4171–4183 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Cai Y., Liu Y., Yan W., Hu Q., Tao J., Zhang M., Shi Z., Tang R., Role of hydroxyapatite nanoparticle size in bone cell proliferation. J. Mater. Chem. 17, 3780–3787 (2007). [Google Scholar]
- 12.Li X., Chen H., Yb3+/Ho3+ Co-doped apatite upconversion nanoparticles to distinguish implanted material from bone tissue. ACS Appl. Mater. Inter. 8, 27458–27464 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Smith B. R., Gambhir S. S., Nanomaterials for in vivo imaging. Chem. Rev. 117, 901–986 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Cawthray J. F., Creagh A. L., Haynes C. A., Orvig C., Ion exchange in hydroxyapatite with lanthanides. lnorg. Chem. 54, 1440–1445 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Olszta M. J., Cheng X., Jee S. S., Kumar R., Kim Y.-Y., Kaufman M. J., Douglas E. P., Gower L. B., Bone structure and formation: A new perspective. Mater. Sci. Eng., R 58, 77–116 (2007). [Google Scholar]
- 16.Tertuliano O. A., Greer J. R., The nanocomposite nature of bone drives its strength and damage resistance. Nat. Mater. 15, 1195–1202 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Karsenty G., Ferron M., The contribution of bone to whole-organism physiology. Nature 481, 314–320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.C. Kaur, in The Scientific Basis of Tissue Transplantation, A. Nather, Ed. (World Scientific, 2001), pp. 97–114. [Google Scholar]
- 19.Henriksen K., Sørensen M. G., Jensen V. K., Dziegiel M. H., Nosjean O., Karsdal M. A., Ion transporters involved in acidification of the resorption lacuna in osteoclasts. Calcif. Tissue Int. 83, 230–242 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Roodman G. D., Cell biology of the osteoclast. Exp. Hematol. 27, 1229–1241 (1999). [DOI] [PubMed] [Google Scholar]
- 21.J. E. Puzas, in Bone Formation, F. Bronner, M. C. Farach-Carson, Eds. (Springer London, 2004), pp. 71–78. [Google Scholar]
- 22.Rosset E. M., Bradshaw A. D., SPARC/osteonectin in mineralized tissue. Matrix Biol. 52-54, 78–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X., Zou Q., Li W., Chen H., Intracellular interaction of hydroxyapatite-based nanocrystals with uniform shape and traceable fluorescence. Inorg. Chem. 57, 13739–13748 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Yu S.-B., Watson A. D., Metal-based x-ray contrast media. Chem. Rev. 99, 2353–2378 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Reznikov N., Chase H., Brumfeld V., Shahar R., Weiner S., The 3D structure of the collagen fibril network in human trabecular bone: Relation to trabecular organization. Bone 71, 189–195 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Zizak I., Roschger P., Paris O., Misof B. M., Berzlanovich A., Bernstorff S., Amenitsch H., Klaushofer K., Fratzl P., Characteristics of mineral particles in the human bone/cartilage interface. J. Struct. Biol. 141, 208–217 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Termine J. D., Kleinman H. K., Whitson S. W., Conn K. M., McGarvey M. L., Martin G. R., Osteonectin, a bone-specific protein linking mineral to collagen. Cell 26, 99–105 (1981). [DOI] [PubMed] [Google Scholar]
- 28.Zoch M. L., Clemens T. L., Riddle R. C., New insights into the biology of osteocalcin. Bone 82, 42–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoang Q. Q., Sicheri F., Howard A. J., Yang D. S. C., Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature 425, 977–980 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Yubao L., Xingdong Z., de Groot K., Hydrolysis and phase transition of alpha-tricalcium phosphate. Biomaterials 18, 737–741 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay6484/DC1
Fig. S1. Crystallization and composition differences of bone apatite and HA-Tb nanocrystals.
Fig. S2. Partial lattice models of HA, HA-Tb, and H2O-substituted HA.