

The dynamic expression of Fgl2 and RvDp5 synergistically programs inflammation resolution to restore homeostasis.
Abstract
The mechanisms that drive programmed resolution of inflammation remain elusive. Here, we report the temporal regulation of soluble (s) and transmembrane (m) fibrinogen-like protein 2 (Fgl2) during inflammation and show that both sFgl2 and mFgl2 correlate with the outcome. The expression and ectodomain shedding of Fgl2 are respectively promoted by miR-466l and metalloproteinases (ADAM10 and ADAM17) during inflammation resolution. Deficiency of Fgl2 enhances polymorphonuclear neutrophil (PMN) infiltration but impairs macrophage (MΦ) maturation and phagocytosis and inhibits the production of n-3 docosapentaenoic acid–derived resolvin 5 (RvDp5). In contrast, administration of sFgl2 blunts PMN infiltration as well as promotes PMN apoptosis and RvDp5 biosynthesis. By activating ALX/FPR2, RvDp5 enhances sFgl2 secretion via ADAM17 and synergistically accelerates resolution of inflammation. These results uncover a previously unknown endogenous programmed mechanism by which Fgl2 regulates resolution of inflammation and shed new light on clinical sepsis treatments.
INTRODUCTION
The ideal outcome of acute inflammation is timely resolution to restore homeostasis, while persistent or uncontrolled stimuli lead to nonresolving inflammation (1). The resolution of acute inflammation is actively driven by a superfamily of essential polyunsaturated fatty acid (PUFA)–derived mediators that include the lipoxins, resolvins, protectins, and maresins, collectively termed specialized proresolving mediators (SPMs), based on their unique structures and potent stereoselective anti-inflammatory [i.e., limiting polymorphonuclear neutrophil (PMN) infiltration] and proresolving [enhancing macrophage (MФ) clearance of microbial particles and apoptotic PMNs] actions (2). The underlying mechanisms by which homeostasis is timely restored remain to be established and are critical for developing new therapeutic strategies to counteract excessive inflammation.
Inflammation and coagulation coexist and closely interact during the development of multitudinous diseases (3, 4). Inflammatory factors can promote the activation of prothrombin, causing severe microvascular thrombosis or disseminated intravascular coagulation, while on the other hand, the process of coagulation itself is closely involved in promoting inflammation. For example, thrombin mediates the adhesion of leukocytes and increases the vascular permeability of endothelial cells and the release of proinflammatory cytokines (3). Hence, anticoagulation is an effective approach to limit inflammation. An essential step for coagulation is the activation of thrombin from prothrombin (5), which depends on the catalysis of prothrombinase, i.e., the transmembrane form of fibrinogen-like protein 2 (mFgl2). The other form of Fgl2 is soluble (sFgl2), which is primarily secreted by regulatory T cells and negatively regulates antigen presentation of dendritic cells and proliferation of effector T cells by binding to the inhibitory Fc receptor FcγRIIB (6). Fgl2 is up-regulated in viral hepatitis, ischemia-reperfusion injury, fulminant hepatitis, and other inflammatory models and plays an important role in the initiation and progression of these inflammatory diseases (7, 8). However, its role in the resolution of inflammation is not currently known.
Here, we report the temporal expression and regulation of sFgl2 and mFgl2 during self-limited (SL) versus delayed-resolving (DR) inflammation. We found that both sFgl2 and mFgl2 correlate with the magnitude and outcome of sepsis and that sFgl2 promotes production of SPM that accelerates resolution of inflammation. These findings reveal a fundamental mechanism that sFgl2 and mFgl2 differentially regulate the resolution of inflammation.
RESULTS
Fgl2 is temporally and differentially regulated during inflammation resolution
To determine the relationship between Fgl2 with sepsis outcome, we collected peripheral blood from patients with or without sepsis upon intensive care unit (ICU) admission within 24 hours. Sepsis patients were divided into resolved (survivors) and in-hospital mortality (nonsurvivors) (Fig. 1, A to C, and table S1). Compared to healthy subjects, the peripheral leukocytes and sFgl2 were increased in sepsis patients, while the sepsis nonsurvivors versus survivors had decreased plasma sFgl2 and up-regulated mFgl2. Together, these findings suggest that Fgl2 may be involved in clinical sepsis outcome.
Fig. 1. Temporal Fgl2 expression in SL versus DR inflammation.
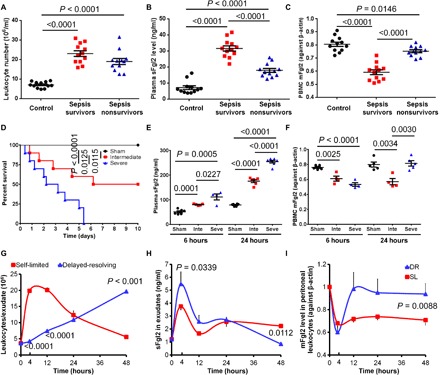
(A to C) Leukocyte numbers (A), plasma sFgl2 (B), and peripheral blood mononuclear cell (PBMC) mFgl2 levels (C) in the peripheral blood of healthy control, sepsis survivors, and nonsurvivors (n = 12 in each group). (D to F) Mice were induced different severity grades of sepsis by CLP as indicated in Materials and Methods (n = 10 in each group). Survival rates (D), plasma sFgl2 (E), and mFgl2 expression in PBMC (F) at indicated intervals. Inte, intermediate; Seve, severe. (G to I) Zym was injected (intraperitoneally) for acute peritonitis into male C57BL/6 mice: SL (1 mg per mouse) and DR (10 mg per mouse). Exudates were collected at indicated intervals. PMNs were enumerated (G). sFgl2 in the supernatant (H) and mFgl2 expression in the peritoneal leukocytes (I) were determined. Error bars represent mean ± SEM. For the survival rates, Mantel-Cox test was applied for the P values.
To explore this involvement, we assessed Fgl2 expression in the cecal ligation and puncture (CLP) mouse sepsis model (9). With increasing extent of CLP, sepsis mice showed increased mortality, hypothermia, blood aerobic bacteria, and peritoneal leukocyte infiltration (Fig. 1D and fig. S1, A to C). The expression of plasma sFgl2 increased, while mFgl2 significantly decreased at 6 hours after CLP. The amplitude correlated with the extent of CLP (Fig. 1, E and F). Of interest, mFgl2 rebounded at 24 hours in the severe versus intermediate CLP group (Fig. 1F), consistent with results from clinical samples (Fig. 1, A to C).
The murine SL [1 mg of zymosan (Zym) intraperitoneally] and DR (10 mg of Zym intraperitoneally) peritonitis models (10) were also used. In SL peritonitis, the infiltration of leukocytes (CD45+) and PMNs (Ly6G+) peaked at ~7 hours followed by a steady decline. Mononuclear cells (monocytes/MΦs; CD11b+Ly6G−) reduced to minimum at ~4 hours but gradually increased subsequently (Fig. 1G and fig. S1D). At 48 hours after injection, the majority (~70%) of SL exudate leukocytes were monocytes and MΦs. In contrast and as expected, DR exudates showed steadily increased PMNs and higher cytokines [e.g., interleukin-10 (IL-10), chemokine (C-X-C motif) ligand 1 (CXCL1), and tumor necrosis factor (TNF)] at 4 hours (fig. S1, D and E). Although the mononuclear cells reduced to baseline levels and started to rebound until ~10 hours, they reached a higher amount at 48 hours than in the SL group (fig. S1D). Of interest, at this juncture, the F4/80+ MΦ percentage in mononuclear cells of the DR group was markedly reduced compared with the SL group (fig. S1F), suggesting that the monocyte-derived mature MΦ proportion was higher in the SL model at the resolution interval.
We next monitored Fgl2 expression in these peritoneal exudates. The sFgl2 levels in the SL group were dramatically up-regulated at 4 hours after Zym injection, but at 12 hours, it decreased close to the level at 0 hour. Afterward, it increased again despite its lower level than that at 4 hours (Fig. 1H). The sFgl2 expression in DR exudates had similar trend during 0 to 24 hours and was higher at 4 hours, but was much lower at 48 hours, as compared with the SL group (Fig. 1H). The leukocytic mFgl2 reduced at 4 hours after both high and low doses of Zym challenge. However, mFgl2 in the DR group rebounded earlier than in the SL group. The leukocytic FGL2 mRNA levels in DR exudates were not higher than those in the SL group (fig. S1G). Nonetheless, leukocytic mFgl2 in DR mice had a lower trend at 4 hours but was significantly higher at 48 hours than in the SL group (Fig. 1I). Together, these results indicated that both sFgl2 and mFgl2 were temporally and differentially regulated during inflammation resolution.
Fgl2 expression is regulated by miR-466l and metalloproteinases
Because the expression pattern of sFgl2 (increased at 4 hours and decreased subsequently sharply at 12 hours and then increased again at the later stage) was similar to that of miR-466l, a key miRNA we identified earlier that promotes both the initiation and resolution of inflammation (10), we wondered whether Fgl2 was regulated by this miRNA. In silico analysis and cotransfection experiments indicated that miR-466l could bind the 3′-untranslated region (3′UTR) of FGL2. Overexpression of miR-466l promoted both sFgl2 secretion and mFgl2 expression in human MΦs (Fig. 2, A to D). However, this posttranscriptional regulatory mechanism could not completely elucidate the differential protein expression of sFgl2 and mFgl2 during the inflammation resolution time course because they were both increased.
Fig. 2. Fgl2 expression is regulated by miR-466l and metalloproteinases.
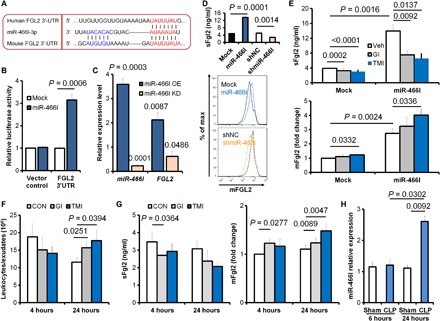
(A) In silico analysis of human and murine Fgl2 binding sites for miR-466l. (B) Mock or miR-466l was co-transfected with 3′UTR-luciferase reporter vector of vehicle control or Fgl2 3′UTR-mutant into mouse peritoneal macrophages (n = 4 independent experiments). Relative increased luciferase activity of respective 3′UTR reporters was normalized against control 3′UTR. (C) After mouse peritoneal MΦs were transfected with mock, miR-466l, shNC, or shmiR-466l, the expressions of miR-466l and FGL2 mRNA were determined with qPCR. Results (n = 4 independent experiments) were expressed as fold change against mock (miR-466l OE) or against shNC (miR-466l KD). (D) sFgl2 in supernatants and mFgl2 expression were determined. (E) After transfection with mock or miR-466l, human MΦs were treated with phosphate-buffered saline (PBS), GI254023X (10 μM), or TMI-1 (10 μM) for 24 hours, and the sFgl2 and mFgl2 expression was determined by enzyme-linked immunosorbent assay (ELISA). (F and G) Mice were challenged intraperitoneally with PBS (vehicle), GI254023X (40 μg/kg), or TMI-1 (40 μg/kg), plus Zym (1 mg per mouse); the peritoneal leukocytes were assessed (F); and the exudate sFgl2 and peritoneal leukocytic mFgl2 were determined with ELISA (G) at 4 and 24 hours. (H) Mice were challenged with/without moderate CLP, and the peritoneal leukocytes were collected for miR-466l determination at indicated time points. Error bars represent mean ± SEM.
Ectodomain shedding refers to extracellular domain proteolytic release from cell membrane molecules, which is mediated mainly by several a disintegrin and metalloproteinases (ADAMs) (11). Among them, ADAM10 and ADAM17 are critical for endothelial permeability, trans-endothelial leukocyte migration, and inflammatory mediator production (12). We found that treatment of metalloproteinase inhibitors GI254023X (ADAM10 inhibitor), TMI-1 (ADAM17 inhibitor), and GW280264X (dual ADAM10 and ADAM17 inhibitor) inhibited sFgl2 secretion and recovered mFgl2 expression in MΦs but not in PMNs (fig. S2, A and B). In addition, TMI-1 decreased sFgl2 but increased mFgl2 in both mock and miR-466l–overexpressing MΦs (Fig. 2E).
Because miR-466l was significantly increased at 4 hours and then decreased at 24 hours in the mice peritonitis model (10), we administered GI254023X and TMI-1 to assess their regulation on Fgl2 in vivo. We observed the leukocyte infiltration with a decreased trend at 4 hours and a promotion at 24 hours in the exudates of SL peritonitis (Fig. 2F). The ADAM-related pro-inflammatory TNF and CXCL1 were both inhibited by GI254023X and TMI-1 at 4 hours. GI254023X enhanced anti-inflammatory IL-10 at 4 hours, while TMI-1 suppressed both TNF and CXCL1 at 24 hours (fig. S2, C and D). GI254023X reduced sFgl2 at 4 hours but increased mFgl2 at 4 and 24 hours after Zym injection. TMI-1 significantly up-regulated mFgl2 at 24 hours (Fig. 2G). Moreover, miR-466l expression was enhanced in the peritoneal leukocytes of CLP mice compared with sham at 24 hours (Fig. 2H). These results demonstrated that the expressions of sFgl2 and mFgl2 upon inflammatory challenge were differentially regulated by miR-466l and ADAMs.
Fgl2 orchestrates inflammation resolution
We next assessed the role of Fgl2 in inflammation resolution. Fgl2 deficiency resulted in decreased survival and thrombosis, but exacerbated abscess formation, hypothermia, bacteremia, inflammatory cell infiltration, and cytokine production in cecal and peritoneum of CLP mice (Fig. 3, A to C, and fig. S3, A to D). The deletion of Fgl2 also consistently increased PMN infiltration and pro-inflammatory cytokines in the peritonitis model (Fig. 3D and fig. S3E). We calculated the resolution indices according to our earlier reports and the introduction of this quantitative evaluation of tissue resolution (10, 13, 14). Fgl2 deficiency prolonged the resolution interval (Ri ~ 17.9 hours) for 4.4 hours (Fig. 3D). Mononuclear cell numbers were also higher in the Fgl2KO exudates; however, monocyte-derived mature MΦs were lower in Fgl2KO exudates, as compared with those in wild-type (WT) exudates (fig. S3, F and G).
Fig. 3. Fgl2 orchestrates inflammation resolution.
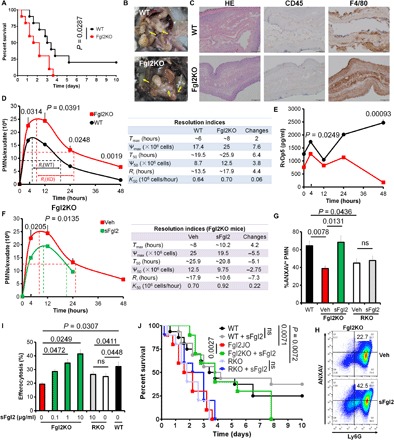
(A to C) Survival rate (A), abscess formation (B), and histological staining for hematoxylin and eosin (H&E), CD45, and F4/80 (C) in WT and Fgl2KO mice challenged with moderate CLP. Mantel-Cox test was applied for the P values of the survival data (n = 10 each group). (D and E) WT and Fgl2KO mice were challenged with Zym (1 mg per mouse; intraperitoneal). Peritoneal PMN numbers at indicated intervals were counted, and resolution indices were calculated (D). Tmax, time point when PMN was infiltrated to maximum; Ѱmax, PMN maximum number; T50, time point when PMNs were reduced to half of Ѱmax; Ѱ50, 50% of Ѱmax; Ri, resolution interval (time interval from Tmax to T50); K50, rate of PMN reduction from Tmax to T50. Quantification of RvDp5 was obtained with a calibration curve (E). (F) Fgl2KO mice were injected intraperitoneally with Zym (1 mg) using PBS (Veh) or sFgl2 (200 ng). Peritoneal PMN numbers at indicated intervals were counted, and resolution indices were calculated as indicated in (B). (G) PMNs from WT, Fgl2KO, and FcγRIIBKO (RKO) mice induced apoptosis by UV for 2 hours with or without sFgl2 treatment (10 μg/ml). Apoptotic PMN (ANXAV+) percentages were analyzed. (H) Fgl2KO mice were challenged intraperitoneally with Zym (1 mg) ± sFgl2 (200 ng) for 12 hours, and the peritoneal apoptotic PMN (ANXAV+Ly6G+) was determined. (I) Murine peritoneal MΦs were isolated from WT, Fgl2KO (red and green), and FcγRIIBKO (RKO) mice. After treatment with or without sFgl2 (10 μg/ml) for 2 hours, MΦs were incubated with carboxyfluorescein diacetate (CFDA)–labeled apoptotic PMNs (1:3) for 1 hour, and fluorescence intensities were determined. Error bars represent mean ± SEM. ns, no significant difference. (J) Sepsis survival of WT, Fgl2KO, and RKO mice with or without sFgl2 administration (400 ng). n = 16 in WT and WT + sFgl2 group; n = 10 in other groups. Mantel-Cox test was applied for the P values. Photo credit: Yongsheng Li, Yu Zhou, and Juan Lei, Clinical Medicine Research Center, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China.
Using ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS), we assessed the metabololipidome of mediators (i.e., eicosanoids and proresolving mediators) using the exudates and found that Fgl2 deficiency resulted in elevated pro-inflammatory mediators TXB2, 5-HETE, and LTB4 but specifically lowered several SPMs that included the recently identified resolvins produced from the precursor n-3 docosapentaenoic acid (DPA), namely, RvDp5 (Fig. 3, D and E, and fig. S4, A to D) (15). Because efferocytosis increases SPM production (10), in our study, compared with the WT group, the production of RvDp5 and LXA4 was significantly lower in the Fgl2KO group during MΦ efferocytosis of apoptotic PMNs (fig. S4E). These results indicated that Fgl2 deficiency promoted inflammation initiation and delayed resolution by reducing the production of proresolving mediators.
To investigate the regulatory mechanism of Fgl2 in resolution, we administrated recombinant sFgl2 into the peritoneum of Fgl2KO mice with Zym. Compared with vehicle, reduced PMN infiltration, shortened Ri (~10.6 hours), and accelerated ΔK50 (~0.22 × 106 cells per hour) were observed (Fig. 3F). In addition, sFgl2 administration at 12 hours after Zym challenge decreased Ri to ~10.8 hours (fig. S5A), illustrating the dual anti-inflammatory and proresolving actions of sFgl2.
After recruitment into tissues triggered by inflammatory stimuli, PMNs subsequently undergo programmed death, release “find-me” signals including adenosine 5′-triphosphate (ATP), and are removed by MΦs (efferocytosis) for resolution (14, 16). We next isolated murine peripheral PMNs and peritoneal MΦs and prepared ultraviolet (UV)–induced apoptotic PMNs (AP-PMNs). We found that both PMNs and MΦs express sFgl2 and mFgl2 and that PMNs showed the lowest expression (fig. S5, B to D). Deficiency of Fgl2 blunted PMN apoptosis and promoted MΦ TNF secretion in vitro, which were reversed by sFgl2 via its receptor FcγRIIB (Fig. 3G and fig. S5, E to G). We also collected the peritoneal exudates at 12 hours after Zym administration with or without sFgl2 treatment and observed that sFgl2 promoted PMN apoptosis in Fgl2KO mice (Fig. 3H). In addition, sFgl2 enhanced MΦ phagocytosis of AP-PMNs and opsonized Zym (Fig. 3I and fig. S5H). Of interest, the peritoneal Fgl2KO MΦs showed lower M2 marker arginase 1 (Arg-1) and higher M1 marker inducible nitric oxide synthase (iNOS), which could be reversed by sFgl2 treatment (fig. S5I). RKO mice phenocopied the Fgl2KO mice that showed shortened survival in the CLP model. Although sFgl2 only showed an improved trend of CLP survival in WT mice, it significantly improved sepsis survival in Fgl2KO mice but not in RKO mice (Fig. 3J). These findings indicated that sFgl2 promoted resolution of inflammation.
sFgl2 promotes RvDp5 biosynthesis
As noted above, both RvDp5 and LXA4 were significantly reduced in Fgl2KO peritoneal exudates during resolution. RvDp5 is a novel SPM derived from n-3 DPA via the catalysis of 12/15-lipoxygenase (12/15-LOX in mice) and 5-LOX (Fig. 4A) (2). We next determined key enzymes involved in the biosynthesis of SPM and pro-inflammatory lipid mediators. Fgl2 deficiency in MΦ inhibited 12/15-LOX, but not 5-LOX, and increased cyclooxygenase-2 (COX-2) and prostaglandin dehydrogenase/eicosanoid oxidoreductase (PGDH/EOR), which were inversely regulated by sFgl2 (Fig. 4B). Consistently, the reduced 12/15-LOX–derived mediators (e.g., RvDp5 and LXA4) in Fgl2KO MΦ were significantly reversed by sFgl2 administration (Fig. 4C). Moreover, a 12/15-LOX inhibitor blunted the sFgl2-induced up-regulation of RvDp5 and LXA4 (Fig. 4D). These results suggested that sFgl2 promoted SPM production by up-regulating 12/15-LOX.
Fig. 4. sFgl2 induces RvDp5 biosynthesis.
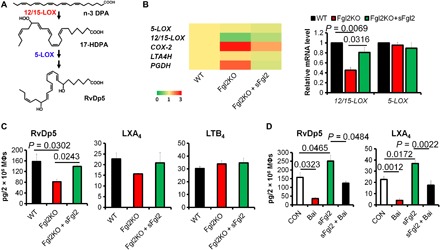
(A) Biosynthesis of RvDp5. (B) mRNA expression of key enzymes in PUFA metabolism. (C) Murine peritoneal MΦs were isolated from WT and Fgl2KO mice. Fgl2KO MΦs were treated with PBS or sFgl2 (10 μg/ml) for 4 hours. Quantification of RvDp5, LXA4, and LTB4 was obtained with UPLC-MS/MS. Results are presented as mean ± SEM (n = 4 to 6). (D) Murine peritoneal MΦs were treated with PBS, baicalein (10 μM), and/or sFgl2 (10 μg/ml) for 4 hours, and the levels of RvDp5 and LXA4 were obtained with UPLC-MS/MS. Error bars represent mean ± SEM. Two-tailed Student’s t test was applied for the P values.
RvDp5 accelerates inflammation resolution by activating ALX/FPR2
We next investigated the actions of the novel resolvin RvDp5 in inflammation. With the Zym-induced SL peritonitis model, we found that RvDp5 significantly reduced PMN infiltration, shortened Ri for ~4 hours, and increased K50 by ~1.2-fold (Fig. 5A). ALX/FPR2 was identified as the mutual G protein–coupled receptor (GPCR) for LXA4 and RvD1 (13, 17). To identify the receptor for RvDp5, the potential binding affinity was analyzed by amber scoring function in DOCK6.5 (18). Intriguingly, we found that RvDp5 and LXA4 shared similar docking modes and were predicted to bind the extracellular domain of ALX/FPR2 (Fig. 5, B and C, and fig. S6A). Compared with the ALX/FPR2-LXA4 system, the ALX/FPR2-RvDp5 complex had lower root mean square deviation values, which suggested that the ALX/FPR2-RvDp5 complex was slightly more stable. These complexes shared similar root mean square fluctuation profiles (fig. S6B). In addition, ALX/FPR2-LXA4 complexes exhibited higher flexibility compared with ALX/FPR2-RvDp5 complexes (fig. S6B). The energy contributions from each residue during molecular dynamics (MD) simulation of ALX/FPR2 were also evaluated (fig. S6C). We found that the electrostatic interactions (∆Eele) and van der Waals interactions (∆Evdw) promoted ligand binding, while the polar solvation free energy (∆EGB) was unfavorable for ligand binding (table S2). The results from GPCR–β-arrestin (17) also delineated that both RvDp5 and LXA4 bound ALX/FPR2 (Fig. 5, D and E). However, LXA4 and RvDp5 did not show additive actions at 10 nM (Fig. 5E, inset). Consistently, both RvDp5 and LXA4 reduced pro-inflammatory IL-1β, MCP-1, and TNF but increased anti-inflammatory IL-10 in peritoneal exudates (Fig. 5F). These results identified that RvDp5 displayed proresolving functions via ALX/FPR2 activation.
Fig. 5. RvDp5 activates ALX/FPR2 to display dual anti-inflammatory and proresolving actions.
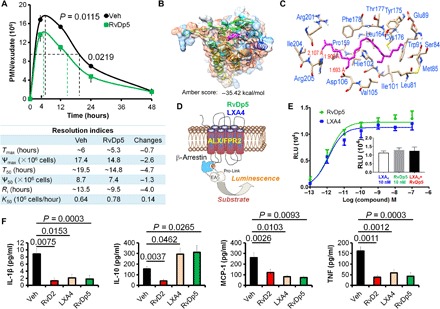
(A) Mice were injected intraperitoneally with Zym (1 mg) using PBS or RvDp5 (200 ng). Infiltrated PMNs were enumerated (upper panel), and resolution indices were calculated as indicated in Fig. 3B (bottom panel). (B) Three-dimensional (3D) chart of RvDp5 binding with ALX/FPR2. (C) Binding mode of RvDp5 structure ZINC35876755 in the binding pocket of ALX/FPR2. Important amino acid residues were shown, and the red lines indicated that the hydrogen bonds formed in the corresponding residues. (D) Diagrammatic principle of β-arrestin system. (E) Ligand (RvDp5 or LXA4)–receptor interaction was monitored in Chinese hamster ovary (CHO) cells using a β-arrestin system overexpressing ALX/FPR2. RLU, relative luminescence unit. (F) Mouse was treated intraperitoneally with Zym (1 mg/mouse; vehicle) with or without 200 ng of RvD2, LXA4, or RvDp5 for 4 hours, and the exudate levels of IL-1β, IL-10, MCP-1, and TNF were determined with ELISA. Error bars represent mean ± SEM. Two-tailed Student’s t test was applied for the P values.
RvDp5 and sFgl2 promote sepsis catabasis
We next determined the temporal Fgl2 expression in peritoneal exudates upon Zym challenge with or without the treatment of RvDp5. We found that RvDp5 significantly increased sFgl2 secretion and reduced leukocytic mFgl2 expression (Fig. 6A and fig. S7, A and B). In PMNs and MΦs, sFgl2 secretion was up-regulated by RvDp5 and RvD1, which was impaired by the ALX/FPR2-selective antagonist WRW4 (Fig. 6B). The ligation of ALX/FPR2 was known to couple protein kinase A (PKA) activation and cyclic adenosine monophosphate (cAMP) production (19). Because ADAM17 could be up-regulated by PKA (20), we speculated whether RvDp5 promoted sFgl2 secretion by ADAMs. Using the PKA inhibitor H89 and selective metalloproteinase inhibitors, we found that the cleavage and secretion of sFgl2 enhanced by RvDp5 were dependent on the PKA-mediated up-regulation of ADAM17, but not on ADAM10 (Fig. 6C and fig. S7C). Because sFgl2 production was found primarily in regulatory T cells (Tregs), we tested Tregs with RvDp5 and found that RvDp5 significantly promoted sFgl2 secretion from Tregs (fig. S7D). Because we showed that sFgl2 promoted PMN apoptosis, we questioned whether this action was dependent on RvDp5. However, RvDp5 did not significantly alter bone marrow–derived PMN apoptosis (fig. S7E). sFgl2 and RvDp5 synergistically enhanced MΦ efferocytosis and IL-10 production, accelerating the resolution of inflammation (Fig. 6, D and E, and fig. S7F), and improved sepsis survival (fig. S7G). As mentioned previously, RvDp5 stimulation of ALX/FPR2 through PKA activates ADAM17 and thus promotes sFgl2 shedding. We wondered whether RvDp5 can also act in an Fgl2 shedding–independent pathway. CLP sepsis experiments showed that RvDp5 improved sepsis survival of RKO mice (fig. S7H), indicating that RvDp5 can also protect sepsis independent of sFgl2. Moreover, inhibition of ALX/FPR2 or 12/15-LOX worsened the sepsis outcome (fig. S7I). Together, these results demonstrated that both Fgl2 and RvDp5 were potent regulators in inflammation resolution (Fig. 6F).
Fig. 6. RvDp5 and sFgl2 synergistically accelerate sepsis catabasis.
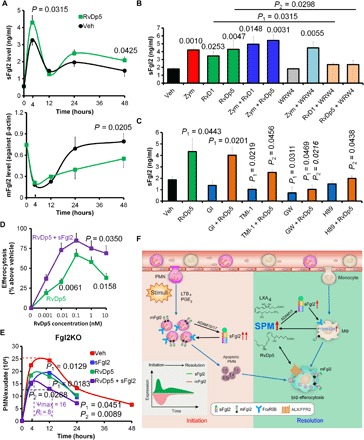
(A) WT C57BL/6 mice were injected intraperitoneally with Zym (1 mg) using PBS or RvDp5 (200 ng), and exudate sFgl2 and peritoneal leukocytic mFgl2 were assessed. (B) Murine MΦs were treated with Zym (100 ng/ml), RvD1 (10 nM), RvDp5 (10 nM), and WRW4 (1 μM) as indicated. The supernatant expression of sFgl2 was detected with ELISA. Error bars represent mean ± SEM. Numbers upon the groups were P values as compared with vehicle. P1 represents the comparison between RvD1 and RvD1 + WRW4, while P2 represents the comparison between RvDp5 and RvDp5 + WRW4. (C) After human MΦs were treated with PBS (Veh), RvDp5 (10 nM) with or without GI254023X (10 μM), TMI-1 (10 μM), GW280264X (10 μM), or H89 (10 μM) for 24 hours, sFgl2 in the supernatant was assessed with ELISA. (D) WT C57BL/6 murine MΦs were treated with RvDp5 (0 to 10 nM) with or without sFgl2 (10 μg/ml) for 1 hour and then coincubated with CFDA-labeled apoptotic PMNs (1:3) for another 1 hour. The percent increases of efferocytosis are shown. (E) Fgl2KO mice were injected intraperitoneally with Zym (1 mg) using PBS, RvDp5 (200 ng), and/or sFgl2 (200 ng). The exudate PMNs were enumerated. Error bars represent mean ± SEM. P1 represents value compared with vehicle; P2 represents value compared with RvDp5 alone. Error bars represent mean ± SEM. (F) Schematic mechanism of Fgl2 and RvDp5 programming inflammation resolution. Inflammatory stimuli trigger the infiltration of PMN and ADAM10/17-mediated Fgl2 shedding. By binding to FcγRIIB, sFgl2 promotes the apoptosis of PMN, which recruit monocytes and MΦs for efferocytosis. The exudate sFgl2 enhances the expression of 12/15-LOX in MΦs and the production of SPM, including RvDp5 and LXA4. RvDp5 and LXA4 activate ALX/FPR2 to promote ADAM17-mediated sFgl2 shedding, thereby synergistically facilitating inflammation resolution.
DISCUSSION
Sepsis is a major clinical challenge. Despite intensive research, its pathophysiology remains incompletely understood. Here, we provide a key mechanistic insight on novel links between sFgl2 and resolvins. The temporal and differential expression of sFgl2 and mFgl2 during the initiation and resolution of inflammation correlates with sepsis survival. Scilicet, the extent and duration of sFgl2 cleavage and mFgl2 reduction upon inflammatory challenge, determines the magnitude and consequence of sepsis.
Our previous results indicated that compared to nonseptic control, both sepsis survivors and nonsurvivors showed a significant increase in both whole blood and plasma miR-466l, a pro-inflammatory and proresolving miRNA that stabilizes downstream targets, upon admission in ICU. Moreover, the sepsis nonsurvivors exhibited a higher miR-466l level than did sepsis survivors (10). Our present results indicated that the expression of Fgl2 was up-regulated by miR-466l, while the ectodomain shedding and secretion of sFgl2 from the cell membrane during the initiation of inflammation were triggered by ADAM10 and ADAM17. Subsequently, sFgl2 promoted PMN apoptosis and clearance by MΦs by activating its receptor FcγRIIB, during which SPMs including LXA4 and RvDp5 were biosynthesized. The novel proresolving RvDp5, in turn, promoted secondary sFgl2 accumulation and reduced leukocytic mFgl2 during the resolution stage by activating the receptor signaling pathway ALX/FPR2-PKA-ADAM17. Conversely, the prothrombinase mFgl2 was sharply reduced upon inflammatory challenge but rebounded subsequently. The earlier increase of mFgl2 and lower sFgl2 indicated the worse outcomes of inflammation. Of note, we demonstrated that RvDp5 per se protects mice against sepsis independent of sFgl2 and the receptor FcγRIIB. Therefore, sFgl2 and RvDp5 synergistically accelerated inflammation resolution and tissue homeostasis. Controlling the temporal expression of sFgl2 and mFgl2 may contribute to the regulation of sepsis catabasis.
Some earlier reports already suggested links between physiologic and pathologic coagulation with the resolution of inflammation and infections. Aspirin displays dual anti-inflammatory and anticoagulation effect by reducing pro-inflammatory [prostaglandin E2 (PGE2)] and procoagulant thromboxane A2 (TXA2) by acetylating COX-2 (21) and promoting SPM production (22). Using both 2D proteomics and lipidomics with self-resolving inflammatory exudates, we identified coagulation factors such as fibrinogen, along with resolvins and protectins biosynthesized from essential fatty acids (13). Moreover, we recently found that removal of adenosine from coagulating blood markedly enhanced the biosynthesis of SPMs (23). RvD4 attenuates the severity of pathological thrombosis (24). In our present study, inflammatory stimuli triggered the infiltration of PMNs that secreted abundant sFgl2 to the exudates, while prothrombinase mFgl2 was temporally down-regulated. Subsequently, sFgl2 promoted PMN apoptosis, which exhibits “find me” signals for monocytes and MΦs. In addition, sFgl2 promoted MΦ maturation and efferocytosis, during which SPMs were biosynthesized. Inefficient shedding of sFgl2 may increase coagulation by membrane Fgl2, but our data showed that sFgl2 improved the resolution of inflammation in Fgl2 knockout mice, which did not express procoagulant membrane Fgl2. It is noteworthy that Fgl2 is also constitutively expressed during embryogenesis (25). Therefore, it may have potential effects in the development and maturation of immune components. Our results suggest that the maturation and phagocytosis activity of macrophages as well as the spontaneous apoptosis of PMN were attenuated in Fgl2KO mice, which contributed to delayed resolution.
In summary, our present results uncovered a new programmed proresolving mechanism involving endogenous Fgl2 and RvDp5 and the identification of differential roles of sFgl2 and mFgl2 in inflammation resolution. Upon the onset of inflammation, shedding and secretion of sFgl2 program the subsequent resolution that requires the reduction and delayed restoration of mFgl2. In future sepsis treatments, timely resolution should be considered, which suggests temporal regulation of Fgl2 given this previously unknown link between Fgl2 and SPM.
MATERIALS AND METHODS
Human subjects and mice
According to 45th Critical Care Congress of the Society of Critical Care Medicine (SCCM) in 2016, sepsis was diagnosed as “life-threatening organ dysfunction caused by dysregulated host response to infection.” The exclusion criteria included the following: age <18 or >65 years, pregnant or lactating women, those who received chemotherapy within the recent 6 months, those who received corticosteroids or other immunosuppressive agents or immunomodulators within the preceding 3 months, those with immune system diseases, HIV-positive patients, those with end-stage liver or renal failure, and those who stayed in the ICU for less than 48 hours. The plasma of survivors and nonsurvivors diagnosed as sepsis and nonseptic patients was obtained within 24-hour ICU admission at the Second Affiliated Hospital of Wenzhou Medical University. All healthy donors and patients gave written consents. The nonseptic controls were healthy volunteers and matched by age and sex with the sepsis patients. Fgl2KO and FcγRIIBKO mice were respectively provided by S. Smiley (The Trudeau Institute, NY, USA) and J. S. Verbeek (Leiden University Medical Center, The Netherlands). C57BL/6 mice were obtained from the Animal Institute of the Academy of Medical Science (Beijing, China). The mice were kept under specific pathogen–free conditions at the Animal Center of the Third Military Medical University. All human and animal experiments met the ethical principles and requirements of our committee and comply with the Declaration of Helsinki. The institutional review board and/or Institutional Animal Care and Use Committee guidelines were followed with human or animal subjects.
Cecal ligation and puncture
CLP was performed in male WT and Fgl2KO C57BL/6 mice according to the protocol reported (9). Ligation of the cecum at designated positions was the major determinant of sepsis severity. The intermediate sepsis was ligated at half the distance between distal pole and the base of cecum (moderate CLP), while severe CLP sepsis comprised ligation of 75% of the cecum. A through-and-through cecal puncture was performed with a 21-gauge needle followed by an additional puncture in the distal tip of the cecum. At indicated intervals, rectal temperature was measured. Peritoneal exudates and blood were collected for respective experiments. Blood bacteria levels were determined by growth on tryptic soy agar plates. Immunohistochemical staining for H&E, CD45 (1:100, catalog no. sc-53665, Santa Cruz Biotechnologies), and F4/80 (1:100, catalog no. 123105, BioLegend) was performed with the cecal tissues from WT and Fgl2KO mice after being challenged with CLP for 24 hours. For the abscess formation experiments, mice were sacrificed at day 7 after CLP.
Murine peritonitis
The SL and DR murine peritonitis models and resolution interval (Ri) were carried out as described (10). Briefly, mice were injected intraperitoneally with 1 ml of Zym at 1 mg per mouse or 10 mg per mouse. For the indicated experiments, 200 ng of RvDp5 (7S,17S-diHDPA, catalog no. 10546, Cayman Chemical Co., Ann Arbor, MI) or 400 ng of recombinant sFgl2 (catalog no. 5257-FL; R&D Systems, Minneapolis, MN, USA) was also injected intraperitoneally. The peritoneum was lavaged at 0, 4, 12, 24, and 48 hours after injection, and the total number of infiltrated cells was assessed using trypan blue. PMNs (CD11b+Ly6G+), monocytes, and macrophage (CD11b+Ly6G−) were enumerated by light microscopy and immunofluorescent staining.
Enzyme-linked immunosorbent assay
Human plasma sFgl2 was determined with a human Fgl2 ELISA kit (catalog no. 436907, BioLegend) according to the manufacturer’s instructions. Mouse peritoneal exudates, plasma, and cell supernatants were used for detecting sFgl2 (catalog no. 437808, BioLegend) and other cytokines [IL-1β, IL-10, MCP-1, CXCL1, IL-6, IL-12p40, TNF, and interferon-γ (IFN-γ); ELISA kits from R&D Systems, Minneapolis, MN, USA]. In some indicated experiments, 10 μM GI254023X (ADAM10 inhibitor; catalog no. SM20789, Sigma-Aldrich), 10 μM TMI-1 (ADAM17 inhibitor; catalog no. 287403-39-8, TOCRIS), 10 μM GW280264X (dual ADAM10 and ADAM17 inhibitor; catalog no. AOB3632, Aobious, Gloucester, MA), 10 μM H89 (PKA inhibitor; catalog no. S1582, SelleckChem, Houston, TX), 10 nM LXA4 (5S,6R,15S-TriHETE; catalog no. 90410, Cayman Chemical), 10 nM RvD1 (7S,8R,17S-TriHETE; catalog no. 10012554, Cayman Chemical), 10 nM RvD2 (7S,16R,17S-TriHETE; catalog no. 10007279, Cayman Chemical), 1 μM baicalein (catalog no. 491-67-8, Adamas), and 1 μM WRW4 (catalog no.344220, Calbiochem) were administrated. For the experiments of mFgl2 determination, cell proteins were collected with a Membrane and Cytoplasmic Protein Extraction kit (catalog no. C510005, Sangon Biotech, Shanghai, China) and assessed with ELISA (catalog no. 436907 for human and catalog no. 437808 for mouse; BioLegend) and were normalized with β-actin (PathScan Total β-Actin Sandwich ELISA Kit, #7880, Cell Signaling Technology).
LC-MS/MS–based lipidomics
Mouse peritoneal exudates and human macrophages with supernatants were extracted for LC-MS/MS–based mediator analysis. We used a UPLC I-Class system (Waters, Milford, MA, USA) equipped with an AB Sciex Instruments 6500 Q-TRAP mass spectrometer (Applied Biosystems, Foster City, CA, USA). Acquisition was carried out in a negative ionization mode. The mobile phase consisted of methanol/water/acetic acid (60:40:0.01, v/v/v) and was ramped to 85:15:0.01 over 30 min and to 100:0:0.01 over the next 5 min at a flow rate of 200 ml/min. Instrument control and data acquisition were performed using Analyst 1.6 software (Applied Biosystems). A minimum of six diagnostic ions and retention time were used for lipid identification (23). Quantification was based on peak area of multiple reaction monitoring transitions and linear calibration curve of each compound.
In silico analysis
The miRBase accession number for mmu-miR-466l-3p is MIMAT0005830. The binding sites of FGL2 3′UTR for miR-466l were obtained using TargetScan (http://www.targetscan.org/), miRDB (http://mirdb.org/miRDB/), and miRanda (http://www.microrna.org/microrna/home.do), with default parameters.
PMN preparation and nucleotide assay
Human and mouse PMNs were isolated from peripheral blood according to the manufacturer’s instructions (TBD Science, Tianjin). To prepare apoptotic PMNs, cells were induced by UV for 4 hours. The concentration of ATP was measured as previously described (16). The supernatants were prepared from apoptotic PMNs cultured in RPMI containing 1% bovine serum albumin and 10 mM Hepes (pH 7.2).
MΦ isolation and transfection and luciferase assay
Human and murine MΦs were prepared and transfected as previously reported (10). For the luciferase assay of 3′UTR vectors of vehicle control and FGL2 (catalog nos. m012 and MT-h07957, Applied Biological Materials Inc.), miR-466l or mock (catalog nos. mm40846 and m003, Applied Biological Materials Inc.) was cotransfected. The luciferase activity was assayed using SuperLight Luciferase reporter assay and measured using a microplate reader (Varioskan Flash, Thermo Fisher Scientific, Shanghai).
MΦ efferocytosis and phagocytosis
MΦs were incubated with RvDp5 or recombinant sFgl2 (catalog no. 5257-FL, R&D Systems) at the indicated dose for 2 hours, followed by incubation with fluorescein isothiocyanate (FITC)–labeled Zym particles at 50:1 (Zym:MΦ) or with CFDA-labeled AP-PMNs at 3:1 (AP-PMNs:MΦs) for 1 hour. Extracellular fluorescence was quenched by trypan blue, and then phagocytosis and efferocytosis were determined by flow cytometry (FCM) using FACSCanto II (BD) or measuring total fluorescence (excitation, 493 nm/emission, 535 nm) using a fluorescent plate reader (Varioskan Flash).
GPCR–β-arrestin system
The interactions between ligands (LXA4 and RvDp5) and ALX/FPR2 were determined with the Beta Arrestin Path Hunter eXpress System (DiscoveRx), as indicated previously (with CHO cells stably transfected with human ALX/FPR2) (16). Cells were plated in 96-well plates 48 hours before experiments. The compounds were incubated with cells for 1 hour at 37°C, and the activation of ALX/FPR2 was assessed by measuring luminescence. The percentage of activity was calculated using the following formula: percent activity = 100% × (mean RLU of test sample − mean RLU of vehicle control)/(mean RLU of vehicle control).
Real-time PCR
Quantitative polymerase chain reaction (PCR) was performed using the SYBR Green System according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA). The expressions of target genes were analyzed using the ΔCt method. The primers for miR-466l (MQP-0101) and 5S ribosomal RNA (rRNA) (MQP-0301) were obtained from Guangzhou RiboBio Co. Ltd. (Guangdong, China). The forward and reverse primers for other genes are as follows:
mFGL2-F GGTGCTCAAAGAAGTGCGGA and mFGL2-R GTTCCTGGACTCTACTGTCCTC; m5-LOX-F CCTATTCCTCCCTGTGTTTCC and m5-LOX-R CACGAGCAGTCCATCATCAC; m12/15-LOX-F GGCTCCAACAACGAGGTCTAC and m12/15-LOX-R CCCAAGGTATTCTGACACATCC; mCOX-2F GGTGCCTGGTCTGATGATGT and mCOX-2-R TGCTGGTTTGGAATAGTTGCT; mLTA4H-F GAGGTCGCGGATACTTGCTC and mLTA4H-R CTCCTGTGACTGGACCGTG; mPGDH-F GGACACACCCATCCTTGAAT and mPGDH-R TCGATGCCGTGATCTTCATA; mβ-ACTIN-F TGACAGGATGCAGAAGGAGA and mβ-ACTIN-R GTACTTGCGCTCAGGAGGAG; hADAM10-F CAACCTACGAATGAAGAGGGACAC and hADAM10-R CCACCACGAGTCTGGATGAATC; hADAM17-F GAAGTGCCAGGAGGCGATTA and hADAM17-R CGGGCACTCACTGCTATTACC; and hβ-ACTIN-F GCGCGGCTACAGCTTCA and hβ-ACTIN-R CTTAATGTCACGCACGATTTCC.
Western blotting
The primary anti-mouse antibodies used were cleaved caspase 3 (Asp175, catalog no. 9661, Cell Signaling Technology, Danvers, MA), ADAM10 (catalog no. AB19026, Merck Millipore, Darmstadt, Germany), ADAM17 (catalog no. AB19027, Merck Millipore), and β-actin (clone 8H10D10, catalog no. 3700, Cell Signaling Technology). The secondary antibody was a conjugated immunoglobulin G (IgG) (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Quantification of bands was achieved by densitometry using the ImageJ software.
Flow cytometry
The FCM was performed as previously reported (10). Peritoneal exudates and cultured PMNs and MΦs were used for determining indicated antigens, including CD11b (clone M1/70, catalog no. 101206, BioLegend, San Diego, CA), F4/80 (clone BM8, catalog nos. 123110 and 123116, BioLegend), Ly6G (clone 1A8, catalog no. 127618, BioLegend), annexin V (catalog no. 640930, BioLegend), and mFgl2 (clone 6D9). The cellular fluorescence was assessed using FACSCanto II (BD), and data were analyzed with FlowJo v10 (Tree Star Inc., Ashland, OR). In Treg experiments, spleen CD4+CD25+ Tregs were isolated by fluorescence-activated cell sorting (FACS) and then treated with or without RvDp5 (10 nM) for 24 hours.
Immunohistochemistry
Mouse cecal tissues were fixed with formaldehyde. Paraffin sections were stained with H&E or subjected to immunohistochemistry for CD45 (30-F11, catalog no. sc-53665, Santa Cruz Biotechnology, Dallas, TX) and F4/80 (catalog no. 123105, BioLegend).
Docking experiment
The 3D structure of ALX/FPR2 was constructed using SWISS-MODEL server (http://swissmodel.expasy.org/), with the structure of the type 1 angiotensin II receptor (Protein Data Bank ID: 4YAY) as template. The structures of RvDp5 (ZINC35876753, ZINC35876755, ZINC35876757, and ZINC35876759) and LXA4 (ZINC03873139, ZINC04556658, ZINC04556659, and ZINC04556660) were downloaded from the ZINC database (http://zinc.docking.org) by querying their Chemical Abstracts Service (CAS) registry number and prepared for dock by added charge with the University of California, San Francisco (UCSF) chimera program. These chemical structures were isomers of RvDp5 and LXA4, respectively. Molecular docking calculations were performed by using the UCSF DOCK6.5 program (18). The molecular surface of the ALX/FPR2 complex was computed with a probe radius of 1.4 Å, and spheres were generated from the molecular surface, with a maximum and minimum radius of 4.0 and 1.4 Å, respectively. The binding sites of RvDp5 and LXA4 with ALX/FPR2 were identified automatically using the largest binding cluster of ALX/FPR2 and located in extracellular domain. The docking pose was ranked on the basis of grid score, and then the amber-based energy scoring function in DOCK6.5 program was used to calculate molecular binding energies.
MD simulation
MD simulation has been widely used for investigating the interaction properties between molecules in a system. In this study, all MD simulations were performed using GROMACS software (version 4.5.5) (26). The ALX/FPR2 protein structure was processed by using tip3p water mode and AMBER99SB force field to generate coordinates and topology files (27). Then, the ligands were dealt with antechamber and tleap tools from AmberTools9 (28), and the ligand’s topology was created by using the ACPYPE tool (29). The simulations were carried out under periodic boundary conditions, with a cubic periodic box setting the minimal distance of 1.0 Å between the protein and edge of the box. To neutralize the system, ions (Na+ and Cl−) were added with a salt concentration of 0.15 M. Then, the system was energy-minimized using steepest descent minimization algorithm until the maximum force under 1000 kcal mol−1 nm−1. Then, we equilibrated the protein-ligand complex with 100-ps NVT equilibration (constant number of particles, volume, and temperature) to stabilize the system at 300 K and NPT (constant number of particles, pressure, and temperature) equilibration within 100 ps. After the equilibration steps, the system was well optimized, and a period of 20-ns MD simulations with a time step of 2 fs at constant pressure (1 atm) and temperature (300 K) was conducted. During the simulation process, energy and trajectory information were collected every 2 ps, long-range electrostatics was investigated by the particle mesh Ewald method (30), and all bond lengths were limited by LINCS algorithm (31).
Binding free energy and energy decomposition analysis
Binding free energy calculation is a powerful tool in providing quantitative measurement of protein-ligand interactions (32). The molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) approach was used to compute binding free energies, as it is often used to study biomolecular complexes (33). In brief, the binding free energy (ΔG binding) was calculated by the equation listed below using an open source tool called g_mmpbsa
| (1) |
ΔEMM consists of the electrostatic and van der Waals energies and is the gas-phase interaction energy between protein and ligand. ΔGsol includes the polar solvation free energy and the nonpolar energy—the former is calculated with the generalized Born approximation model and the latter is obtained as a function of the solvent-accessible surface area. TΔS indicates the changing of conformational entropy upon ligand binding. It is acceptable to exclude the entropy contribution (TΔS) in practice if ligands have similar structures and binding modes (34).
Energy decomposition analysis was performed using the mm_pbsa.pl. program (34). The residue-ligand interaction often contains four parts: van der Waals contribution (ΔGvdw), electrostatic contribution (ΔGele), the polar part of desolvation (ΔGGB), and the nonpolar part of desolvation (ΔGSA)
| (2) |
Statistical analysis
Statistical analysis was performed with Excel (Microsoft) and Prism software (GraphPad Software). Error bars represent means ± SEM. All representative experimental findings were verified in at least three independent experiments. Data were verified to be in a normal distribution. Results are expressed as mean ± SEM. Two-tailed Student’s t test was used to compare differences between groups. Analysis of variance (ANOVA) was applied to perform multiple comparisons. Mantel-Cox test was applied to compare survival rates between groups. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank C. Hu, R. Xin, and L. Jiang (Third Military Medical University) for the FCM and immunohistochemistry assistance. Funding: The work was supported by the National Natural Science Foundation of China (81671573) and Entrepreneurship & Innovation Program for Chongqing Overseas Returnees (no. cx2017016). C.N.S. was supported by NIH R01 GM038765. Author contributions: Y.L. conceived, designed, and supervised the study. Y.L., Y.Z., J.L., and Q.X. performed both CLP and peritonitis experiments. Q.X., S.J., J.G., and X.L. provided clinical samples and information and analyzed data. B.G. provided Fgl2KO and RKO mice and analyzed data. L.W. performed bioinformatics analysis and participated in the preparation of figures. Y.L., X.W., and J.Z. performed LC-MS/MS experiments. G.Y., Q.Z., H.Z., X.Z., and J.W. contributed to FCM experiments and data analysis. X.L., H.M., and D.Y. contributed to data discussion. Y.L., H.M., and C.N.S. analyzed data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax0629/DC1
Table S1. Characteristics of patients.
Table S2. The predicted binding free energy components for the ALX/FPR2-RvDp5 and ALX/FPR2-LXA4 complexes using the MM/PBSA method.
Fig. S1. Inflammation profiles in SL versus DR inflammation.
Fig. S2. Fgl2 expression is regulated by miR-466l and metalloproteinases.
Fig. S3. Deficiency of Fgl2 promotes inflammation initiation and delays resolution.
Fig. S4. Fgl2 deficiency reduces RvDp5 production.
Fig. S5. Fgl2 regulates PMN apoptosis and phagocytes of MΦs.
Fig. S6. An overview of binding modes for the four isomers of RvDp5 and LXA4 in the binding pocket of ALX/FPR2.
Fig. S7. RvDp5 modulates Fgl2 and improves sepsis survival.
REFERENCES AND NOTES
- 1.Nathan C., Ding A., Nonresolving inflammation. Cell 140, 871–882 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Serhan C. N., Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levi M., van der Poll T., Büller H. R., Bidirectional relation between inflammation and coagulation. Circulation 109, 2698–2704 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Marx J. L., Coagulation as a common thread in disease. Science 218, 145–146 (1982). [DOI] [PubMed] [Google Scholar]
- 5.Coughlin S. R., Thrombin signalling and protease-activated receptors. Nature 407, 258–264 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Shalev I., Liu H., Koscik C., Bartczak A., Javadi M., Wong K. M., Maknojia A., He W., Liu M. F., Diao J., Winter E., Manuel J., McCarthy D., Cattral M., Gommerman J., Clark D. A., Phillips M. J., Gorczynski R. R., Zhang L., Downey G., Grant D., Cybulsky M. I., Levy G., Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J. Immunol. 180, 249–260 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Liu J., Tan Y., Zhang J., Zou L., Deng G., Xu X., Wang F., Ma Z., Zhang J., Zhao T., Liu Y., Li Y., Zhu B., Guo B., C5aR, TNF-α, and FGL2 contribute to coagulation and complement activation in virus-induced fulminant hepatitis. J. Hepatol. 62, 354–362 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Selzner N., Liu H., Boehnert M. U., Adeyi O. A., Shalev I., Bartczak A. M., Xue-Zhong M., Manuel J., Rotstein O. D., McGilvray I. D., Grant D. R., Phillips M. J., Levy G. A., Selzner M., FGL2/fibroleukin mediates hepatic reperfusion injury by induction of sinusoidal endothelial cell and hepatocyte apoptosis in mice. J. Hepatol. 56, 153–159 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A., Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Dalli J., Chiang N., Baron R. M., Quintana C., Serhan C. N., Plasticity of leukocytic exudates in resolving acute inflammation is regulated by microRNA and proresolving mediators. Immunity 39, 885–898 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khokha R., Murthy A., Weiss A., Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Pruessmeyer J., Hess F. M., Alert H., Groth E., Pasqualon T., Schwarz N., Nyamoya S., Kollert J., van der Vorst E., Donners M., Martin C., Uhlig S., Saftig P., Dreymueller D., Ludwig A., Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood 123, 4077–4088 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Bannenberg G. L., Chiang N., Ariel A., Arita M., Tjonahen E., Gotlinger K. H., Hong S., Serhan C. N., Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Chiang N., Fredman G., Bäckhed F., Oh S. F., Vickery T., Schmidt B. A., Serhan C. N., Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalli J., Colas R. A., Serhan C. N., Novel n-3 immunoresolvents: Structures and actions. Sci. Rep. 3, 1940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elliott M. R., Chekeni F. B., Trampont P. C., Lazarowski E. R., Kadl A., Walk S. F., Park D., Woodson R. I., Ostankovich M., Sharma P., Lysiak J. J., Harden T. K., Leitinger N., Ravichandran K. S., Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnamoorthy S., Recchiuti A., Chiang N., Yacoubian S., Lee C. H., Yang R., Petasis N. A., Serhan C. N., Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 1660–1665 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang P. T., Brozell S. R., Mukherjee S., Pettersen E. F., Meng E. C., Thomas V., Rizzo R. C., Case D. A., James T. L., Kuntz I. D., DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 15, 1219–1230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Zein N., Badran B., Sariban E., VIP differentially activates β2 integrins, CR1, and matrix metalloproteinase-9 in human monocytes through cAMP/PKA, EPAC, and PI-3K signaling pathways via VIP receptor type 1 and FPRL1. J. Leukoc. Biol. 83, 972–981 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Leemhuis J., Boutillier S., Schmidt G., Meyer D. K., The protein kinase A inhibitor H89 acts on cell morphology by inhibiting Rho kinase. J. Pharmacol. Exp. Ther. 300, 1000–1007 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Capone M. L., Tacconelli S., Sciulli M. G., Grana M., Ricciotti E., Minuz P., Di Gregorio P., Merciaro G., Patrono C., Patrignani P., Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation 109, 1468–1471 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Serhan C. N., Fredman G., Yang R., Karamnov S., Belayev L. S., Bazan N. G., Zhu M., Winkler J. W., Petasis N. A., Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 18, 976–987 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norris P. C., Libreros S., Chiang N., Serhan C. N., A cluster of immunoresolvents links coagulation to innate host defense in human blood. Sci. Signal. 10, eaan1471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherpokova D., Jouvene C. C., Libreros S., DeRoo E., Chu L., de la Rosa X., Norris P., Wagner D. D., Serhan C. N., Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu J., Qu D., Bartczak A., Phillips M. J., Manuel J., He W., Koscik C., Mendicino M., Zhang L., Clark D. A., Grant D. R., Backx P. H., Levy G. A., Adamson S. L., Fgl2 deficiency causes neonatal death and cardiac dysfunction during embryonic and postnatal development in mice. Physiol. Genomics 31, 53–62 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E., GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C., Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D. Case, D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, D. A. Pearlman, M. Crowley, R. C. Walker, W. Zhang, B. Wang, S. Hayik, A. Roitberg, G. Seabra, K. F. Wong, F. Paesani, X. Wu, S. Brozell, V. Tsui, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, P. Beroza, D. H. Mathews, C. Schafmeister, W. S. Ross, P. A. Kollman, AMBER 9 (University of California, San Francisco, 2006). [Google Scholar]
- 29.Sousa da Silva A. W., Vranken W. F., ACPYPE—AnteChamber PYthon Parser interfacE. BMC. Res. Notes 5, 367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 31.Hess B., P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Wang W., Kollman P. A., Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 303, 567–582 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Massova I., Kollman P. A., Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspec. Drug Discov. Design 18, 113–135 (2000). [Google Scholar]
- 34.Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D. A., Cheatham T. E., Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax0629/DC1
Table S1. Characteristics of patients.
Table S2. The predicted binding free energy components for the ALX/FPR2-RvDp5 and ALX/FPR2-LXA4 complexes using the MM/PBSA method.
Fig. S1. Inflammation profiles in SL versus DR inflammation.
Fig. S2. Fgl2 expression is regulated by miR-466l and metalloproteinases.
Fig. S3. Deficiency of Fgl2 promotes inflammation initiation and delays resolution.
Fig. S4. Fgl2 deficiency reduces RvDp5 production.
Fig. S5. Fgl2 regulates PMN apoptosis and phagocytes of MΦs.
Fig. S6. An overview of binding modes for the four isomers of RvDp5 and LXA4 in the binding pocket of ALX/FPR2.
Fig. S7. RvDp5 modulates Fgl2 and improves sepsis survival.