

This work determines the interactions between spCas9/sgRNA and DNA that dictate spCas9 binding and dissociation.
Abstract
Cas9 is an RNA-guided endonuclease that targets complementary DNA for cleavage and has been repurposed for many biological usages. Cas9 activities are governed by its direct interactions with DNA. However, information about this interplay and the mechanism involved in its direction of Cas9 activity remain obscure. Using a single-molecule approach, we probed Cas9/sgRNA/DNA interactions along the DNA sequence and found two stable interactions flanking the protospacer adjacent motif (PAM). Unexpectedly, one of them is located approximately 14 base pairs downstream of the PAM (post-PAM interaction), which is beyond the apparent footprint of Cas9 on DNA. Loss or occupation of this interaction site on DNA impairs Cas9 binding and cleavage. Consistently, a downstream helicase could readily displace DNA-bound Cas9 by disrupting this relatively weak post-PAM interaction. Our work identifies a critical interaction of Cas9 with DNA that dictates its binding and dissociation, which may suggest distinct strategies to modulate Cas9 activity.
INTRODUCTION
CRISPR-Cas imparts bacteria and archaea with adaptive immunity against invasive viruses and plasmids (1). In the type II CRISPR-Cas system, the Cas9 protein from Streptococcus pyogenes (spCas9), which is in a complex with two RNAs (CRISPR RNA and trans-activating RNA), targets complementary 20–base pair (bp) sequences adjacent to a protospacer adjacent motif (PAM) in foreign DNA for site-specific double-stranded cleavage (2, 3). These two noncoding RNAs can be fused into a single guide RNA (sgRNA), while fully functional Cas9-mediated sequence-specific DNA cleavage is retained (2). Because of the simplicity and specificity of programmable DNA recognition and cleavage by spCas9, it has been widely repurposed for genome editing, transcriptional perturbation, and genomic imaging in various organisms (4). Increasing its fidelity and minimizing off-target cleavage to ensure its efficient application in medicine and biology have been the focus of considerable research efforts (5, 6). These improvements demand a clear understanding of the interplay between spCas9 and DNA. Both biochemical and structural studies have shown that the nuclease activity of spCas9/sgRNA requires PAM recognition, DNA duplex bending and unwinding, almost complete RNA-DNA complementarity, and subsequent subdomain conformational rearrangement (7–11). All these prerequisites are largely dictated by the critical and specific interactions of Cas9 with the target DNA. Thus, information about these interactions would aid in the understanding of the specificity of Cas9 in DNA targeting and cleavage and the development of Cas9 derivatives.
One of the features that distinguishes spCas9 from other endonucleases is its stable binding to its target DNA after cleavage (7, 12, 13). In vitro studies have demonstrated that spCas9 can bind to the DNA target for hours without dissociation (7). This is consistent with in vivo studies showing that the spCas9 in cells remains associated with the DNA target for several hours until DNA replication (14, 15). The long lifetime of the spCas9-DNA complex reflects the strong interactions between the two components and results in the enzyme being a single-turnover nuclease, limiting the usage of each spCas9 protein. In addition, stable binding of spCas9 to the DNA target could impair its use in genomic editing by impeding subsequent repair by DNA repair pathways. This is substantiated by the finding that mammalian cells require much more time to repair lesions generated by spCas9 compared with DNA double-strand breaks (DSBs) caused by ionizing radiation (16). Thus, it is expected that increasing the slow off-rate of the binding of spCas9 to DNA would improve its efficiency as a genome-editing tool. A recent report demonstrated that RNA polymerase (RNAP) can act as a scavenger to displace DNA-bound Cas9, leading to an increase in genome-editing efficiency (17). Thus, an understanding of the nature and duration of Cas9/sgRNA/DNA interactions as well as the mechanism involved in Cas9 displacement could suggest distinct approaches that could be used to increase its effectiveness in applications.
In this work, we used an optical tweezer–based DNA unzipping technique to determine the interactions between spCas9/sgRNA and DNA. We identified two stable interactions of spCas9 with the target DNA that flanked the PAM as well as an intermittent interaction near the PAM-distal region. The stable interaction downstream of the PAM is relatively weaker than the other interaction inside the 20-bp region; yet, it is essential for Cas9 binding, cleavage, and dissociation, and its disruption leads to the immediate collapse of the ternary complex. Consistently, a helicase on the downstream side of the PAM can thus more readily displace the protein by breaking this relatively weak interaction than a helicase on the upstream side. In conclusion, we directly determined both the locations and the strength of the interactions between spCas9/sgRNA and the target DNA and revealed how these interactions dictate its binding and dissociation.
RESULTS
Positions and strength of Cas9/sgRNA/DNA interactions
To probe the interactions of Cas9/sgRNA and DNA, we used a single-molecule optical tweezer technique to mechanically unzip double-stranded (ds) DNA through a bound Cas9/sgRNA complex. This single-molecule approach has proven to be a unique and versatile tool for the accurate and precise determination of the positions and the strength of DNA-protein interactions (figs. S1 and S2) (18, 19). To this end, we first constructed a Y-shaped DNA template consisting of two arms and a trunk (fig. S1) (20). This template was suspended between an optically trapped microsphere and a glass coverslip surface via two special labeled arm ends (Fig. 1A). By programming the sgRNA, Cas9/sgRNA was uniquely positioned on the trunk and flanked by relatively long segments of DNA. Moving the coverslip away from the optical trap resulted in the unzipping of the double helix of the DNA trunk. The difference between the measured force and that corresponding to the unzipping of naked DNA, which is a sequence-dependent baseline of approximately 18 pN, serves as a signal of protein-DNA interactions that can be used to reveal both the location and the strength of the binding of an associated Cas9/sgRNA (Fig. 1B).
Fig. 1. Experimental configuration for single-molecule unzipping experiments and dCas9/sgRNA/DNA interaction mapping.

(A) Cartoon illustrating the single-molecule DNA unzipping experiment used for the detection of the interactions between dCas9/sgRNA and DNA. The sequence of the steps of the unzipping of the DNA template is numbered as illustrated. (B and E) Representative traces of forward (black) and reverse unzipping (blue) in the presence of dCas9 showing the force versus number of base pairs unzipped. The naked DNA unzipping signatures are also presented for comparison (gray). The light-yellow rectangle shows the expected Cas9 binding position. Red arrows indicate the force peaks. Insets: Zoom in of the regions with increases in force. (C and F) Histogram of the disruption forces of dCas9/sgRNA. The data and their Gaussian fits are shown. (D and G) Histogram of the positions of dCas9/sgRNA and target DNA interactions along the DNA sequence. The data and their Gaussian fits are shown. The black and blue triangles indicate the pre- and post-PAM interactions, respectively.
We first used a catalytically dead version of spCas9 (hereafter referred to as dCas9) with an sgRNA (sgRNA-1) whose complementary DNA target is located between position +20 (the PAM-distal side) and +1 (the PAM-proximal side) on the DNA trunk (Fig. 1A and table S1) (2). The PAM sequences were sequentially designated as 0, −1, and −2 for the sake of data presentation (Fig. 1A). In this experiment, DNA tethers with attached microspheres in a chamber were formed first. Then, dCas9/sgRNA complexes were introduced into the chamber, and the unbound proteins and sgRNAs were washed out after a short period of incubation (~30 s) (all subsequent unzipping experiments were performed after the washout unless otherwise stated). The experiment was performed by unzipping a single DNA molecule starting from the upstream side of the PAM (termed forward unzipping) (Fig. 1B). The unzipping force initially was similar to that of the corresponding naked DNA. However, as the unzipping fork encountered the expected position of the dCas9/sgRNA, the detected forces rose above those of the naked DNA baseline for all examined traces (n = 104), after which the forces continued to be similar to those of the corresponding naked DNA (Fig. 1B). Among the traces, 89 showed a single force peak, and the rest presented two peaks at the expected Cas9 binding position (Fig. 1B and fig. S3). In the control experiments, which used either sgRNA or dCas9, no such rise in force was observed after the washout of the proteins or the sgRNAs. Random off-target binding along the DNA template was only detectable if free dCas9 was present in the chamber (no washout). Therefore, we attributed the rise in force exclusively to the disruption of a stably formed dCas9/sgRNA/DNA complex at the expected position. We next carefully analyzed the positions and the strength of the interactions of Cas9 with the target DNA, as indicated by the rise in force. As the traces with single peak were predominant, we analyzed them first, and the traces with two peaks were analyzed later. As expected for a thermally activated off-equilibrium process, the magnitude of the single peak rise in force varied from trace to trace and averaged 38 ± 12 pN (mean ± SD) (Fig. 1C). The position of the rise in force was located 3.2 ± 4.8 bp (mean ± SD) away from the PAM-proximal region, which was within the 20-bp RNA-DNA complementarity region (Fig. 1D). This interaction, termed the pre-PAM interaction, was expected as spCas9 cleaves both DNA strands +3 bp upstream of the PAM (2).
Next, we unzipped the DNA molecule with the associated dCas9/sgRNA from the downstream side of the PAM (termed reverse unzipping). The unzipping signature of the trunk DNA with bound dCas9/sgRNA/DNA complex was readily detected and compared with that of the naked DNA (Fig. 1E). A relatively small disruption force of approximately 25 pN was observed exclusively at the position of −16.4 ± 4.7 bp (mean ± SD, n = 53), indicating that the interaction was located approximately 14 bp (without counting the PAM itself) downstream of the PAM (termed the post-PAM interaction) (Fig. 1, F and G). Exonuclease III footprinting assays further corroborated the existence of this interaction (fig. S4). This interaction site was completely unexpected, as it is beyond the apparent footprint of Cas9 on the target DNA (21). Moreover, following the disruption of this post-PAM interaction, the pre-PAM interaction was expected to be intact, as its disruption force is higher than that of the post-PAM interaction. Unexpectedly, it was not detected at all in the examined traces, indicating the immediate collapse of the ternary complex upon the disruption of this post-PAM interaction. These findings suggest that the unexpected post-PAM interaction is essential for the stable binding of Cas9/sgRNA to the target DNA. Experiments with another sgRNA (sgRNA-2) confirmed the presence of these two interactions, reflecting that these detected interactions are conserved in the ternary complex (fig. S5, A to C).
To determine whether these two interactions are also observed for other forms of spCas9, we repeated the experiments by replacing dCas9 with wild-type Cas9 (wtCas9). The two interactions flanking the PAM were also detected in these unzipping experiments in both directions (fig. S5, D to F). However, the unzipping force dropped to zero immediately after the disruption of these interactions due to the breakage of the dsDNA induced by the endonuclease activity of wtCas9. The positions of these two interactions were similar to those obtained with dCas9, yet the forces were slightly higher (fig. S5F). Consistently, the post-PAM interaction was also weaker than the pre-PAM interaction with wtCas9. The detection of the rise in force in both unzipping directions demonstrated the stable binding of wtCas9 to the target DNA after cleavage, which was in agreement with the previous findings (7, 13).
In conclusion, two interactions that flank the PAM exist between Cas9/sgRNA and the target DNA, and they are highly stable regardless of DNA cleavage. The disruption of the post-PAM interaction leads to the immediate collapse of the ternary complex.
Effect of mismatches on Cas9/sgRNA/DNA interactions
Previous studies have demonstrated that DNA binding by Cas9 is far more promiscuous than DNA cleavage and that targets with up to eight mismatches can still be tolerated by this enzyme (22). We, thus, examined how DNA targets with imperfect RNA/DNA complementarity affect the interactions of the ternary complex. As the complementarity near the PAM-proximal region is crucial for the binding of Cas9/sgRNA, we introduced mismatches at the PAM-distal end. We first repeated the unzipping experiments with a DNA template containing six PAM-distal mismatches (denoted as D15-20mm; table S1). All examined DNA unzipping traces (n = 28) were found to associate with the dCas9 protein at the expected position. When we performed the forward unzipping experiments, two types of unzipping signatures were recorded. Forty-three percent of the traces showed a single rise in force at approximately +6.6 ± 3.7 bp with an average of 40 pN that resembled the pre-PAM interaction with the fully matched DNA/sgRNA (Figs. 1B and 2, A and G). In addition to this force peak, the remaining traces also showed an additional small peak prior to the larger peak at +15.3 ± 1.0 bp (mean ± SD), with an average of 24 pN, which was rarely detected with fully matched DNA/sgRNA (Fig. 2, B and G, and fig. S3). This force peak at +15 bp is also within the 20-bp RNA-DNA complementary region and indicates the existence of an intermittent interaction. To confirm these results and examine the relationship between this intermittent interaction and the number of RNA-DNA mismatches, we performed unzipping experiments with a DNA template containing 10 PAM-distal mismatches at the binding site (denoted as D11-20mm; table S1). Only 54% of all examined traces (n = 26) were found to associate with dCas9 at the expected position in the forward unzipping experiment (Fig. 2H). These findings were in agreement with those of previous studies in which the stable binding of Cas9 decreased as the number of mismatches increased (7, 22). For the dCas9-bound DNA templates, the unzipping signatures had characteristics that were essentially identical to those of the D15-20mm template in that there were two types of unzipping signatures with similar populations for each type that had interactions with similar positions and strengths (Fig. 2, D, E, and G). Next, we carried out reverse unzipping experiments with the two partially mismatched DNA/sgRNAs. A single rise in force was detected at the position approximately −14 bp (without counting the PAM) downstream of the PAM, which resembled that found in the unzipping signature of the fully matched DNA/sgRNA (Figs. 1G and 2, C, F, and G). Consistently, interactions within the RNA/DNA complementary region were not detected after the disruption of this post-PAM interaction. We have also introduced mismatches into sgRNAs and obtained similar results with mismatches within the DNA templates (fig. S6). In conclusion, stable pre- and post-PAM interactions are observed for DNA-bound dCas9 with imperfect RNA/DNA complementarity, and an additional intermittent interaction between dCas9 and DNA appears frequently near the PAM-distal region.
Fig. 2. Interactions of dCas9/sgRNA with a PAM-distal mismatched DNA target.
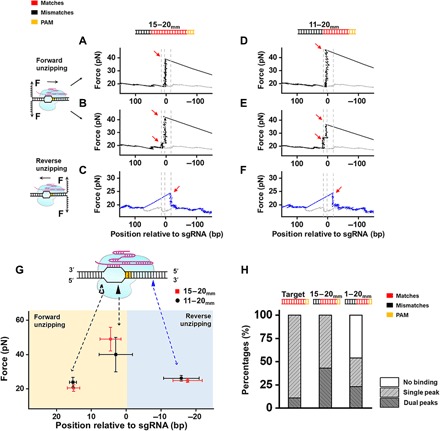
(A to F) Representative traces of forward and reverse DNA unzipping with the D15-20mm (A, B, and C) and the D11-20mm templates (D, E, and F). The forward and reverse unzipping forces versus the number of base pairs are presented in black and blue, respectively. The naked DNA unzipping signatures are also presented for comparison (gray). Red arrows indicate the force peaks. The forward unzipping forces populated two signatures: one peak and dual peaks. (G) Positions and forces of the three interactions between Cas9/sgRNA and mismatched DNA: pre-, post-, and intermittent interaction. The hollow triangle indicates the transient interaction, and the black and blue triangles indicate the pre- and post-PAM interactions, respectively. (H) Fractions of dCas9-bound DNA at the expected position with fully matched (target) and mismatched DNAs (D15-20mm and D11-20mm), as revealed by forward unzipping.
Post-PAM interaction is essential for the stable binding and cleavage of Cas9
The finding that the post-PAM interaction is highly reserved for different forms of Cas9 and that its disruption immediately collapses the ternary complex implied that this interaction might be essential for the stable binding of Cas9 to DNA. To verify this, we first examined whether removing this interaction site from DNA could affect dCas9 binding. To this end, we constructed a series of DNA templates with various DNA lengths downstream of the PAM (denoted as Post-X, where X represents the number of DNA base pairs downstream of the PAM) (Fig. 3A). Using these DNA templates, we revisited the single-molecule forward unzipping assay to measure the binding efficiency of dCas9 at the target site. A series of concentrations of the dCas9 protein was incubated in a chamber for 10 min to determine the dissociation constant (Kd) of dCas9 (Fig. 3B). We found that when the DNA length downstream of the PAM is shorter than 30 bp (X < 30), the Kd of dCas9 at the target site markedly increased as X decreased (Fig. 3B). Notably, the Kd of dCas9 binding of the Post-5 DNA template is 300 times higher than that of the Post-800 DNA template. By conducting electrophoretic mobility shift assays (EMSAs), we further confirmed our findings and excluded the possibility that the observed low affinity of dCas9 on the shortened DNA templates is because the binding is too weak to be detected in our DNA unzipping assays (fig. S7). These data demonstrated that the binding efficiency of dCas9 at the target DNA site is notably reduced when the DNA length downstream of the PAM is shortened. Considering that the post-PAM interaction site is located 14 bp downstream of the PAM, these results corroborate our speculation that the post-PAM interaction is essential for Cas9 binding. Consistently, the cleavage efficiency of wtCas9 also correlated with the DNA length downstream of the PAM (Fig. 3C). Similar observations were also made with another sequence, making it less likely that the effect originates from an unusual sequence (fig. S8A). It is noteworthy that wtCas9 cleavage activity was completely abolished for the Post-0 DNA template.
Fig. 3. Post-PAM interaction is essential for Cas9 binding and cleavage.
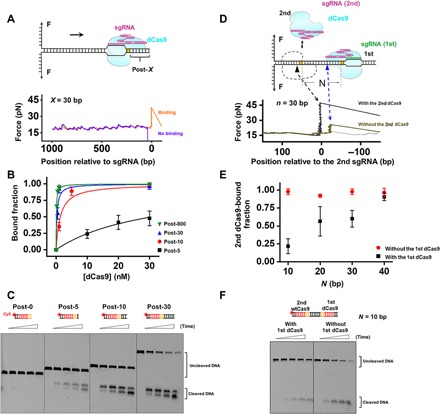
(A) Schematic diagram of the unzipping of DNA templates with various DNA lengths downstream of the PAM (represented by X). Representative traces of forward unzipping of the “Post-30” DNA templates. (B) Fraction of dCas9-bound DNA as a function of dCas9 concentration. DNA templates with various DNA lengths are shown in different colors. (C) A representative gel showing Cas9 cleavage of DNA templates with various DNA lengths downstream of the PAM. Reactions were quenched at five time points (0, 5, 10, 15, and 30 min). (D) Schematic diagram of the Cas9 sequential binding assay. Representative traces of forward unzipping of the PAM-in DNA template with 30-bp dsDNA remaining between the two PAMs show the force versus the number of base pairs unzipped. These traces were obtained when both the 1st and the 2nd dCas9 were present. The naked DNA unzipping signatures are also presented for comparison (gray). (E) Fraction of the second dCas9-bound DNA in the presence and absence of the 1st dCas9. N represents the number of bases between the two dCas9 exclusive PAM regions. (F) A representative gel showing Cas9 cleavage of a PAM-in DNA template with 10 bp of separation between the two PAMs. Reactions were quenched at five time points (0, 1, 2, 3, and 5 min).
In vivo, Cas9 binding sites are often fully or partially occupied by DNA binding proteins, such as histones. The finding that the lack of a post-PAM interaction site in the DNA impairs Cas9 binding motivated us to ask whether occupation of this site by other DNA binding proteins could also lead to similar results. To examine this, we designed a dCas9 sequential binding assay in which the ability of dCas9 to bind to a target site adjacent to a preassociated dCas9 was examined. To do so, we constructed DNA templates containing two adjacent DNA target sites for two sgRNAs in a “PAM-in” orientation with various DNA lengths between the two PAMs (denoted as “N”; table S2). In this sequential binding assay, we introduced the first dCas9 protein with an sgRNA that was associated with the target site into the chamber. After 10 min of incubation, the unbound proteins and sgRNAs were flushed out, and the second dCas9 protein with another sgRNA whose target site was adjacent to the first bound dCas9 was introduced (Fig. 3D). After flushing out the free proteins and sgRNAs, we mechanically unzipped the DNA template from the upstream side of the second dCas9 protein to examine whether it was associated with its target site. The pre-PAM interaction of the second dCas9 protein was expected to be detected if it was bound to the target. Otherwise, the post-PAM interaction of the first bound dCas9 would be detected (Fig. 3D). In control experiments in which the first dCas9 protein was not introduced, the second dCas9 protein was found to efficiently bind to its target site in the experimental conditions used for all DNA templates (Fig. 3E). However, whereas the binding of the second dCas9 protein was barely affected by the association of the first dCas9 protein with the 40-bp-length PAM template, it was significantly decreased when the DNA length between the two PAMs was shortened to less than 30 bp (Fig. 3E). In line with the results for dCas9 binding, we found that the wtCas9 cleavage efficiency was also impaired by the occupation of the DNA by the adjacent dCas9 when the DNA length between the two PAMs was shorter than 30 bp (Fig. 3F and fig. S8, B and C). These data imply that efficient binding and cleavage of the dCas9 protein require at least 15 bp of DNA downstream of the PAM. These findings confirm our speculation that occupation of the post-PAM interaction site could also impair Cas9 activity and strengthen the conclusion that the post-PAM interaction is essential for stable Cas9 binding.
A helicase more readily disrupts the weak post-PAM interaction
Both previous results and our data showed that, after cleavage, spCas9 remains bound to the DNA for a long time without dissociation (fig. S5, D and E) (7, 14, 15). We next aimed to investigate how the detected interactions of Cas9 with DNA govern its dissociation. Cas9-induced DNA DSBs are repaired by nonhomologous end joining (NHEJ) or homology-directed repair (HDR) (12). These repair pathways commonly require helicases or translocases to first displace the bound protein and expose the DSB termini (23). Our single-molecule study demonstrated that the post-PAM interaction is essential for its stable binding but is relatively weak. We thus speculated that it should be easier to displace tightly bound Cas9 from the downstream side of the PAM than from the upstream side. To verify this hypothesis, we chose to use a core fragment of the Bloom syndrome helicase (BLM642–1290, hereafter referred to as BLM), which is a typical homologous recombination–associated helicase, to examine the displacement of the dCas9/sgRNA complex. A previously developed single-molecule optical tweezer assay was used to detect the helicase unwinding activity in real time (20, 24, 25). Briefly, helicase-catalyzed unwinding of a DNA fork junction was monitored via the increase in the single-stranded DNA (ssDNA) length under a constant force that was not sufficient to mechanically unzip the fork junction. We found that the BLM helicase could smoothly unwind naked DNA without obvious pauses in both directions under an assisting force of 12 pN (Fig. 4, A and C). After prebinding of dCas9/sgRNA to the DNA template, we recorded the trajectory of the unwinding of BLM toward the bound dCas9 from both directions. The smooth unwinding of the BLM helicase toward the dCas9 protein from the upstream side of the PAM was found to be interrupted by pauses at the expected dCas9 binding position in all examined traces (n = 34) (Fig. 4A). Eventually, all helicases exited the pause (~51 s) (Fig. 4B) and then proceeded at their initial speeds, indicating the displacement of the dCas9 protein from the DNA template. A control experiment showed that BLM can unwind a DNA-RNA hybrid template without interruption or obvious pauses (fig. S9), and thus, excluded the possibility that the observed pauses with dCas9 were a result of the inability of BLM to unwind the hybrid formed between the sgRNA and the target DNA strand. Therefore, the pauses were attributed to the collision of the BLM helicase with the tightly bound Cas9 protein. We next monitored BLM unwinding toward dCas9 from the downstream side of the PAM and found that all examined traces (n = 42) showed continuous unwinding after relatively shorter pauses (~17 s) (Fig. 4, C and D). These results suggest that a downstream helicase can more readily disrupt DNA-bound Cas9 protein by disrupting the relatively weak post-PAM interaction, which is in agreement with our DNA unzipping results (Fig. 1).
Fig. 4. BLM unwinds through DNA-bound dCas9.
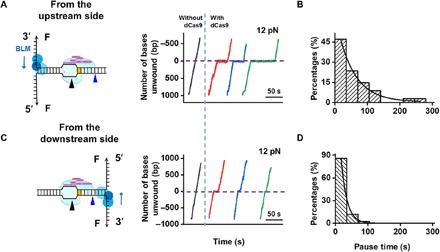
(A and C) The BLM unwinding was initiated from either the upstream (A) or downstream (C) side of the PAM. Representative traces showed the number of unwound base pairs versus time under an assisting force of 12 pN in the presence or absence of prebound dCas9. For clarity, the traces have been shifted along the time axis. The dashed lines indicate the expected dCas9 binding position. (B and D) Histogram showing the pause time of BLM at the expected dCas9 binding position when unwinding started on the upstream (B) or downstream (D) side of the PAM. These time distributions followed a single exponential distribution with average times of 51 and 17 s, respectively.
Our finding that DNA-bound dCas9 is more readily displaced by a downstream helicase is in contrast with a recent report showing that only a downstream RNAP is incapable of displacing Cas9, whereas an upstream RNAP can do so (17). This discrepancy raised the possibility that different DNA-based motors may react differently upon collision with the DNA-bound Cas9 protein. To address this, we chose to use another motor, Phi29 DNA polymerase (DNAP), to examine the outcomes of its collision with a DNA-bound dCas9 protein in both directions. We found that dCas9 can be a strong barrier to a downstream replicating DNAP, whereas an upstream replicating DNAP can easily remove bound dCas9 protein (fig. S10). Phi29 DNAP is more prone to activating its exonuclease activity upon collision with the dCas9 protein from the upstream side. These observations are in contrast to the results obtained with the BLM helicase and are in agreement with the RNAP report (17), suggesting that the mechanism that governs the displacement of the DNA-bound Cas9 protein might be protein dependent.
DISCUSSION
This study presents a high-resolution quantitative map of Cas9/sgRNA/DNA interactions. It not only provides the precise locations of these interactions at near base pair resolution but also quantitatively assays their strength and dynamics (Figs. 1 and 2). The Cas9/sgRNA/DNA interaction map reveals the existence of two major interactions that lie approximately 14 bp downstream of the PAM and at 3 bp within the PAM-proximal region (Fig. 1). The strongest interaction appears at 3 bp, where endonucleolytic cleavage occurs (2). It is thus highly likely that this interaction might be directly involved in regulating the cleavage of dsDNA. Unlike the pre-PAM interaction, the post-PAM interaction was completely unexpected. Although an atomic force microscopy assay revealed a wide Cas9 footprint on the DNA (26, 27), according to the crystal structure of DNA/sgRNA-bound spCas9, this protein only covers a few base pairs of the target DNA downstream of the PAM (21). Thus, the 14-bp post-PAM interaction site can be assumed to be completely outside the apparent footprint of spCas9 on the target DNA. It is quite possible that the downstream DNA may bend and make contact with the protein, leading to an additional interaction outside of its apparent footprint. This particular DNA configuration may serve as a scaffold for Cas9 to efficiently bind and cleave the target DNA. This speculation is supported by our findings that the loss or occupation of this interaction site on the DNA impairs Cas9 binding and cleavage (Fig. 3). Previous studies have shown that Cas9 is unable to access and cleave nucleosomal DNA (28, 29). In addition to that of the target DNA sequences, the occupation of the post-PAM interaction site in nucleosomal DNA by histones may also be an impediment to access by Cas9. In addition, the existence of a post-PAM interaction site may also explain our previous finding that Cas9 nickase/paired sgRNA complexes with a “PAM-out” orientation that is separated by 10 to 30 bp have higher cleavage activity than Cas9 nickase/paired sgRNA complexes with a PAM-in orientation because not enough space exists in the DNA at the post-PAM interaction sites for binding of the latter complex (30). The requirement of the post-PAM interaction of Cas9 for efficient binding and cleavage may guide the selection of proper in vivo on-target DNA sites during future applications.
Information about the Cas9/sgRNA/DNA interaction has important implications for how Cas9 might be effectively displaced from the DNA template after cleavage in order for the resulting DSB to be repaired afterward. DNA-based molecular motors involved in the NHEJ and HDR pathways, such as helicases, are responsible for the displacement of the bound Cas9 protein (12, 31). The mechanical unzipping experiments described here mimic the actions of these molecular motors, which disengage Cas9 and displace it from the template. The weak post-PAM interaction can potentially be modulated to alter the Cas9 dissociation rate. Using the BLM helicase as an example, we provide a proof of concept that Cas9 is more readily displaced from the post-PAM side (Fig. 4). However, an advancing DNAP on the downstream side of the PAM is highly prone to being blocked by the DNA-bound Cas9 (fig. S10). This is in agreement with a recent study that showed that T7 RNAP is incapable of displacing the Cas9 protein from the downstream side of the PAM (17). The difference in the response of Cas9 to DNAP or RNAP as opposed to BLM could result from either of the two nonexclusive alternatives. One possibility is that, by displacing a DNA-bound Cas9 protein, DNAP and RNAP may serve as single-use enzymes if DNAP exonuclease activity or RNAP backtracking is activated upon collision, which would prevent the motors from interacting with the DNA-bound protein. As shown here (fig. S10), these proteins more likely switch to backward movement modes upon collision with the Cas9 protein from the downstream side of the PAM, possibly due to the likely configurational arrangement of the DNA-spCas9 complex induced by the post-PAM interaction. In contrast, the helicase can repeatedly strike the bound protein, facilitating the passage of the helicase from the weak interaction side. Alternatively, both RNAP and DNAP are strand-bias motors that can synthesize RNA or DNA only in the 5′ to 3′ direction. When they encounter the Cas9/sgRNA complex from the PAM-distal end, the existing sgRNA is located on the same strand as the synthesized RNA transcript, which may assist them in removing the bound Cas9 protein. These findings suggest that the effective use of Cas9 should consider both its binding orientation on the DNA and the DNA-based molecular motors that may remove it.
In addition to the two aforementioned stable interactions, a transient interaction at approximately +15 nucleotides (nt) relative to the PAM was also frequently detected with Cas9 targets with imperfect RNA-DNA complementarity (Fig. 2). A previous study that used kinetic Monte Carlo simulations also predicted the existence of a similar interaction at this region, and this interaction was speculated to stabilize the Cas9 conformational change required for further cleavage (26). However, the +15 nt interaction was barely observed in our study when the sgRNA was fully matched with the DNA template for both Cas9 and dCas9 (Fig. 1 and fig. S3). Recently, a series of studies confirmed that a conformational rearrangement of the Cas9 HNH domain occurs during cleavage, before which a conformational checkpoint between DNA binding and cleavage exists (9, 10, 32, 33). This transient interaction possibly serves to mediate sensing of RNA-DNA complementarity and govern HNH domain mobility. We found that this interaction appears less frequently when PAM-distal matches exist. It can be reasoned that the PAM-distal matches would enhance DNA unwinding and that the fully unwound state promotes less frequent detection of RNA-DNA complementarity. Alternatively, this interaction may reflect the driving force required for the Cas9 protein to unwind the dsDNA near the PAM-distal region, as the DNA target is in an incomplete unwinding state in these conditions (34).
Overall, we provide a mechanistic perspective on Cas9 binding, cleavage, and dissociation. This information has important implications for the use of Cas9 as a genome engineering technology and might suggest distinct avenues that could be used to improve Cas9 efficiency.
MATERIALS AND METHODS
Preparation of DNA templates and sgRNAs
The DNA template for the single-molecule unzipping assay consisted of three pieces (two arms and a trunk) and was prepared as previously described (fig. S1) (35). Arm 1 was polymerase chain reaction (PCR) amplified from plasmid pBR322 using a digoxigenin-labeled primer. The resulting DNA fragment was digested with Bst XI (NEB) to create an overhang and was subsequently annealed to a short DNA with a complementary overhang formed by adapter 1 (5′-CGTTACGTCATTCTATACACTGTACAGGTTACACTG-3′) and adapter 2 (5′-/phos/GTAACCTGTACAGTGTATAGAATGACGTAACGCGCAATCATCGATATCTCGTAATCACGTGCAAGGCCTA-3′). Arm 2 was PCR amplified from plasmid pBR322 using a biotin-labeled primer. The resulting DNA fragment was digested with Bst XI (NEB) to create an overhang and was subsequently annealed to adapter 3 (5′-/phos/CGATGCAGTACCGAGCTCATCCAATTCTACATGCCGC-3′) and adapter 4 (5′-/phos/GCCTTGCACGTGATTACGAGATATCGATGATTGCGGCGGCATGTAGAATTGGATGAGCTCGGTACTGCATCGTGAC-3′). Adapter 2 from arm 1 and adapter 4 from arm 2 were partially complementary to each other and were annealed to create a short 35-bp trunk with a 3-bp overhang for the trunk ligation. The 1795-bp trunk containing the target DNA sequence was amplified from the plasmid pEGFP-N1 and digested with Alw NI (NEB). The trunks containing the partially matched DNA sequences were ligation products of two DNA segments that were amplified from the plasmid pEGFP-N1. The upstream DNA segment was digested with Alw NI (NEB) and Bsa I (NEB), while the downstream segment was digested with Bas I (NEB) (fig. S1B). The trunks containing the two DNA target sequences were amplified from the modified plasmid pHY42. The final product was produced by ligating the arms with the trunk at a 1:4 ratio. Hairpin-capped trunk was constructed as previously described (18, 19).
The sgRNAs were prepared as follows. The pUC57-sgRNA expression vectors were linearized by Bsa I (NEB) and transcribed using the T7 High Efficiency Transcription Kit (TransGen Biotech). The sgRNAs were then purified using the EasyPure RNA Purification Kit (TransGen Biotech). The sequences of the sgRNAs, the adapters, and the primers used in this study are listed in table S1.
Expression and purification of proteins
All spCas9s were expressed and purified as described previously (2). A pET-based expression vector, which consisted of a sequence encoding Cas9 (Cas9 residues 1 to 1368 from S. pyogenes) and an N-terminal His6-tag followed by a peptide sequence containing a tobacco etch virus protease cleavage site, was used. The proteins were expressed in Escherichia coli strain BL21 Rosetta 2 (DE3) (TransGen Biotech) cells that were grown in LB at 37°C for a few hours. When the optical density at 600 nm reached 0.6, protein expression was performed at 18°C for 12 to 16 hours following induction with 0.2 mM isopropyl β-d-1-thiogalactopyranoside. The medium was then discarded, and the cells were harvested. The harvested cells were lysed in 20 mM tris-HCl (pH 8.0), 250 mM NaCl, 5 mM imidazole (pH 8.0), and 1 mM phenylmethylsulfonyl fluoride and passed through a homogenizer three times at ~1000 bar. The lysed dilution was then ultracentrifuged at 13,000g for 30 to 60 min, and the clarified supernatant from the cell lysate was separated from the cellular debris and bound in batch to Ni-NTA Agarose (Qiagen). The resin was washed extensively with 20 mM tris-HCl (pH 8.0), 250 mM NaCl, and 10 mM imidazole (pH 8.0), and the bound protein was eluted in a single step with 20 mM tris-HCl (pH 8.0), 250 mM NaCl, and 250 mM imidazole (pH 8.0). The dialysis of Cas9 in dialysis buffer [20 mM Hepes-KOH (pH 7.5), 150 mM KCl, 10% (v/v) glycerol, 1 mM dithiothreitol (DTT), and 1 mM EDTA] was performed overnight at 4°C. Cas9 was further purified with a HiTrap SP HP Sepharose column (GE Healthcare) and was subject to gel filtration chromatography with a Superdex 200 16/60 column (GE Healthcare) in Cas9 storage buffer [20 mM Hepes-KOH (pH 7.5), 500 mM KCl, and 1 mM DTT], and it was then stored at −80°C. The truncation mutant BLM642–1290 (core-BLM) encompassing the region homologous to the RecQ catalytic core was purified as previously described (36). Phi29 DNAP was purchased from NEB (M0269S).
Single-molecule optical tweezer DNA unzipping assay
An M-trap optical tweezer microscope from LUMICKS (Amsterdam, Netherlands) was used to perform the single-molecule optical tweezer assays. The sample chamber preparation was similar to that previously described (35). Briefly, glass coverslips were cleaned and functionalized with partially biotinylated polyethylene glycol (Laysan Bio) as described previously (37), followed by coating with streptavidin (Thermo Fisher Scientific). DNA tethers were then formed by incubation with biotin-tagged DNA. Antidigoxigenin-coated 0.48-mm polystyrene microspheres (Polysciences) were then added to the chamber. Cas9 and sgRNA were preassembled by mixing them at a ratio of 1:2.5 in unzipping buffer [50 mM tris-HCl (pH 7.9), 100 mM NaCl, and 10 mM MgCl2]. This solution was then diluted to obtain the final experimental concentration of 30 nM. The resulting solution was added to the chamber just prior to data acquisition.
The experiments were conducted in a climate-controlled room at a temperature of 23.3°C; however, owing to the local laser trap heating, the temperature increased slightly to 25° ± 1°C. Each experiment was conducted by mechanically unzipping the dsDNA at a slow velocity of 50 nm/s to probe the potential interactions at the fork.
Bulk DNA cleavage experiments
For the bulk DNA cleavage assay, 1 μM wtCas9 was first complexed with sgRNA at a 1:5 ratio at room temperature for 10 min in reaction buffer. The complexed wtCas9 (100 nM) was incubated with annealed Cy5-DNA (1 nM) for the indicated time at room temperature (table S2). The reactions were stopped by adding loading dye containing 96% formamide and 40 mM EDTA and heating them to 95°C for 5 min, followed by slow cooling, and they were analyzed by 12% denaturing polyacrylamide gel electrophoresis and phosphorimaging.
Helicase unwinding and DNAP strand displacement replication assays
The helicase unwinding experiments were conducted as follows (35). First, the dCas9/sgRNA complex was added to the chamber and incubated for 5 min. Second, 25 μl of 100 nM BLM helicase in unwinding buffer [25 mM tris-HCl (pH 7.5), 100 mM NaCl, 1 mM MgCl2, 0.1 g/ml BSA, 2 mM adenosine triphosphate, and 3 mM DTT] was added to the chamber before data acquisition. Last, the DNA tether was stretched until the force reached 12 pN; the force was maintained at a constant level, while the helicase unwound the dsDNA. The DNA length was recorded in real time.
A Phi29 DNAP strand displacement replication assay was conducted similarly. In brief, 25 μl of 30 nM Phi29 DNAP in the replication buffer [50 mM tris-HCl (pH 7.5), 10 mM MgCl2, 10 mM (NH4)2SO4, and 4 mM DTT] was added to the chamber before data acquisition. During the experiment, several hundred base pairs of dsDNA were mechanically unzipped to produce a priming template for the polymerase. The DNA length was maintained until the force dropped below a threshold, indicating that the polymerase was unwinding the DNA fork. Last, a constant force of 15 pN was maintained, while the polymerase unwound and replicated the dsDNA.
Data collection and analysis
Single-molecule data were obtained at 5 kHz and later filtered at 50 Hz. The acquired data signals were converted into force and DNA extension values as previously described (18). The elasticity parameters of both dsDNA and ssDNA were necessary for data conversion and were obtained from the DNA force-extension measurements (fig. S2) (38). In the unzipping experiments, one separated base pair generated two nucleotides of ssDNA. Accordingly, the real-time DNA extension in nanometer was converted into the number of base pairs unwound based on the elastic parameters of ssDNA under our experimental conditions (39). As described previously (18), to improve the positional precision and accuracy, the curves showing the force versus the number of base pairs unzipped were aligned to the theoretical curve by the cross-correlation of a region before and after the ternary complex disruption. To account for minor instrumental drift, trapping bead size variations, and DNA linker variations, the alignment allowed for a small additive shift (<5 bp) and multiplicative linear stretch (<2%) using algorithms similar to those previously described (18).
Supplementary Material
Acknowledgments
We thank J. Chen and H. Ma (ShanghaiTech University) for the helpful discussions, technical assistance, and critical reading of the manuscript. We also thank all the staff of the molecular and cell biology core facility of the School of Life Science and Technology at ShanghaiTech University for their technical support. Funding: This work was supported by the National Key R&D Program of China (2016YFA0500902 and 2017YFA0106700), the Natural Science Foundation of Shanghai (19ZR1434100), the ShanghaiTech University Startup funding, the National Science Fund for Excellent Young Scholars (31622039), the Science Foundation for Distinguished Young Scholars of Jiangsu Province (BK20160045), and the National Natural Science Foundation of China (31571372). Author contributions: B.Su. designed the experiments. Q.Z. and F.W. performed the experiments and analyzed the data. S.Z., L.B., X.H., B.Su., and B.Sh. contributed to the data analysis and interpretation. Q.Z., J.J., and B.Sh. purified all Cas9 proteins. Y.L., M.L., and X.-G.X. purified the BLM helicase. B.Su., Q.Z., and B.Sh. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaaw9807/DC1
Fig. S1. DNA template design for single-molecule unzipping assay.
Fig. S2. Characterization of the DNA elasticity parameters and the accuracy and precision of the unzipping method.
Fig. S3. A representative unzipping trace of on-target dCas9 with two peaks.
Fig. S4. Footprinting experiments with DNA bound by dCas9/sgRNA.
Fig. S5. Mapping of dCas9/sgRNA-2/DNA and wtCas9/sgRNA-1/DNA interactions.
Fig. S6. Forward unzipping with R15-20mm and R11-20mm sgRNAs.
Fig. S7. EMSA confirmed the effect of the post-PAM DNA length on Cas9 binding.
Fig. S8. DNA cleavage of wtCas9 with various DNA templates.
Fig. S9. BLM unwinding through DNA/RNA hybrid chain.
Fig. S10. Phi29 DNAP replicates through DNA-bound dCas9.
Table S1. Sequences of sgRNAs and DNA.
Table S2. Sequences of oligos used for bulk DNA cleavage assays.
REFERENCES AND NOTES
- 1.Marraffini L. A., Sontheimer E. J., CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11, 181–190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mojica F. J. M., Díez-Villaseñor C., García-Martínez J., Almendros C., Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Knott G. J., Doudna J. A., CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinstiver B. P., Pattanayak V., Prew M. S., Tsai S. Q., Nguyen N. T., Zheng Z., Joung J. K., High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slaymaker I. M., Gao L. Y., Zetsche B., Scott D. A., Yan W. X., Zhang F., Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sternberg S. H., Redding S., Jinek M., Greene E. C., Doudna J. A., DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sternberg S. H., LaFrance B., Kaplan M., Doucina J. A., Conformational control of DNA target cleavage by CRISPR–Cas9. Nature 527, 110–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dagdas Y. S., Chen J. S., Sternberg S. H., Doudna J. A., Yildiz A., A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv. 3, eaao0027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J. S., Dagdas Y. S., Kleinstiver B. P., Welch M. M., Sousa A. A., Harrington L. B., Sternberg S. H., Joung J. K., Yildiz A., Doudna J. A., Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim Y., Bak S. Y., Sung K., Jeong E., Lee S. H., Kim J.-S., Bae S., Kim S. K., Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat. Commun. 7, 13350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L., Corn J. E., Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Newton M. D., Taylor B. J., Driessen R. P. C., Roos L., Cvetesic N., Allyjaun S., Lenhard B., Cuomo M. E., Rueda D. S., DNA stretching induces Cas9 off-target activity. Nat. Struct. Mol. Biol. 26, 185–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones D., Unoson C., Leroy P., Curic V., Elf J., Kinetics of dCas9 target search in Escherichia Coli. Biophys. J. 112, 314a (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma H., Tu L.-C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T., CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 214, 529–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S., Kim D., Cho S. W., Kim J., Kim J.-S., Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke R., Heler R., MacDougall M. S., Yeo N. C., Chavez A., Regan M., Hanakahi L., Church G. M., Marraffini L. A., Merrill B. J., Enhanced bacterial immunity and mammalian genome editing via RNA-polymerase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol. Cell 71, 42–55.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall M. A., Shundrovsky A., Bai L., Fulbright R. M., Lis J. T., Wang M. D., High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng C. A., Fazal F. M., Block S. M., Real-time observation of polymerase-promoter contact remodeling during transcription initiation. Nat. Commun. 8, 1178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun B., Pandey M., Inman J. T., Yang Y., Kashlev M., Patel S. S., Wang M. D., T7 replisome directly overcomes DNA damage. Nat. Commun. 6, 10260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang F., Taylor D. W., Chen J. S., Kornfeld J. E., Zhou K., Thompson A. J., Nogales E., Doudna J. A., Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh D., Sternberg S. H., Fei J. Y., Doudna J. A., Ha T., Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat. Commun. 7, 12778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myler L. R., Gallardo I. F., Soniat M. M., Deshpande R. A., Gonzalez X. B., Kim Y., Paull T. T., Finkelstein I. J., Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell 67, 891–898.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun B., Johnson D. S., Patel G., Smith B. Y., Pandey M., Patel S. S., Wang M. D., ATP-induced helicase slippage reveals highly coordinated subunits. Nature 478, 132–135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun B., Singh A., Sultana S., Inman J. T., Patel S. S., Wang M. D., Helicase promotes replication re-initiation from an RNA transcript. Nat. Commun. 9, 2306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Josephs E. A., Kocak D. D., Fitzgibbon C. J., McMenemy J., Gersbach C. A., Marszalek P. E., Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res. 43, 8924–8941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata M., Nishimasu H., Kodera N., Hirano S., Ando T., Uchihashi T., Nureki O., Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 8, 1430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarrington R. M., Verma S., Schwartz S., Trautman J. K., Carroll D., Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. U.S.A. 115, 9351–9358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isaac R. S., Jiang F., Doudna J. A., Lim W. A., Narlikar G. J., Almeida R., Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. eLife 5, e13450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen B., Zhang W. S., Zhang J., Zhou J. K., Wang J. Y., Chen L., Wang L., Hodgkins A., Iyer V., Huang X. X., Skarnes W. C., Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 11, 399–402 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Maruyama T., Dougan S. K., Truttmann M. C., Bilate A. M., Ingram J. R., Ploegh H. L., Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M., Peng S., Sun R., Lin J., Wang N., Chen C., The conformational dynamics of Cas9 governing DNA cleavage are revealed by single-molecule FRET. Cell Rep. 22, 372–382 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Osuka S., Isomura K., Kajimoto S., Komori T., Nishimasu H., Shima T., Nureki O., Uemura S., Real-time observation of flexible domain movements in CRISPR-Cas9. EMBO J. 37, e96941 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh D., Wang Y. B., Mallon J., Yang O., Fei J. Y., Poddar A., Ceylan D., Bailey S., Ha T., Mechanisms of improved specificity of engineered Cas9s revealed by single-molecule FRET analysis. Nat. Struct. Mol. Biol. 25, 347–354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun B., Wang M. D., Single-molecule optical-trapping techniques to study molecular mechanisms of a replisome. Methods Enzymol. 582, 55–84 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Janscak P., Garcia P. L., Hamburger F., Makuta Y., Shiraishi K., Imai Y., Ikeda H., Bickle T. A., Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J. Mol. Biol. 330, 29–42 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Yardimci H., Loveland A. B., van Oijen A. M., Walter J. C., Single-molecule analysis of DNA replication in Xenopus egg extracts. Methods 57, 179–186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bustamante C., Marko J. F., Siggia E. D., Smith S., Entropic elasticity of lambda-phage DNA. Science 265, 1599–1600 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Smith S. B., Cui Y., Bustamante C., Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science 271, 795–799 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaaw9807/DC1
Fig. S1. DNA template design for single-molecule unzipping assay.
Fig. S2. Characterization of the DNA elasticity parameters and the accuracy and precision of the unzipping method.
Fig. S3. A representative unzipping trace of on-target dCas9 with two peaks.
Fig. S4. Footprinting experiments with DNA bound by dCas9/sgRNA.
Fig. S5. Mapping of dCas9/sgRNA-2/DNA and wtCas9/sgRNA-1/DNA interactions.
Fig. S6. Forward unzipping with R15-20mm and R11-20mm sgRNAs.
Fig. S7. EMSA confirmed the effect of the post-PAM DNA length on Cas9 binding.
Fig. S8. DNA cleavage of wtCas9 with various DNA templates.
Fig. S9. BLM unwinding through DNA/RNA hybrid chain.
Fig. S10. Phi29 DNAP replicates through DNA-bound dCas9.
Table S1. Sequences of sgRNAs and DNA.
Table S2. Sequences of oligos used for bulk DNA cleavage assays.