

Abstract
Rhabdomyosarcoma (RMS) is a rare oral malignant soft tissue tumor whose pathological features may influence the clinical behavior, treatment and prognosis of the lesion. We report a case of a 13-year-old female patient, presenting an asymptomatic polypoid swelling in the left buccal mucosa that was approximately 2.5 cm in diameter and 3 months evolution. The presumptive diagnosis was fibrous hyperplasia and an excisional biopsy was carried out. Pathologic analysis revealed proliferation of predominantly ovoid cells, with eosinophilic cytoplasm and pleomorphic nuclei, arranged in subepithelial cambium layer. The mucosal surface presented a papillary–verrucous appearance. Immunohistochemical analysis revealed intense positivity for desmin, myogenin and Ki-67. The diagnosis was of embryonal RMS (botryoid variant). The patient was subjected to complementary chemotherapy and radiotherapy, with no evidence of recurrence or metastatic disease after 12 months follow-up. A discussion on the clinical, histopathological, immunohistochemical and therapeutic aspects of botryoid RMS will be provided.
Keywords: Pediatric rhabdomyosarcoma, Rhabdomyosarcoma, Head and neck neoplasms, Radiotherapy
History
A 13-year-old white female patient sought the service of the stomatology clinic at Tiradentes University (Aracaju/SE) with chief complaint of swelling in the mouth.
Clinical Findings
On intraoral examination, an asymptomatic polypoid swelling measuring about 2.5 cm in diameter was seen in the left buccal mucosa, covered by normal-colored mucosa, evolution time of 3 months and absence of lymphadenopathy. The lesion was mobile and pediculated, with no hardening of surround tissues (Fig. 1). Previous medical history was not contributory. With the clinical differential diagnosis of a reactive or benign mesenchymal lesion, an excisional biopsy was performed under local anesthesia, and the mass was easily removed.
Fig. 1.
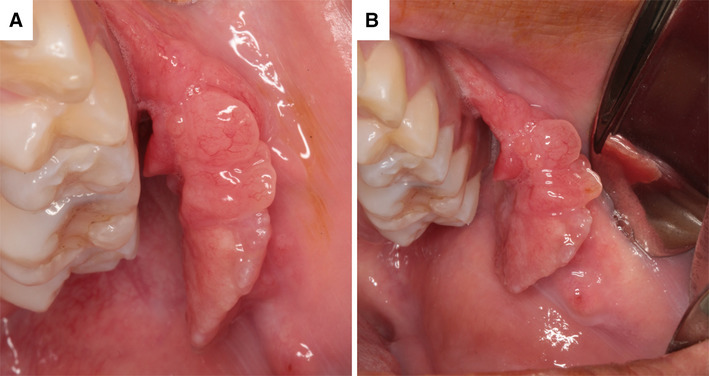
a, b Intraoral aspects showing polypoid pediculated mass involving the left buccal mucosa
Diagnosis
The surgical specimen was submitted to the Service of Oral Pathology of the School of Dentistry at Tiradentes University. Gross examination revealed a well circumscribed distinctly polypoid tumor with firm and rubbery consistency and a gray-white surface. On a cross-section, tumor showed a glistening white surface with patchy areas of hemorrhage (Fig. 2). Microscopic examination revealed a proliferation of predominantly ovoid cells, with eosinophilic cytoplasm and pleomorphic nuclei, arranged in subepithelial cambium layer. Proliferative cells exhibit, in some areas, an organoid arrangement, with densely cellular areas interspersed by hypocellular and myxoid regions. Classical areas of reactional hyperplasia of extranodal lymphoid tissue are also observed. The mucosal surface presented a polypoid appearance (Fig. 3). Based on these findings, the initial histologic diagnosis was embryonal rhabdomyosarcoma (RMS) (botryoid subtype).
Fig. 2.

Macroscopic aspect of the surgical specimen
Fig. 3.
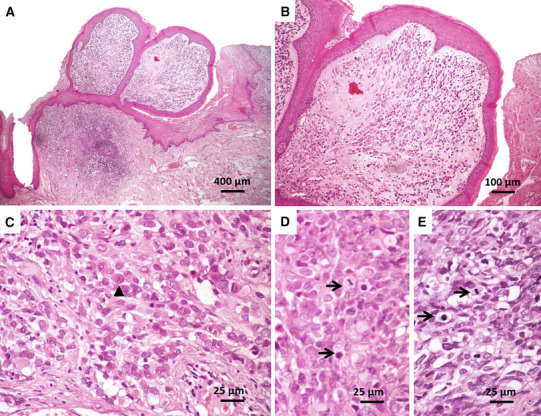
Pathological features of botryoid RMS. a, b Botryoid RMS, organized into polypoid structures lined by squamous epithelium with underlying cellular aggregates (cambium layer). c Rhabdomyoblasts (indicated by arrowheads), d, e mitotic figures (arrows) were seen throughout the tumor parenchymal cells (HE)
Immunohistochemical analysis was subsequently performed for confirmation of the rhabdoid phenotype of tumor cells. The tumor cells were strongly and diffusely positive for desmin (Fig. 4a) and myogenin (Fig. 4b). Positivity for Ki-67 was approximately 80% of parenchymal cells (Fig. 4c). Based on the combined clinical, morphologic and immunohistochemical findings, the final diagnosis was of embryonal RMS (botryoid subtype).
Fig. 4.

Immunohistochemical profile of botryoid RMS. a Tumor cells showing intense and diffusely positivity for desmin and b myogenin. c Tumor cells showing intense positivity for Ki-67 (over 80%) (LSAB, ×400)
Treatment
The patient was referred to an oncology center (A.C. Camargo Cancer Center, São Paulo, Brazil) and imaging examinations, such as magnetic resonance imaging and bone scintillography, was performed and revealed no evidence of regional or distant metastasis. Pre-treatment staging of RMS was determined by the tumor location (head and neck), tumor size (> 5 cm in diameter), nodal involvement (none) and distant metastases (none) [1]. The patient was placed into a clinical group based on the Intergroup Rhabdomyosarcoma Study Group (IRS) [1]/Children’s Oncology Group (COG) guidelines [2]. Clinical groups are determined by the presence of residual disease following surgery and dictate if further treatment is indicated. The patient was categorized as Group III (localized tumor, with gross residual disease after grossly incomplete removal, or biopsy only). Based on the IRS/COG guidelines [1], the patient was classified as low risk, subset 2, and subjected to surgical enlargement of the edges of the lesion and complementary radiotherapy (3600 cGy for 4 weeks) and triple-agent chemotherapy consisting of vincristine, dactinomycin, and cyclophosphamide. After 12 months of follow-up, the patient remained with no evidence of recurrence or metastatic disease.
Discussion
RMSs constitute the most common malignant musculoskeletal tumors in children and adolescents, exhibit a male predilection, and occur particularly in children younger than 5 years of age, but a bimodal age distribution was observed, with a larger peak between 0 and 5 years and a smaller in adolescence [3, 4].
Approximately 40% of cases of RMS are diagnosed in the head and neck region [5], with the orbit, face, and nasal cavity regarded as the most commonly affected locations [6]. RMS is particularly rare in the oral and perioral regions, and when it occurs intraorally, the soft palate and tongue are anatomical sites more frequently involved [7].
It is believed that RMS originate from the malignant transformation of undifferentiated primitive mesenchymal cells that exhibit a compromise regarding rhabdomyoblastic differentiation, justifying the development of this neoplasm in anatomical sites devoid of skeletal muscle [7]. There are no etiological factors clearly related to the development of RMS; however, the recent discovery of genetic abnormalities may help to better understand the etiopathogenesis of this lesion, such as the frequent chromosomal translocation t(2;13)(q35;q14), involving PAX3 and FKHR genes, consistently identified in alveolar RMSs [8].
Clinical signs and symptoms mainly depend on the affected anatomical sites and vary considerably [5]. Although some cases are asymptomatic, such as ours, the typical clinical presentation in the head and neck region is a painless swelling that may display rapid growth and ulceration, usually causes facial asymmetry, and thus becomes alarming. Other complaints may also be observed, including proptosis, nasal obstruction and nasal discharge [9].
Clinically, despite the classic polypoid appearance, the botryoid subtype in the buccal mucosa may be difficult to distinguish from other soft tissue tumors frequently observed in this region. Reactive and benign mesenchymal lesions with a polypoid aspect should be considered in the differential diagnosis. Furthermore, due to relative rarity of RMS in oral cavity and nonspecific clinical presentation, misdiagnosis may occur, leading to an incorrect therapeutic approach and significant delay in achieving the correct diagnosis [5, 10].
In our case, the submucosal presentation, slow growth pattern, with no hardening of surround tissues and the clearly defined limits of the tumor suggested a benign process. Therefore, our diagnostic hypothesis included mainly conditions commonly observed in young patients such as reactive proliferative processes and mesenchymal tumors, and the patient was misdiagnosed with fibrous hyperplasia.
The fibrous hyperplasia is the most common benign soft tissue tumor seen in the oral cavity, representing a reactive response to local irritation or masticatory trauma. The most commonly affected site is the buccal mucosa along the line of occlusion; however, it can occur anywhere in the oral cavity, such as the labial mucosa, tongue, and gingiva. Clinically, it manifests as asymptomatic, moderately firm, smooth-surfaced, pink, sessile or pedunculated nodules, as in our case. They rarely occur before the fourth decade of life and do not show sex predilection [11]. In the current case, the patient reported in this study was in her second decade of life, a finding that contrasts with the affected age group described in the literature. Conservative surgical excision is the treatment of choice and recurrence is rare [11]. Because it is the most common lesion of the buccal mucosa, it would be a dentists’ main choice as a provisional diagnosis.
Furthermore, the rare occurrence of RMS in the oral cavity, particularly in the buccal mucosa, and the similarity with other reactive or benign mesenchymal lesions make the clinical diagnosis challenging. In fact, several cases reported in the literature emphasize the similarity of RMS with periodontal lesions of inflammatory origin [10], benign reactive proliferative processes [12], and infectious lesions [5], which makes the pathological analysis of such oral lesions imperative in order to provide the proper management of the tumors.
Four histologic variants of RMS have been described. The embryonal subtype (EMS) corresponds to > 60% of the cases [13] and is particularly common in the orbital region of children. When it involves bodily cavities, it may give rise to the so-called botryoid variant [14]; the spindle cell subtype has previously been considered a variant of the embryonal RMS, but it is now recognized as a separate subtype [15]. The alveolar subtype represents approximately 30% of the cases, tends to occur on the soft tissues of the extremities and affects patients with a slightly higher mean age [5]; the pleomorphic subtype is more rare, and usually affects patients over 45 years and comprises about 5% of all cases diagnosed [5, 16].
The histopathological aspects of this study led to the classification of the tumor as an embryonal variant of RMS. In fact, a number of studies have pointed to this histological subtype as the most common form of RMS in the head and neck region in children and adolescents [5, 9]. Histologically, embryonal RMS is characterized by varying degrees of rhabdomyoblastic differentiation of tumor cells [17]. On the one hand, the lesion is composed of poorly differentiated small round or oval cells, resembling normal embryonal muscle cells, containing a sparse and finely granular eosinophilic cytoplasm with few cells exhibiting transverse striations. On the other hand, as seen in the current case, a well-differentiated elongated or polygonal rhabdomyoblastic cells, occasionally bi- or multinucleated, containing a more vesiculated nucleus and presenting varying degrees of cellular atypia, as well as mitosis figures, may be observed. Tumor stroma is usually scarce and exhibits collagen fibrils loosely or densely arranged, creating areas with a myxoid appearance [5, 17]. The botryoid subtype of the embryonal variant represents a descriptive term for cases affecting bodily cavities and macroscopically resembling grape clusters. Botryoid RMS which is often submucosal, polypoid, and requires the presence of a condensed layer of neoplastic cells beneath epithelium (cambium layer) [14], as demonstrated in the current case.
Immunohistochemistry is particularly useful in establishing a correct diagnosis. Positive immunohistochemical reactions for desmin, myogenin, and Myo-D1 are of great value to confirm the diagnosis, especially in undifferentiated cases, and to exclude other neoplasms with parenchymal cells demonstrating rhabdomyoblast-like features [3, 5, 14]. The intense and diffuse positivity for desmin and myogenin in the current case supports our diagnosis.
Prognosis of the lesion depends on age of the patient, location, clinical staging, and histologic and genetic characteristics of the tumor. A combination of these characteristics serves to stratify RMSs into low-risk, intermediate-risk, and high-risk groups. Treatment regimens include surgical resection, chemotherapy, and radiotherapy [5, 13, 18] and are based on guidelines from the Intergroup Rhabdomyosarcoma Study (IRS) grouping [1, 19]. In cases where the anatomical location allows total resection of the tumor, surgery is indicated, followed by multiagent chemotherapy and radiotherapy. Where free surgical margins are not possible to be obtained, chemotherapy and postoperative radiotherapy are employed [5]. In our case, surgery is indicated followed by radio- and chemotherapy. Current chemotherapy regimens consist of a varied combination of vincristine, actinomycin-D, cyclophosphamide, and doxorubicin. The most common cause of death is tumor progression and involvement of adjacent structures. In relation to distant metastases, the most commonly involved sites are the lung, lymph node, and bone marrow [7].
The primary location of the disease can significantly influence the outcome of the patients. According to the site of diagnosis, head and neck RMS can be divided into three subtypes: parameningeal, non-parameningeal, and orbital [3]. Parameningeal areas (nasal cavity, paranasal sinuses, mastoid area, and infratemporal fossa) tend to present a worse prognosis than non-parameningeal cases (oral cavity, oropharynx, face, cheek, parotid region, and soft tissues of the neck), which may be a consequence of the impossibility of obtaining a complete resection of the lesion and due to their proximity of the intracranial area [5, 20].
Overall, embryonal RMS presents a better prognosis than all other subtypes, having a 66% 5-year survival rate while botryoid variant has a nearly 90% 5-year survival [17], that makes the precise distinction between these variants a particularly important issue. Furthermore, the size of the lesion in the moment of diagnosis also represents an important prognostic factor, once lesions larger than 5 cm present a worse prognosis; similarly, adult patients have lower survival rates than infants [5]. Considering the histological subtype (botryoid type), tumor localization (head and neck), age (13 years) and tumor size (2.5 cm in diameter), the prognosis of this case can be considered as favorable. According to the IRS/COG criteria, she is expected to have a 5-year survival of greater than 87% with an approximately 8% risk of local/regional recurrence [19]. After 12 months of follow-up, the patient remained with no evidence of recurrence or metastatic disease.
In conclusion, botryoid type RMS is a soft tissue sarcoma that usually affects the head and neck region. Recent improvements in therapeutic approaches have significantly increased survival rates. However, due to rare occurrence in the oral cavity and similarities to benign tumors and inflammatory lesions frequently observed in this region, misdiagnosis may occur, causing delay in the diagnosis and leading to an incorrect therapeutic approach. Furthermore, although very rare, the botryoid RMS should be considered in the differential diagnosis of reactive and benign mesenchymal lesions with a polypoid aspect in children and adolescents.
References
- 1.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) soft-tissue Sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walterhouse D, Pappo AS, Meza JL, et al. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the children’s oncology group. J Clin Oncol. 2014;32:3547–3552. doi: 10.1200/JCO.2014.55.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Andrade CR, dos Santos Trento G, Jeremias F, et al. Rabdomyosarcoma of the Mandible. J Craniofac Surg. 2017 doi: 10.1097/SCS.0000000000004154. [DOI] [PubMed] [Google Scholar]
- 4.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer. 2009;115(18):4218–4226. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pontes F, de Oliveira J, de Souza L, et al. Clinicopathological analysis of head and neck rhabdomyosarcoma: a series of 10 cases and literature review head and neck rhabdomyosarcoma. Med Oral Patol Oral y Cir Bucal. 2018 doi: 10.4317/medoral.22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters SM, Kunkle T, Perrino MA, Philipone EM, Yoon AJ. Mandibular embryonal rhabdomyosarcoma with cartilaginous metaplasia: report of a case and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;124(6):e288–e293. doi: 10.1016/j.oooo.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Datta S, Ray JG, Deb T, Patsa S. Embryonal rhabdomyosarcoma: a rare oral tumor. J Oral Maxillofac Pathol. 2016;20(3):527–531. doi: 10.4103/0973-029X.190959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biegel JA, Nycum LM, Valentine V, Barr FG, Shapiro DN. Detection of the t(2;13)(q35;q14) and PAX3-FKHR fusion in alveolar rhabdomyosarcoma by fluorescence in situ hybridization. Genes Chromosom Cancer. 1995;12(3):186–192. doi: 10.1002/gcc.2870120305. [DOI] [PubMed] [Google Scholar]
- 9.Moretti G, Guimarães R, Oliveira KMD, Sanjar F, Voegels RL. Rhabdomyosarcoma of the head and neck: 24 cases and literature review. Braz J Otorhinolaryngol. 2010;76:533–537. doi: 10.1590/S1808-86942010000400020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Motallebnejad M, Aminishakib P, Derakhshan S, Karimi A. Rhabdomyosarcoma of the maxillary gingiva. Dent Res J. 2018;15(1):80–83. doi: 10.4103/1735-3327.223619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutra KL, Longo L, Grando LJ, Rivero ERC. Incidence of reactive hyperplastic lesions in the oral cavity: a 10 year retrospective study in Santa Catarina, Brazil. Braz J Otorhinolaryngol. 2018 doi: 10.1016/j.bjorl.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chi AC, Barnes JD, Budnick S, Agresta SV, Neville B. Rhabdomyosarcoma of the maxillary gingiva. J Periodontol. 2007;78(9):1839–1845. doi: 10.1902/jop.2007.060454. [DOI] [PubMed] [Google Scholar]
- 13.Radzikowska J, Kukwa W, Kukwa A, et al. Management of pediatric head and neck rhabdomyosarcoma: a case-series of 36 patients. Oncol Lett. 2016;12(5):3555–3562. doi: 10.3892/ol.2016.5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly BK, Kim A, Peña MT, Dong TA, Rossi C, Murnick JG, et al. Rhabdomyosarcoma of the head and neck in children: review and update. Int J Pediatr Otorhinolaryngol. 2015;79:1477–1483. doi: 10.1016/j.ijporl.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 15.Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer. 2014;120(12):1763–1774. doi: 10.1002/cncr.28657. [DOI] [PubMed] [Google Scholar]
- 16.Zhang WL, Zhang Y, Huang DS, Guo F, Han T, Hong L, et al. Clinical character of pediatric head and neck rhabdomyosarcomas: a 7-year retrospective study. Asian Pac J Cancer Prev. 2013;14:4089–4093. doi: 10.7314/APJCP.2013.14.7.4089. [DOI] [PubMed] [Google Scholar]
- 17.McInturff M, Adamson A, Donaldson C, Nelson BL. Embryonal rhabdomyosarcoma of the oral cavity. Head Neck Pathol. 2017;11(3):385–388. doi: 10.1007/s12105-016-0761-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casey DL, Wolden SL. Rhabdomyosarcoma of the head and neck: a multimodal approach. J Neurol Surg B. 2018;79(1):58–64. doi: 10.1055/s-0037-1617450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkins DS, Gupta AA, Rudzinski E. What’s new in the biology and treatment of pediatric rhabdomyosarcoma? Curr Opin Pediatr. 2014;26(1):50–56. doi: 10.1097/MOP.0000000000000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen E, Ricciotti R, Futran N, Oda D. Head and neck rhabdomyosarcoma: clinical and pathologic characterization of seven cases. Head Neck Pathol. 2017;11(3):321–326. doi: 10.1007/s12105-016-0771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]