

Abstract
Both arylsulfonyl and alkylsulfonyl azides can be effectively activated by the cobalt(II) complexes of D2-symmetric chiral amidoporphyrins for enantioselective radical 1,5-C–H amination to stereoselectively construct 5-membered cyclic sulfonamides. In addition to C–H bonds with varied electronic properties, the Co(II)-based metalloradical system features chemoselective amination of allylic C–H bonds and is compatible with heteroaryl groups, producing functionalized 5-membered chiral cyclic sulfonamides in high yields with high enantioselectivities. The unique profile of reactivity and selectivity of the Co(II)-catalyzed C–H amination is attributed to its underlying stepwise radical mechanism, which is supported by several lines of experimental evidence.
Graphical Abstract
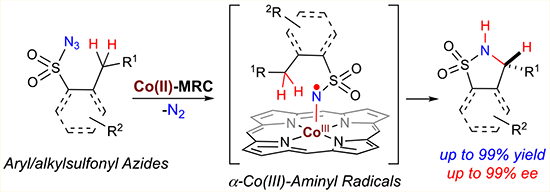
INTRODUCTION
Radical reactions have been increasingly exploited as new synthetic tools, which are complementary to ionic reactions, for molecular construction of organic compounds.1 With the aim of addressing the outstanding issues in controlling reactivity and stereoselectivity of radical species,2 metalloradical catalysis (MRC) has been recently introduced as a conceptually different approach to explore the catalytic application of open-shell metalloradical complexes for both initiation and regulation of homolytic radical reactions.3–5 To this end, cobalt(II) complexes of porphyrins ([Co(Por)]), as stable 15e metalloradicals, have demonstrated the capability of activating organic azides and diazo compounds to generate the fundamentally new α-Co(III)-aminyl radicals and α-Co(III)-alkyl radicals, respectively, upon the release of N2.3,6,7 While the N- and C-centered organic radicals are stabilized by the Co(III) complexes, they retain the ability to undergo common reactions of free radicals, such as radical addition and H-atom abstraction. More importantly, the reactivity and selectivity of these and subsequent radical reactions can be effectively controlled by the environment of the porphyrin ligands, leading to new catalytic radical processes for stereoselective organic transformations.8–12 In particular, Co(II)-based metalloradical catalysis (Co(II)-MRC) has been successfully applied to intramolecular radical C–H amination with different types of organic azides as radical precursors.8 Previously, the Co(II) complex of tetraphenylporphyrin, [Co(TPP)], was shown to catalyze intramolecular C–H amination of arylsulfonyl azides to generate 5-membered cyclic sulfonamides but required elevated reaction temperature.8a Subsequent employment of amidoporphyrins as supporting ligands has enhanced the catalytic activity of the Co(II)-based metalloradical system for intramolecular C–H amination of sulfamoyl azides, even at lower temperature.8b,e This ligand-accelerated catalysis is attributed to the potential hydrogen-bonding interactions between the S=O acceptor of the sulfonyl unit and the N–H donor of the amide moiety of the porphyrin ligand in the α-Co(III)-aminyl radical intermediates.8b,e Assuming the similar ligand acceleration effect, we hoped to enhance the Co(II)-catalyzed system for 1,5-C–H amination of sulfonyl azides by employing amidoporphyrins as supporting ligands (Scheme 1). Besides arylsulfonyl azides, such enhancement might even enable the activation of the more challenging alkylsulfonyl azides for productive C–H amination. Furthermore, considering the availability of D2-symmetric chiral amidoporphyrins with tunable electronic, steric and chiral environments,8e,9b,10a,11a–c the Co(II)-catalyzed C–H amination could be potentially rendered as an enantioselective radical process (Scheme 1). While corresponding α-Co(III)-aminyl radical intermediates I generated from metalloradical activation of sulfonyl azides were anticipated to undergo 1,5-H-atom abstraction, it was unclear if the two prochiral faces of the C-centered radical in the resulting ε-Co(III)-alkyl radical intermediates II, could be effectively discriminated for controlling enantioselectivity of the C–N bond formation during the succeeding 5-exo-tet radical cyclization (Scheme 1). If appropriate chiral amidoporphyrin ligands could be identified to achieve the enantiocontrol, then such asymmetric catalytic process for radical C–H amination would be desirable as it allows for stereoselective construction of 5-membered cyclic sulfonamides (also known as sultams), which are both medicinally important and synthetically useful (Figure 1),13 from readily available sulfonyl azides.
Scheme 1.

Intramolecular Radical C(sp3)–H Amination of Sulfonyl Azides via Co(II)-Based Metalloradical Catalysis
Figure 1.

Selective examples of bioactive and synthetically useful molecules containing cyclic sulfonamide motifs.
Catalytic intramolecular C(sp3)–H amination, especially its enantioselective variant, represents an attractive strategy for the construction of N-heterocyclic structures.14 Despite considerable advancements in recent years,15–17 so far, there are only two previous reports on stereoselective formation of chiral cyclic sulfonamides via asymmetric intramolecular C–H amination.18 While the first catalytic system based on Rh(II)2 complexes of chiral tetracarboxylates achieved only moderate asymmetric induction,18a the more recent Ir(III)-based system with the use of chiral salens could catalyze intramolecular amination of arylsulfonyl azides to form benzofused cyclic sulfonamides with high enantioselectivity.18b However, the Ir(III)-catalyzed amination was mainly restricted to arylsulfonyl azides with only one example of cyclic alkylsulfonyl azide.18b Evidently, there is still a need to develop new catalytic systems, especially using base metal catalysts, for asymmetric intramolecular C–H amination that is generally applicable for stereoselective synthesis of valuable chiral cyclic sulfonamides with diverse structural features. Herein, we wish to report the development of Co(II) complexes of D2-symmetric chiral amidoporphyrins ([Co(D2-Por*)]) as effective metalloradical catalysts for enantioselective radical 1,5-C–H amination of sulfonyl azides. In addition to arylsulfonyl azides, the Co(II)-catalyzed asymmetric system is also applicable to the more challenging alkylsulfonyl azides with different types of C–H bonds, generating chiral 5-membered cyclic sulfonamides in high yields with high enantioselectivities. As additional practical attributes associated with the use of organic azides, the Co(II)-based amination system can be operated under neutral and nonoxidative conditions without the need of any additives, generating the environmentally benign N2 gas as the only byproduct. Furthermore, we present several lines of experimental evidence that shed light on the unique radical mechanism of this metalloradical system for C–H amination.
RESULTS AND DISCUSSION
Asymmetric 1,5-C–H Amination of Arylsulfonyl Azides.
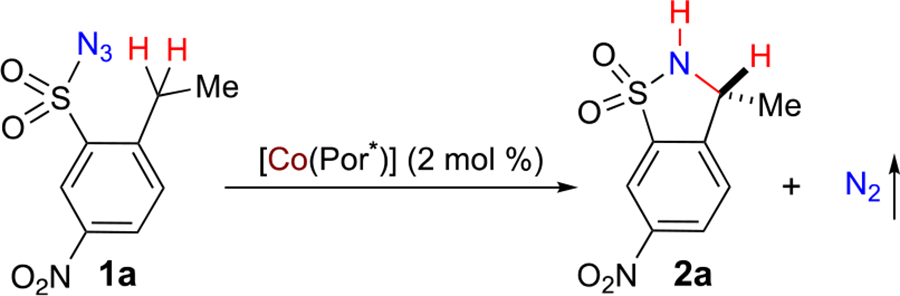
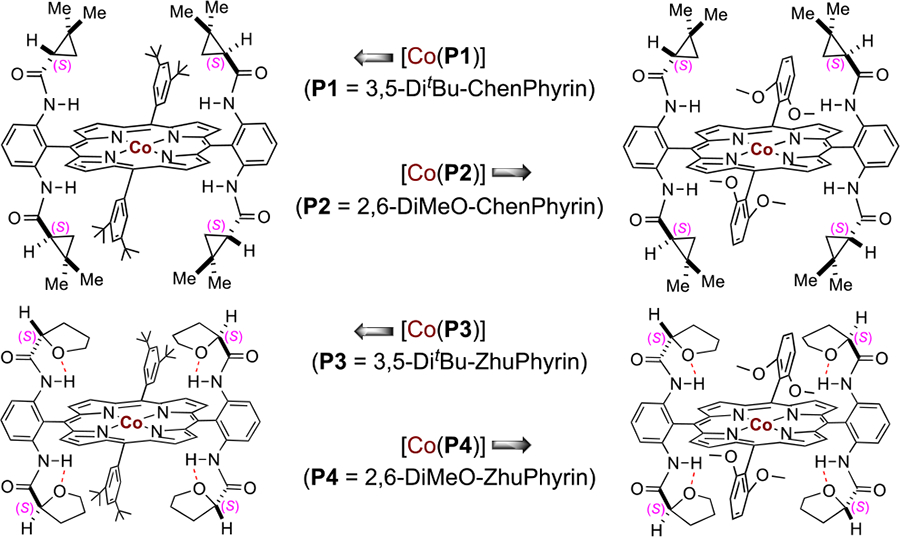
As the initial efforts for this project, we focused on identification of a suitable D2-symmetric chiral amidoporphyrin ligand to support the Co(II)-based catalytic system for intramolecular C–H amination with potential asymmetric induction. 2-Ethyl-5-nitrobenzenesulfonyl azide (1a), which was shown to be a challenging substrate for the existing Ir(III)-based system because of the presence of the electron-withdrawing nitro group,18b was chosen as a test substrate for the Co(II)-based metalloradical system (Table 1). On the basis of the previously established conditions,8a it was found that first-generation chiral metalloradical catalyst [Co(P1)] (P1 = 3,5-DitBu-ChenPhyrin)11a could effectively catalyze the intramolecular amination reaction of azide 1a, generating 5-membered benzofused cyclic sulfonamide 2a as the exclusive product in nearly quantitative yield with low but significant asymmetric induction (Table 1, entry 1). Switching to catalyst [Co(P2)] (P2 = 2,6-DiMeO-ChenPhyrin), bearing the same chiral amide units but with more sterically hindered achiral meso-aryl groups, led to only slight improvement in the enantioselectivity while maintaining the quantitative yield (Table 1, entry 2). On the hypothesis that the low asymmetric induction may be attributed to the relative flexibility of the chiral arms in ChenPhyrin catalysts, we decided to test Co(II) complexes of D2-symmetric chiral amidoporphyrins containing chiral amide units with increased conformational rigidity. Indeed, with the use of [Co(P3)] (P3 = 3,5-DitBu-ZhuPhyrin) as the catalyst, which was previously shown to possess a more rigid conformation as a result of the unique intramolecular O⋯H–N hydrogen-bonding interactions in the (S)-(−)-2-tetrahydrofurancarboxamide units,11b asymmetric induction was significantly improved without affecting the excellent yield (Table 1, entry 3). Encouraged by the positive outcome, catalyst [Co(P4)] (P4 = 2,6-DiMeO-ZhuPhyrin),11b which incorporates more sterically bulky achiral meso-aryl groups into the structure to further enhance the rigidity of the chiral environment, was employed for the amination reaction. Gratifyingly, desired 1,5-C–H amination product 2a was produced with 84% ee in still quantitative yield (Table 1, entry4). The enantioselectivity with [Co(P4)] could be further improved by lowering the reaction temperature (Table 1, entries 5 and 6). However, a much lower yield was observed along with the higher enantioselectivity for the reaction at 40 °C (entry 6). Among the various solvents screened (see Table S1), chloroform was identified as the optimal medium for enantioselectivity (Table 1, entry 7). In chloroform, when the reaction was performed at 50 °C with 4 mol % of [Co(P4)], desired benzofused cyclic sulfonamide 2a was produced in 96% yield with 92% ee (Table 1, entry 8).
Table 1.
Ligand Effect on Co(II)-Based Metalloradical System for Asymmetric 1,5-C–H Amination of Arylsulfonyl Azidea
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | temp (°C) | solvent | yield (%) | ee (%) |
| 1 | [Co(P1)] | 80 | chlorobenzene | 99 | 28 |
| 2 | [Co(P2)] | 80 | chlorobenzene | 99 | 32 |
| 3 | [Co(P3)] | 80 | chlorobenzene | 99 | 49 |
| 4 | [Co(P4)] | 80 | chlorobenzene | 99 | 84 |
| 5 | [Co(P4)] | 60 | chlorobenzene | 99 | 86 |
| 6 | [Co(P4)] | 40 | chlorobenzene | 63 | 88 |
| 7 | [Co(P4)] | 60 | chloroform | 70 | 89 |
| 8b | [Co(P4)] | 50 | chloroform | 96 | 92 |
Reactions were carried out for 18 h on 0.10 mmol scale under N2; [1a] = 0.25 M. Isolated yields Enantiomeric excess determined by chiral HPLC.
4 mol % catalyst for 48 h.
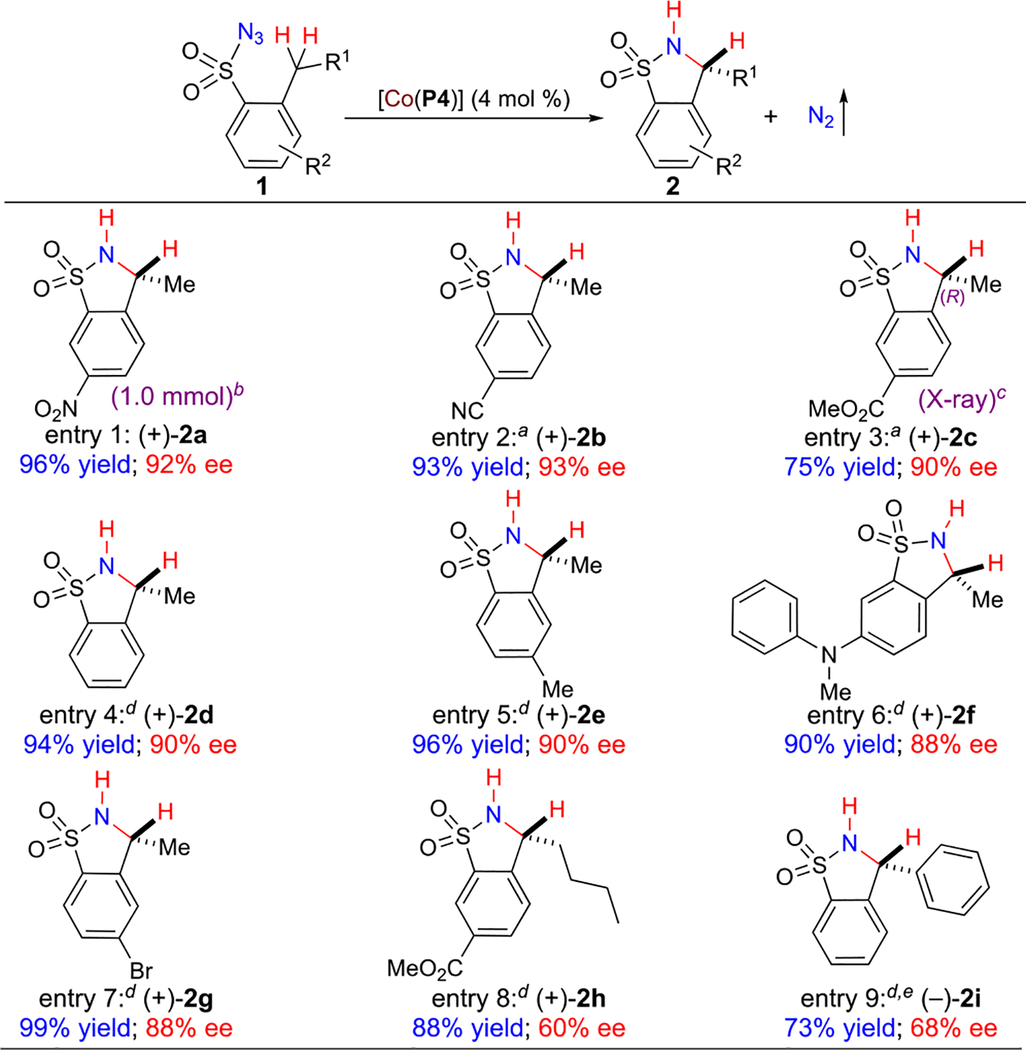
Metalloradical catalyst [Co(P4)] was shown to be generally effective for asymmetric intramolecular C–H amination of various arylsulfonyl azides under the optimized conditions (Table 2). First, it is worth noting that the same high yield and enantioselectivity for formation of benzofused cyclic sulfonamide 2a could also be obtained when the reaction with 1a was scaled up 10 times to 1.0 mmol (Table 2, entry 1). In addition to the nitro group, the Co(II)-based metalloradical amination could also be effectively applied to 2-ethylarenesulfonyl azides bearing other electron-withdrawing substituents such as cyano (1b) and ester (1c) groups, allowing for high-yielding production of functionalized benzofused cyclic sulfonamides 2b and 2c in a highly asymmetric manner (Table 2, entries 2 and 3). The absolute configuration of the newly generated chiral center in 2c was established as (R) by X-ray crystal structural analysis. At a slightly elevated temperature (80 °C), 2-ethylbenzenesulfonyl azide (1d) and its derivatives containing different aryl substituents such as electron-donating alkyl (1e) and amino (1f) groups as well as halogen (1g) atoms were also suitable substrates for the amination system, giving corresponding 5-membered benzofused cyclic sulfonamides 2d–2g in high yields with high enantioselectivities (Table 2, entries 4–7). The complete regioselectivity at the benzylic C–H position by [Co(P4)]-catalyzed amination was further highlighted with the high-yielding formation of 5-membered benzofused cyclic sulfonamide 2h from 2-pentyl-5-(methoxycarbonyl)benzenesulfonyl azide (1h), albeit with lower enantioselectivity (Table 2, entry 8). The 6-membered benzofused cyclic sulfonamide from potential amination of the homobenzylic C–H bonds in 1h was not observed. As expected, C–H bonds at bis-benzylic positions could also be aminated by [Co(P4)] as exemplified with the reaction of 2-benzyl benzenesulfonyl azide (1i), resulting in formation of desired 2i, but in a relatively lower yield with moderate enantioselectivity (Table 2, entry 9).
Table 2.
[Co(P4)]-Catalyzed Enantioselective 1,5-C–H Amination of Arylsulfonyl Azidesa
|
Reactions were carried out in chloroform at 50 °C for 48 h on 0.10 mmol scale under N2; [1] = 0.25 M. Isolated yields enantiomeric excess determined by chiral HPLC.
At 1.0 mmol scale.
Absolute configuration determined by X-ray analysis.
At 80 °C.
In chlorobenzene.
Asymmetric 1,5-C–H Amination of Alkylsulfonyl Azides.
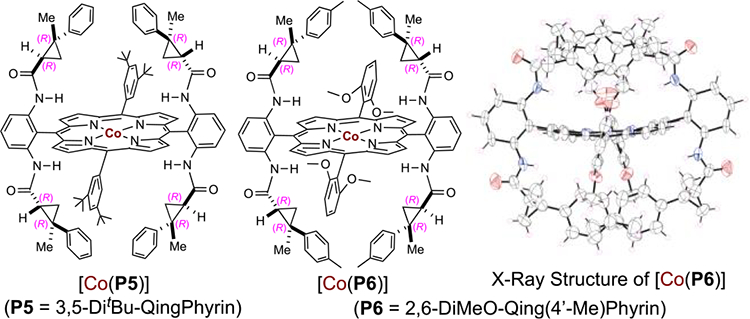
Subsequent experiments indicated that asymmetric intramolecular C–H amination of alkylsulfonyl azides behaves differently from arylsulfonyl azides, presumably due to the flexible nature of the linear alkyl chain. As illustrated in Table 3 with substrate 3-phenylpropylsulfonyl azide (3a), catalyst [Co(P4)] was much less effective in catalyzing the reaction of this alkylsulfonyl azide, affording 5-membered cyclic sulfonamide 4a in only 10% yield although with 90% ee (Table 3, entry 1). Catalyst [Co(P1)] was found to be much more effective for this reaction, generating 4a in 97% yield but with only 48% ee (Table 3, entry 2). When second-generation metalloradical catalyst [Co(P5)] (P5 = 3,5-DitBu-QingPhyrin)11c was used, the enantioselectivity of this heterocyclization reaction was increased to 69% ee while maintaining an excellent yield of 99%. These results prompted us to develop new metalloradical catalysts with combined features of these existing catalysts. This effort led to the design and synthesis of D2-symmetric chiral amidoporphyrin 2,6-DiMeO-Qing(4′-Me)Phyrin (P6). Its cobalt(II) complex [Co(P6)], whose structure was established by X-ray crystallographic analysis, was found to be a superior metalloradical catalyst for asymmetric C–H amination of alkylsulfonyl azide 3a. The [Co(P6)]-catalyzed reaction displayed both high efficiency and excellent stereoselectivity, affording cyclic sulfonamide 4a in 92% yield with 94% ee (Table 3, entry 4). With [Co(P6)] as the optimized catalyst, further solvent evaluation was performed for intramolecular C–H amination of the alkylsulfonyl azide (see Table S2). Among the various solvents evaluated, benzene was identified as the optimal medium as it gave the highest enantioselectivity (Table 3, entries 4–6).
Table 3.
Ligand Effect on Co(II)-Catalyzed Asymmetric 1,5-C–H Amination of Alkylsulfonyl Azides 3aa
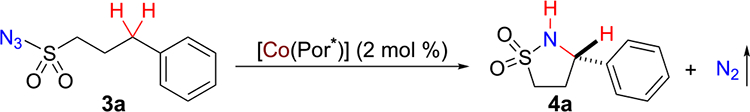 | ||||
|---|---|---|---|---|
| entry | catalyst | solvent | yield (%) | ee (%) |
| 1 | [Co(P4)] | benzene | 10 | 90 |
| 2 | [Co(P1)] | benzene | 97 | 48 |
| 3 | [Co(P5)] | benzene | 99 | 69 |
| 4 | [Co(P6)] | benzene | 92 | 94 |
| 5 | [Co(P6)] | chlorobenzene | 95 | 91 |
| 6 | [Co(P6)] | chloroform | 95 | 84 |
Reactions were carried out at 40 °C for 18 h on 0.25 mmol scale under N2; [3a] = 0.10 M; isolated yields enantiomeric excess determined by chiral HPLC.
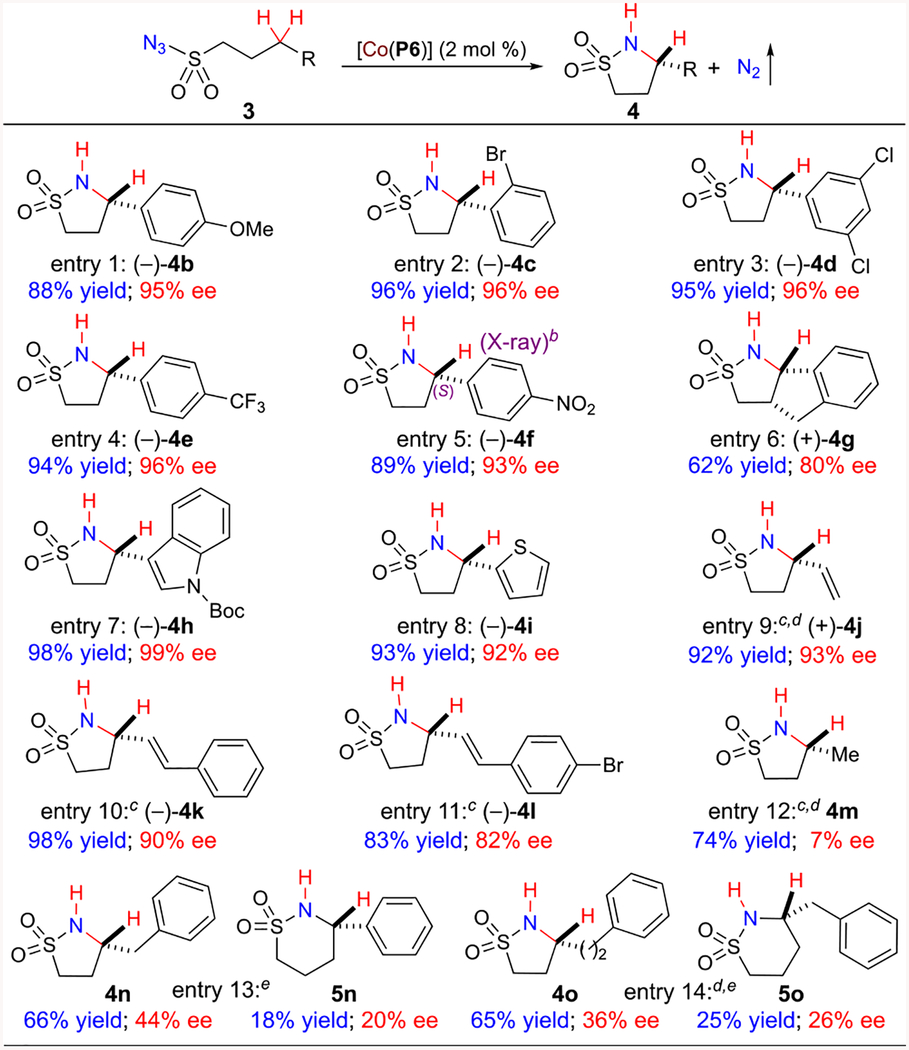
The new metalloradical catalyst [Co(P6)] proved to be generally effective for asymmetric intramolecular C–H amination of alkylsulfonyl azides as demonstrated with reactions of different propylsulfonyl azide derivatives 3 under the optimized conditions (Table 4). Like the amination reaction of 3a, the [Co(P6)]-based catalytic system performed equally well for 3-arylpropylsulfonyl azides 3b–3d with various aryl substituents at the ortho-, meta-, or para-positions, producing corresponding 5-membered cyclic sulfonamides 4b–4d in high yields with excellent enantioselectivities (Table 4, entries 1–3). Resembling the [Co(P4)]-based catalytic system for arylsulfonyl azides 1 (Table 2), the [Co(P6)]-catalyzed amination could also be applicable for electron-deficient C–H bonds, as exemplified with enantioselective formation of the desired cyclic sulfonamides 4e and 4f from azide substrates bearing electron-withdrawing aryl substituents such as trifluoromethyl (3e) and nitro (3f) groups (Table 4, entries 4 and 5). The absolute configuration of the newly generated chiral center in cyclic sulfonamide 4f was established as (S) by X-ray crystal structural analysis. Additionally, indane-derived substrate 3g was successfully desymmetrized by [Co(P6)]-based amination to give cis-fused cyclic sulfonamide 4g in moderate yield with good enantioselectivity (Table 4, entry 6). Alkylsulfonyl azides derived from indole (3h) and thiophene (3i) could also be intramolecularly aminated to furnish desired heteroarene-containing cyclic sulfonamides 4h and 4i, respectively (Table 4, entries 7 and 8). Furthermore, the Co(II)-based system was shown to be capable of catalyzing intramolecular amination of allylic C–H bonds chemoselectively without any complication from the competitive intramolecular aziridination of C=C bonds. For example, sulfonyl azides 3j–3l derived from both terminal and internal olefins could be chemoselectively aminated at the allylic C–H positions, providing corresponding vinyl-substituted cyclic sulfonamides 4j–4l in high yields with high enantioselectivities (Table 4, entries 9–11). The [Co(P6)]-based system could be also extended for amination of nonactivated C–H substrates such as sulfonyl azides 3m–3o, affording 5-membered cyclic sulfonamides 4m–4o in good yields (Table 4, entries 12–14). While the asymmetric induction was low for these reactions, the enantioselectivities of 4n and 4o were significantly higher than that of 4m, indicating the positive effect of distal phenyl substituent. More importantly, the formation of 5-membered cyclic sulfonamide 4n as the major product (66% yield) with 6-membered cyclic sulfonamide 5n as the minor product (18% yield) from the reaction of azide 3n illustrated that the metalloradical catalyst [Co(P6)] could regioselectively aminate the stronger homobenzylic over the weaker benzylic C–H bonds (Table 4, entry 13). The regioselective amination of stronger (bishomobenzylic) over weaker C–H bonds (homobenzylic and benzylic) by [Co(P6)] was also clearly demonstrated from the formation of 5-membered cyclic sulfonamide 4o as the major product (65% yield) with 6-membered cyclic sulfonamide 5o as the minor product (25% yield) without observation of any 7-membered product from the reaction of azide 3o (Table 4, entry 14). This remarkable regioselectivity indicated that corresponding α-Co(III)-aminyl radicals I (Scheme 1) inside the catalyst [Co(P6)] was positioned to have a kinetic preference for 1,5- over 1,6- and 1,7-H abstraction.
Table 4.
[Co(P6)]-Catalyzed Enantioselective 1,5-C–H Amination of Alkylsulfonyl Azidesa
|
Reactions were carried out in benzene at 40 °C for 18 h on 0.25 mmol scale under N2; [3] = 0.10 M; isolated yields enantiomeric excess determined by chiral HPLC.
Absolute configuration determined by X-ray analysis.
Carried out on 0.10 mmol scale at 0 °C for 24 h using 5 mol % [Co(P6)] in PhCl.
Enantiomeric excess determined via derivatization.
Carried out for 48 h.
Mechanistic Studies of 1,5-C–H Amination of Sulfonyl Azides.
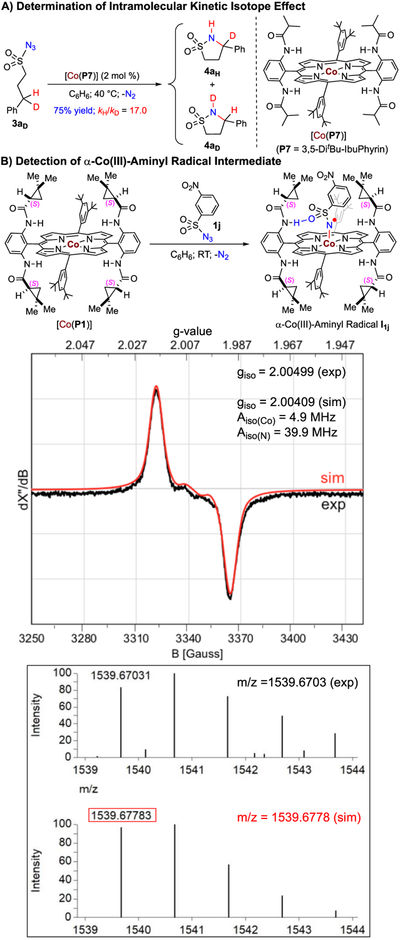
The profile of reactivity and selectivity exhibited by the Co(II)-based system for 1,5-C–H amination is consistent with the proposed stepwise radical mechanism that involves α-Co(III)-aminyl radicals I and ε-Co(III)-alkyl radicals II as the key intermediates (Scheme 1).6 To obtain direct evidence for the proposed mechanism, a set of mechanistic experiments was carried out (Scheme 2). First, monodeuterated 3-phenylpropylsulfonyl azide 3aD was used as the substrate to determine the kinetic isotopic effect (KIE) for the Co(II)-catalyzed intramolecular C–H amination (Scheme 2A). Using the Co(II) complex of D2h-symmetric amidoporphyrin 3,5-DitBu-IbuPhyrin,9a [Co(P7)], as an achiral catalyst, the reaction of azide 3aD provided a mixture of cyclic sulfonamides 4aH and 4aD, which resulted from 1,5-C–H and 1,5-C–D amination, respectively, in 75% combined yield. Analysis of the product mixture by 1H NMR provided a value of 17.0 for intramolecular KIE. This high value of primary KIE agrees well with the proposed step of C–H bond cleavage via H-atom abstraction by α-Co(III)-aminyl radical intermediate I (Scheme 1). Furthermore, in an effort to directly detect radical intermediate I, 3-nitrobenzenesulfonyl azide (1j), which resembles azide 1a but lacks C(sp3)H bonds for further H-atom abstraction, was chosen to react with [Co(P1)] for the generation of corresponding α-Co(III)-aminyl radical I1j (Scheme 2B). The isotropic EPR spectrum of the reaction solution of [Co(P1)] with 1j was recorded at room temperature (Scheme 2B; see the Supporting Information for details), displaying the well-resolved signals that are characteristic of α-Co(III)-aminyl radicals.6a,b In accord with the spin translocation from the Co(II) ion to the N atom during the step of metalloradical activation of the azide (Scheme 1), the observed isotropic g value of 2.00499 evidently indicated the existence of organic radicals. Furthermore, the experimental spectrum could be fittingly simulated on the basis of couplings by both 14N (I = 1) and 59Co (I = 7/2) with hyperfine coupling constants of 39.9 MHz and 4.9 Hz, respectively. Moreover, α-Co(III)-aminyl radical I1j from the reaction mixture of [Co(P1)] with azide 1j could also be detected by high-resolution mass spectrometry (HRMS) with ESI ionization. The obtained spectrum (Scheme 2B; see SI for details) clearly revealed a signal corresponding to [Co(P1)-(NSO2C6H4NO2)]+ (m/z = 1539.6703), which resulted from the neutral α-Co(III)-aminyl radical I1j by the loss of one electron. Both the exact mass and the pattern of isotope distribution determined by ESI-HRMS matched nicely with those calculated from the formula [Co(P1)(NSO2C6H4-NO2)]+
Scheme 2.
Kinetic Isotope Effect of Catalytic 1,5-C–H Amination and Spectroscopic Detection of α-Co(III)-Aminyl Radical
Further experiments were designed to trap and probe the ε-Co(III)-alkyl radical intermediates II generated from the subsequent step of 1,5-H-atom abstraction by α-Co(III)-aminyl radical intermediates I (Scheme 1). To this end, the amination of allylic C–H bonds of azide (E)-3k by [Co(P6)] was performed in the presence of TEMPO (Scheme 3A). Remarkably, even in the presence of a large excess of TEMPO (5.0 equiv), the 5-membered cyclic sulfonamide (E)-4k was still produced as the major product in 72% yield, indicating highly facile formation of the C–N bond via 5-exo-tet radical cyclization of ε-Co(III)-allylic radical II(E)-3k. Concomitantly, the reaction produced (E)-5k as a minor product in 10% yield, which was presumably formed from the TEMPO-trapped intermediate III(E)-3k. The fact that there was no asymmetric induction in (E)-5k while observing significant enantioselectivity for (E)-4k in 72% ee indicates markedly different environment for the C–O and C–N bond formation processes. The existence of ε-Co(III)-allylic radical intermediates II could be further probed via olefin isomerization during the catalytic reaction of azide (Z)-3k by [Co(P6)] (Scheme 3B). Like the reaction of (E)-3k, the 1,5-amination of allylic C–H bonds of (Z)-3k resulted in exclusive formation of (E)-4k in 86% yield with 82% ee without forming any (Z)-4k, representing a rare example of enantioselective allylic C–H amination with diastereoconvergence. This result suggests that the C–N bond formation via 5-exo-tet radical cyclization occurred much faster in the isomerized radical II(E)-3k than the initially formed ε-Co(III)-allylic radical II(Z)-3k, presumably due to the steric difference between the (E)- and (Z)-olefin units. When the cyclohexene-based azide 3p that contains an endocyclic (Z)-olefin was employed as the substrate, the 1,5-amination of the racemic tertiary allylic C–H bond could be effectively catalyzed by [Co(P6)] to afford the spirobicyclic product (Z)-4p in an excellent yield (Scheme 3C). While the (Z)-configuration of the olefin was retained owing to incapable isomerization of the locked endocyclic C=C bond, the fact that (Z)-4p was obtained in 95% yield with low but significant enantioselectivity (14% ee) implies the intermediacy of the tertiary allylic radicals II(Si)-3p and II(Re)-3p resulted from 1,5-H-atom abstraction from the racemic tertiary C–H bond. Furthermore, the catalytic reaction of azide 3q by [Co(P7)] was performed to probe the existence of the corresponding ε-Co(III)-alkyl radical II3q through ring-opening (Scheme 3D). Besides the construction of cyclic sulfonamide 4q in 50% yield from ε-Co(III)-alkyl radical II3q, the reaction produced sulfonamide 6q in 30% yield directly from α-Co(II)-aminyl radical I3q. Additionally, detailed analysis of the reaction mixture by 1H NMR revealed the formation of acyclic sulfonamide 5q as the third product in 5% yield. The identity of product 5q was further confirmed through an independent synthesis (see SI). Compound 5q was likely originated from homoallylic radical intermediate III3q that was generated from ring-opening of the cyclopropylcarbinyl radical II3q. Collectively, these experimental results (Schemes 2 and 3) provided convincing evidence for the proposed stepwise radical mechanism of the Co(II)-catalyzed 1,5-C–H amination of sulfonyl azides (Scheme 1).
Scheme 3.

Trapping and Probing of ε-Co(III)-Alkyl Radicals after 1,5-H Atom Abstraction in Co(II)-Catalyzed Amination
Scalability and Utility of Co(II)-Catalyzed Asymmetric 1,5-C–H Amination of Sulfonyl Azides.
The successful development of asymmetric intramolecular C–H amination of both arylsulfonyl and alkylsulfonyl azides via Co(II)-based metalloradical catalysis provides a practical method to access optically active cyclic sulfonamides bearing various functionalities, which should find applications in research and development of pharmaceuticals. To demonstrate the practicality of the Co(II)-based system for 1,5-C–H amination, the catalytic reaction of 3-phenylpropylsulfonyl azide (3a) by [Co(P6)] was scaled up 40 times from 0.25 to 10 mmol under the identical conditions (Scheme 4A). Desired 5-membered cyclic sulfonamide 4a was produced in multigram quantities in 96% yield with 94% ee, which was similar to those (92% yield; 94% ee) obtained from the corresponding small-scale reaction (Table 3, entry 4). To showcase its synthetic utility, resulting enantioenriched cyclic sulfonamide 4a was employed as the key chiral synthon for the synthesis of compound 7a in view of recent attention to the broad enzyme inhibitory properties of enantiopure fused-cyclic sulfonamides.13,19 As depicted in Scheme 4B, the fused-tricyclic sulfonamide 7a could be effectively synthesized from 4a through a 4-step sequence of N-methylation,13m selenylation,20 elimination,21 and Diels–Alder reactions22 in an overall 64% yield without the erosion of the original enantiomeric purity. The stereochemistry of endo-stereoisomer 7a was unambiguously established by X-ray structural analysis (Scheme 4B).
Scheme 4.

Applications of Co(II)-Catalyzed Enantioselective 1,5-C–H Amination of Sulfonyl Azides
CONCLUSIONS
In summary, we have developed Co(II)-based metalloradical systems for asymmetric 1,5-C–H amination of both arylsulfonyl and alkylsulfonyl azides under neutral and nonoxidative conditions. In addition to benzylic C–H bonds with varied steric and electronic properties, the Co(II)-catalyzed enantioselective radical amination is highlighted with chemoselective amination of allylic C–H bonds as well as compatibility with heteroaryl groups, allowing for stereoselective construction of functionalized 5-membered cyclic sulfonamides in high yields with excellent enantioselectivities. This work represents, to date, one of the most general and selective catalytic systems for asymmetric intramolecular C–H amination of sulfonyl azides. The unique profile of reactivity and selectivity of the Co(II)-based asymmetric 1,5-C–H amination is attributed to the underlying stepwise radical mechanism, which is well-supported by several lines of experimental evidence. Practically, we have demonstrated that the Co(II)-based radical C–H amination protocol can be operated on a multigram-scale under the same mild conditions without affecting the high yield and excellent enantioselectivity. To showcase its synthetic utility, the Co(II)-catalyzed enantioselective radical amination has been applied as the key step for the highly effective synthesis of the fused-tricyclic sulfonamide molecule.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (R01-GM098777) and that in part by NSF (CHE-1900375).
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08894.
Experimental details and analytical data for all new compounds (PDF)
Crystallography data for 2c, [Co(P6)], 4f, and endo-7a (ZIP)
Chemdraw files for 2c, [Co(P6)], 4f, and endo-7a (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).Select books:; (a) Chatgilialoglu C; Studer A Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, 2012. [Google Scholar]; (b) Curran DP; Porter NA; Giese B Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications; John Wiley & Sons, 2008. [Google Scholar]; (c) Zard SZ Radical Reactions in Organic Synthesis; Oxford University Press, 2003. [Google Scholar]; For recent reviews, see the following:; (d) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (e) Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]; (f) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Quiclet-Sire B; Zard SZ Fun with Radicals: Some New Perspectives for Organic Synthesis. Pure Appl. Chem 2010, 83, 519–551. [Google Scholar]; (h) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (i) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- (2).For select examples on approaches to control radical reactivity and stereoselectivity, see the following:; (a) Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huo HH; Shen XD; Wang CY; Zhang LL; Röse P; Chen LA; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (c) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ Iron-Catalyzed Intermolecular [2 + 2] Cycloadditions of Unactivated Alkenes. Science 2015, 349, 960–693. [DOI] [PubMed] [Google Scholar]; (d) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Funken N; Mühlhaus F; Gansäuer A General, Highly Selective Synthesis of 1,3- and 1,4-Difunctionalized Building Blocks by Regiodivergent Epoxide Opening. Angew. Chem., Int. Ed 2016, 55, 12030–12034. [DOI] [PubMed] [Google Scholar]; (g) Brill ZG; Grover HK; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science 2016, 352, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kern N; Plesniak MP; McDouall JJW; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (i) Morrill C; Jensen C; Just-Baringo X; Grogan G; Turner NJ; Procter DJ Biocatalytic Conversion of Cyclic Ketones Bearing α-Quaternary Stereocenters into Lactones in an Enantioselective Radical Approach to Medium-Sized Carbocycles. Angew. Chem., Int. Ed 2018, 57, 3692–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Huang H-M; McDouall JJW; Procter DJ SmI2-Catalysed Cyclization Cascades by Radical Relay. Nat. Catal 2019, 2, 211–218. [Google Scholar]
- (3).For select reviews and highlights on Co(II)-based MRC, see the following:; (a) Huang H-M; Garduño-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (b) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (c) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar]; (d) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (e) Che CM; Lo VKY; Zhou CY; Huang JS Selective Functionalisation of Saturated C–H Bonds with Metalloporphyrin Catalysts. Chem. Soc. Rev 2011, 40, 1950–1975. [DOI] [PubMed] [Google Scholar]; (f) Driver TG Recent Advances in Transition Metal-Catalyzed N-Atom Transfer Reactions of Azides. Org. Biomol. Chem 2010, 8, 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Fantauzzi S; Caselli A; Gallo E Nitrene Transfer Reactions Mediated by Metalloporphyrin Complexes. Dalton Trans. 2009, 5434–5443. [DOI] [PubMed] [Google Scholar]; (h) Doyle MP Exceptional Selectivity in Cyclopropanation Reactions Catalyzed by Chiral Cobalt(II)–Porphyrin Catalysts. Angew. Chem., Int. Ed 2009, 48, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For select examples of Ti(III)-based radical processes, see the following:; (a) Nugent WA; RajanBabu TV Titanium(III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]; (b) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Rinker B; Pierobon M; Grimme S; Gerenkamp M; Mück-Lichtenfeld C A Radical Tandem Reaction with Homolytic Cleavage of a Ti–O Bond. Angew. Chem., Int. Ed 2003, 42, 3687–3690. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Fan C-A; Keller F; Keil J Titanocene-Catalyzed Regiodivergent Epoxide Openings. J. Am. Chem. Soc 2007, 129, 3484–3485. [DOI] [PubMed] [Google Scholar]; (e) Gansäuer A; Fleckhaus A; Lafont MA; Okkel A; Kotsis K; Anoop A; Neese F Catalysis via Homolytic Substitutions with C–O and Ti–O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc 2009, 131, 16989–16999. [DOI] [PubMed] [Google Scholar]; (f) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Cationic Titanocene(III) Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (g) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers RA II Tuning the redox properties of the Titanocene(III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (h) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (i) Yao C; Dahmen T; Gansäuer A;Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (j) Ye K-Y; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- (5).For select examples of catalytic radical processes involving metalloradical intermediates, see the following:; (a) Smith DM; Pulling ME; Norton JR Tin-Free and Catalytic Radical Cyclizations. J. Am. Chem. Soc 2007, 129, 770–771. [DOI] [PubMed] [Google Scholar]; (b) Estes DP; Norton JR; Jockusch S; Sattler W Mechanisms by which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J. Am. Chem. Soc 2012, 134, 15512–15518. [DOI] [PubMed] [Google Scholar]; (c) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (d) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H·Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]
- (6).For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of α-Co(III)-aminyl radical intermediates (also known as Co(III)-nitrene radicals), see the following:; (a) Lyaskovskyy V; Suarez AIO; Lu H; Jiang H; Zhang XP; de Bruin B Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)-Nitrene Intermediates. J. Am. Chem. Soc 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]; (b) Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related DFT studies on the radical mechanism of [Co(Por)]-catalyzed olefin aziridination and ligand hydrogen-bonding effect, see the following:; (c) Hopmann KH; Ghosh A Mechanism of Cobalt-Porphyrin–Catalyzed Aziridination. ACS Catal. 2011, 1, 597–600. [Google Scholar]
- (7).For detailed studies on the radical mechanism involving α-Co(III)-alkyl radical intermediates (also known as Co(III)-carbene radicals), see the following:; (a) Dzik WI; Xu X; Zhang XP; Reek JNH; de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]; (b) Lu H; Dzik WI; Xu X; Wojtas L; de Bruin B; Zhang XP Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (8).For select examples of radical C–H amination via Co(II)-based MRC, see the following:; (a) Ruppel JV; Kamble RM; Zhang XP Cobalt-Catalyzed Intramolecular C–H Amination with Arylsulfonyl Azides. Org. Lett 2007, 9, 4889–4892. [DOI] [PubMed] [Google Scholar]; (b) Lu H; Jiang H; Wojtas L; Zhang XP Selective Intramolecular C–H Amination through the Metalloradical Activation of Azides: Synthesis of 1,3-Diamines under Neutral and Nonoxidative Conditions. Angew. Chem., Int. Ed 2010, 49, 10192–10196. [DOI] [PubMed] [Google Scholar]; (c) Zardi P; Intrieri D; Caselli A; Gallo E Co(porphyrin)-Catalysed Amination of 1,2-Dihydronaphthalene Derivatives by Aryl Azides. J. Organomet. Chem 2012, 716, 269–274. [Google Scholar]; (d) Kuijpers PF; Tiekink MJ; Breukelaar WB; Broere DLJ; van Leest NP; van der Vlugt JI; Reek JNH; de Bruin B Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C–H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem. - Eur. J 2017, 23, 7945–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]; For select example on asymmetric radical C–H amination via Co(II)-MRC, see the following:; (e) Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For select examples of radical olefin aziridination via Co(II)-based MRC, see the following:; (a) Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]; (b) Jiang H; Lang K; Lu H; Wojtas L; Zhang XP Asymmetric Radical Bicyclization of Allyl Azidoformates via Cobalt(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 9164–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For select examples of radical C–H alkylation via Co(II)-based MRC, see the following:; (a) Wang Y; Wen X; Cui X; Zhang XP Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C–H Alkylation. J. Am. Chem. Soc 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karns AS; Goswami M; de Bruin B Catalytic Synthesis of Indolines by Hydrogen Atom Transfer to Cobalt(III)-Carbene Radicals. Chem. - Eur. J 2018, 24, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For select examples of radical olefin cyclopropanation via Co(II)-based MRC, see the following:; (a) Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]; (b) Zhu S; Ruppel JV; Lu H; Wojtas L; Zhang XP Cobalt-Catalyzed Asymmetric Cyclopropanation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc 2008, 130, 5042–5043. [DOI] [PubMed] [Google Scholar]; (c) Xu X; Lu H; Ruppel JV; Cui X; Lopez de Mesa S; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]; (d) Reddy AR; Hao F; Wu K; Zhou C-Y; Che CM Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem., Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]; (e) Goswami M; de Bruin B; Dzik WI Difluorocarbene Transfer from a Cobalt Complex to an Electron-Deficient Alkene. Chem. Commun 2017, 53, 4382–4385. [DOI] [PubMed] [Google Scholar]; (f) Chirila A; Gopal Das B; Paul ND; de Bruin B Diastereoselective Radical-Type Cyclopropanation of Electron-Deficient Alkenes Mediated by the Highly Active Cobalt(II) Tetramethyltetraaza[14]annulene Catalyst. ChemCatChem 2017, 9, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Roy S; Das SK; Chattopadhyay B Cobalt(II)-based Metalloradical Activation of 2-(Diazomethyl)pyridines for Radical Transannulation and Cyclopropanation. Angew. Chem., Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]
- (12).For other transformations via MRC, see the following:; (a) Cui X; Xu X; Wojtas L; Kim MM; Zhang XP Regioselective Synthesis of Multisubstituted Furans via Metalloradical Cyclization of Alkynes with alpha-Diazocarbonyls: Construction of Functionalized alpha-Oligofurans. J. Am. Chem. Soc 2012, 134, 19981–19984. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paul ND; Mandal S; Otte M; Cui X; Zhang XP; de Bruin B Metalloradical Approach to 2H-Chromenes. J. Am. Chem. Soc 2014, 136, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Das BG; Chirila A; Tromp M; Reek JNH; de Bruin B CoIII-Carbene Radical Approach to Substituted 1H-Indenes. J. Am. Chem. Soc 2016, 138, 8968–8975. [DOI] [PubMed] [Google Scholar]; (d) Gu Z-Y; Liu Y; Wang F; Bao X; Wang S-Y; Ji S-J Cobalt(II)-Catalyzed Synthesis of Sulfonyl Guanidines via Nitrene Radical Coupling with Isonitriles: A Combined Experimental and Computational Study. ACS Catal. 2017, 7, 3893–3899. [Google Scholar]; (e) Liu J; Hu L; Wang L; Chen H; Deng L An Iron(II) Ylide Complex as a Masked Open-Shell Iron Alkylidene Species in Its Alkylidene-Transfer Reactions with Alkenes. J. Am. Chem. Soc 2017, 139, 3876–3888. [DOI] [PubMed] [Google Scholar]; (f) Roy S; Khatua H; Das SK; Chattopadhyay B Iron(II)-Based Metalloradical Activation: Switch from Traditional Click Chemistry to Denitrogenative Annulation. Angew. Chem., Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]
- (13).For select reviews, see the following:; (a) Majumdar KC; Mondal S Recent Developments in the Synthesis of Fused Sultams. Chem. Rev 2011, 111, 7749–7773. [DOI] [PubMed] [Google Scholar]; For select examples, see the following:; (b) Duan J; Chen L; Cherney RJ; Decicco CP; Voss ME Novel Cyclic Sulfonamide Derivatives as Metalloproteinase Inhibitors. WO9941246A1, 1999.; (c) Tozer MJ; Kalindjian SB; Linney ID; Steel KIM; Pether MJ; Cooke T Preparation of 1H-4(5)-Substituted Imidazoles as Histamine H3 Receptor Ligands. WO9905114A2, 1999.; (d) Wells GJ; Tao M; Josef KA; Bihovsky R 1,2-Benzothiazine 1,1-Dioxide P2–P3 Peptide Mimetic Aldehyde Calpain I Inhibitors. J. Med. Chem 2001, 44, 3488–3503. [DOI] [PubMed] [Google Scholar]; (e) Cherney RJ; Duan JJW; Voss ME; Chen L; Wang L; Meyer DT; Wasserman ZR; Hardman KD; Liu R-Q; Covington MB; Qian M; Mandlekar S; Christ DD; Trzaskos JM; Newton RC; Magolda RL; Wexler RR; Decicco CP Design, Synthesis, and Evaluation of Benzothiadiazepine Hydroxamates as Selective Tumor Necrosis Factor-α Converting Enzyme Inhibitors. J. Med. Chem 2003, 46, 1811–1823. [DOI] [PubMed] [Google Scholar]; (f) Baker DC; Mayasundari A; Mao J; Johnson SC; Yan S Methods of synthesizing sultams and anti-viral compositions. U.S. Patent 6562850B1, 2003.; (g) Singh SK; Reddy PG; Rao KS; Lohray BB; Misra P; Rajjak SA; Rao YK; Venkateswarlu A Polar Substitutions in the Benzenesulfonamide Ring of Celecoxib Afford a Potent 1,5-Diarylpyrazole Class of COX-2 Inhibitors. Bioorg. Med. Chem. Lett 2004, 14, 499–504. [DOI] [PubMed] [Google Scholar]; (h) Valente C; Guedes RC; Moreira R; Iley J; Gut J; Rosenthal PJ Dipeptide Vinyl Sultams: Synthesis via the Wittig–Horner Reaction and Activity Against Papain, Falcipain-2 and Plasmodium Falciparum. Bioorg. Med. Chem. Lett 2006, 16, 4115–4119. [DOI] [PubMed] [Google Scholar]; (i) Zeng W; Chemler SR Copper(II)-Catalyzed Enantioselective Intramolecular Carboamination of Alkenes. J. Am. Chem. Soc 2007, 129, 12948–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Jiménez-Hopkins M; Hanson PR An RCM Strategy to Stereodiverse δ-Sultam Scaffolds. Org. Lett 2008, 10, 2223–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rolfe A; Young K; Hanson PR Domino Heck–Aza-Michael Reactions: A One-Pot, Sequential Three-Component Approach to 1,1-Dioxido-1,2-Benzisothiazoline-3-Acetic Acid. Eur. J. Org. Chem 2008, 2008, 5254–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Rommel M; Fukuzumi T; Bode JW Cyclic Ketimines as Superior Electrophiles for NHC-Catalyzed Homoenolate Additions with Broad Scope and Low Catalyst Loadings. J. Am. Chem. Soc 2008, 130, 17266–17267. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Yu C-B; Wang D-W; Zhou Y-G Highly Enantioselective Synthesis of Sultams via Pd-Catalyzed Hydrogenation. J. Org. Chem 2009, 74, 5633–5635. [DOI] [PubMed] [Google Scholar]; (n) Ghosh AK; Kulkarni S; Anderson DD; Hong L; Baldridge A; Wang Y-F; Chumanevich AA; Kovalevsky AY; Tojo Y; Amano M; Koh Y; Tang J; Weber IT; Mitsuya H Design, Synthesis, Protein–Ligand X-ray Structure, and Biological Evaluation of a Series of Novel Macrocyclic Human Immunodeficiency Virus-1 Protease Inhibitors to Combat Drug Resistance. J. Med. Chem 2009, 52, 7689–7705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Hanessian S; Larsson A; Fex T; Knecht W; Blomberg N Design and Synthesis of Macrocyclic Indoles Targeting Blood Coagulation Cascade Factor XIa. Bioorg. Med. Chem. Lett 2010, 20, 6925–6928. [DOI] [PubMed] [Google Scholar]; (p) Swiderska N; Hawcutt D; Eaton V; Stockton F; Kumar R; Kneen R; Appleton R Sulthiame in Refractory Paediatric Epilepsies: An Experience of an ‘Old’ Antiepileptic Drug in a Tertiary Paediatric Neurology Unit. Seizure 2011, 20, 805–808. [DOI] [PubMed] [Google Scholar]; (q) Bodil Van Niel M; Fauber B; Gaines S; Gobbi A; Rene O; Vesey D; Ward S Aryl Sultam Derivatives as RORc Modulators and Their Preparation. WO2014009447A1, 2014.; (r) Aldrich LN; Kuo S-Y; Castoreno AB; Goel G; Kuballa P; Rees MG; Seashore-Ludlow BA; Cheah JH; Latorre IJ; Schreiber SL; Shamji AF; Xavier RJ Discovery of a Small-Molecule Probe for V-ATPase Function. J. Am. Chem. Soc 2015, 137, 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Osborne CA; Endean TBD; Jarvo ER Silver-Catalyzed Enantioselective Propargylation Reactions of N-Sulfonylketimines. Org. Lett 2015, 17, 5340–5343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Shan W; Balog A; Nation A; Zhu X; Chen J; Cvijic ME; Geng J; Rizzo CA; Spires T; Attar RM; Obermeier M; Traeger S; Dai J; Zhang Y; Galella M; Trainor G; Vite GD; Gavai AV [2.2.1]-Bicyclic Sultams as Potent Androgen Receptor Antagonists. Bioorg. Med. Chem. Lett 2016, 26, 5707–5711. [DOI] [PubMed] [Google Scholar]; (u) Kiran INC; Reddy RS; Lagishetti C; Xu H; Wang Z; He Y Selective Aza Diels–Alder and Domino [4 + 2]/[2 + 2] Cycloaddition Reactions of Arynes with N-Sulfonyl Ketimines. J. Org. Chem 2017, 82, 1823–1832. [DOI] [PubMed] [Google Scholar]; (v) Khalifa A; Evans P The Titanium-Mediated Double Reductive Cleavage of Cyclic Sulfonamides for the Synthesis of Aryl Pyrrolidines. J. Org. Chem 2019, 84, 2969–2975. [DOI] [PubMed] [Google Scholar]
- (14).For select reviews, see the following:; (a) Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (b) Alderson JM; Corbin JR; Schomaker JM Tunable, Chemo- and Site-Selective Nitrene Transfer Reactions through the Rational Design of Silver(I) Catalysts. Acc. Chem. Res 2017, 50, 2147–2158. [DOI] [PubMed] [Google Scholar]; (c) Collet F; Lescot C; Dauban P Catalytic C–H Amination: the Stereoselectivity Issue. Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]
- (15).For select examples of nonasymmetric intramolecular C–H amination, see the following:; (a) Huard K; Lebel H N-Tosyloxycarbamates as Reagents in Rhodium-Catalyzed C–H Amination Reactions. Chem.-Eur. J 2008, 14, 6222–6230. [DOI] [PubMed] [Google Scholar]; (b) Hennessy ET; Betley TA Complex N-Heterocycle Synthesis via Iron-Catalyzed, Direct C–H Bond Amination. Science 2013, 340, 591–595. [DOI] [PubMed] [Google Scholar]; (c) Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM Ligand-Controlled, Tunable Silver-Catalyzed C–H Amination. J. Am. Chem. Soc 2014, 136, 16720–16723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hong SY; Park Y; Hwang Y; Kim YB; Baik M-H; Chang S Selective Formation of γ-Lactams via C–H Amidation Enabled by Tailored Iridium Catalysts. Science 2018, 359, 1016–1021. [DOI] [PubMed] [Google Scholar]; (e) Azek E; Khalifa M; Bartholoméüs J; Ernzerhof M; Lebel H Rhodium(II)-Catalyzed C–H Aminations using N-Mesyloxycarbamates: Reaction Pathway and By-Product Formation. Chem. Sci 2019, 10, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Baek Y; Betley TA Catalytic C–H Amination Mediated by Dipyrrin Cobalt Imidos. J. Am. Chem. Soc 2019, 141, 7797–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]; For metal-free C(sp3)–H radical amination, see the following:; (g) Evoniuk CJ; Gomes G. d. P.; Hill SP; Fujita S; Hanson K; Alabugin IV Coupling N–H Deprotonation, C–H Activation, and Oxidation: Metal-Free C(sp3)–H Aminations with Unprotected Anilines. J. Am. Chem. Soc 2017, 139, 16210–16221. [DOI] [PubMed] [Google Scholar]; (h) Bafaluy D; Muñoz-Molina JM; Funes-Ardoiz I; Herold S; de Aguirre AJ; Zhang H; Maseras F; Belderrain TR; Pérez PJ; Muñiz K Copper-Catalyzed N–F Bond Activation for Uniform Intramolecular C–H Amination Yielding Pyrrolidines and Piperidines. Angew. Chem., Int. Ed 2019, 58, 8912–8916. [DOI] [PubMed] [Google Scholar]
- (16).For select examples of intramolecular asymmetric C–H amination, see the following:; (a) Liang J-L; Yuan S-X; Huang J-S; Yu W-Y; Che C-M Highly Diastereo- and Enantioselective Intramolecular Amidation of Saturated C–H Bonds Catalyzed by Ruthenium Porphyrins. Angew. Chem., Int. Ed 2002, 41, 3465–3468. [DOI] [PubMed] [Google Scholar]; (b) Milczek E; Boudet N; Blakey S Enantioselective C–H Amination Using Cationic Ruthenium(II)–Pybox Catalysts. Angew. Chem., Int. Ed 2008, 47, 6825–6828. [DOI] [PubMed] [Google Scholar]; (c) Wang H; Park Y; Bai Z; Chang S; He G; Chen G Iridium-Catalyzed Enantioselective C(sp3)–H Amidation Controlled by Attractive Noncovalent Interactions. J. Am. Chem. Soc 2019, 141, 7194–7201. [DOI] [PubMed] [Google Scholar]; (d) Zhou Z; Chen S; Qin J; Nie X; Zheng X; Harms K; Riedel R; Houk KN; Meggers E Catalytic Enantioselective Intramolecular C(sp3)–H Amination of 2-Azidoacetamides. Angew. Chem., Int. Ed 2019, 58, 1088–1093. [DOI] [PubMed] [Google Scholar]; (e) Xing Q; Chan C-M; Yeung Y-W; Yu W-Y Ruthenium(II)-Catalyzed Enantioselective γ-Lactams Formation by Intramolecular C–H Amidation of 1,4,2-Dioxazol-5-Ones. J. Am. Chem. Soc 2019, 141, 3849–3853. [DOI] [PubMed] [Google Scholar]
- (17).For a recent example of intermolecular asymmetric C–H amination, see the following:; Nasrallah A; Boquet V; Hecker A; Retailleau P; Darses B; Dauban P Catalytic Enantioselective Intermolecular Benzylic C(sp3)–H Amination. Angew. Chem., Int. Ed 2019, 58, 8192–8196. [DOI] [PubMed] [Google Scholar]
- (18).For the first example of asymmetric cyclic sulfonamide formation via intramolecular C–H amination, see the following:; (a) Fruit C; Müller P Intramolecular Asymmetric Amidations of Sulfonamides and Sulfamates Catalyzed by Chiral Dirhodium(II) Complexes. Helv. Chim. Acta 2004, 87, 1607–1615. [Google Scholar]; For the only example of highly asymmetric benzofused cyclic sulfonamide formation via intramolecular C–H amination, see the following:; (b) Ichinose M; Suematsu H; Yasutomi Y; Nishioka Y; Uchida T; Katsuki T Enantioselective Intramolecular Benzylic C–H Bond Amination: Efficient Synthesis of Optically Active Benzosultams. Angew. Chem., Int. Ed 2011, 50, 9884–9887. [DOI] [PubMed] [Google Scholar]
- (19).For select examples of synthesis and bioactivity studies on chiral fused-cyclic sulfonamides, see the following:; (a) Samarakoon TB; Loh JK; Rolfe A; Le LS; Yoon SY; Lushington GH; Hanson PR A Modular Reaction Pairing Approach to the Diversity-Oriented Synthesis of Fused- and Bridged-Polycyclic Sultams. Org. Lett 2011, 13, 5148–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yin X; Zheng Y; Feng X; Jiang K; Wei X-Z; Gao N; Chen Y-C Asymmetric [5 + 3] Formal Cycloadditions with Cyclic Enones through Cascade Dienamine–Dienamine Catalysis. Angew. Chem., Int. Ed 2014, 53, 6245–6248. [DOI] [PubMed] [Google Scholar]; (c) Loh JK; Asad N; Samarakoon TB; Hanson PR Modular, One-Pot, Sequential Aziridine Ring Opening–SNAr Strategy to 7-, 10-, and 11-Membered Benzo-Fused Sultams. J. Org. Chem 2015, 80, 9926–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also ref 13t.
- (20).Brattesani DN; Heathcock CH Sulfenylation and Selenylation of Nitriles. Tetrahedron Lett. 1974, 15, 2279–2282. [Google Scholar]
- (21).Grieco PA; Gilman S; Nishizawa M A Facile One-Step Synthesis of Alkyl Aryl Selenides from Alcohols. J. Org. Chem 1976, 41, 1485–1486. [Google Scholar]
- (22).Wanner J; Harned AM; Probst DA; Poon KWC; Klein TA; Snelgrove KA; Hanson PR A Dual Metathesis Route to Oligomeric Sulfonamides. Tetrahedron Lett. 2002, 43, 917–921. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.