

Abstract
Despite the success of antiretroviral therapies, there is no cure for HIV-1 infection due to the establishment of a long-lived latent reservoir that fuels viral rebound upon treatment interruption. “Shock-and-kill’ strategies to diminish the latent reservoir have had modest impact on the reservoir leading to considerations of alternative approaches to target HIV-1 proviruses. This review explores approaches to target HIV-1 transcription as a way to block the provirus expression.
Graphical Abstract
Introduction
Antiretroviral therapy suppresses HIV-1 replication and decreases morbidity and mortality of HIV-associated diseases; however, viral replication rapidly rebounds once treatment is discontinued indicating the presence of a long-lived latently infected HIV reservoir. This long-lived reservoir presents a major barrier to curing HIV-1 infection [1-6].
Latently infected cells have decreased or inactive proviral transcription resulting from multiple biochemical mechanisms that include chromatin organization, lack of key transcription factors, fluctuations in Tat expression and/or inefficient proviral transcriptional elongation [7-13]. One strategy for reducing the HIV-1 reservoir is “shock and kill” in which latency reversing compounds induce HIV-1 expression and the reservoir would be eliminated by immune clearance and HIV-1 triggered cell death [14-17]. Compounds that target chromatin remodeling and/or act as pan-T cell activators have been examined for their potential to reverse latency and purge HIV-1 but have failed to decrease the reservoir size in HIV-1 infected patients [14,18,19]. The lack of success of latency reversing agents may reflect the paucity of latently infected cells, which are estimated to be approximately one in 106 cells. Challenges include targeting such a small population of latent cells as well as finding treatments that can effectively deliver both shock and kill signals. Furthermore, an absence of latency markers and the DNA background signal associated with abundant defective proviral DNAs that are resistant to induction, create difficulties for monitoring reservoir size following treatments [20-22*]. The lack of efficacy of latency reversal also reflects the complexity of the reservoir which includes multiple cell types in different tissues including targeting HIV-1 provirus harbored in CD4+ T memory subsets, macrophages, dendritic cells and other myeloid cells in lymph nodes, gastrointestinal tissues, and the brain [1,23,24**].The poor outcomes of latency reversal strategies have shifted cure strategies away from broad activation of T cells to those that specifically target HIV-1 proviruses including long-term repression of infected cells [18,19,25,26]. Drugging HIV-1 specific transcription has been a long-standing interest and it is impossible to extensively review the large literature focusing on latency, Tat function, and molecular mechanisms of HIV transcription. We will highlight a subset of more recent attempts to engineer RNAs and proteins to target HIV provirus transcription as a potential treatment strategy.
Transcriptional regulation and mechanisms that contribute to HIV latency
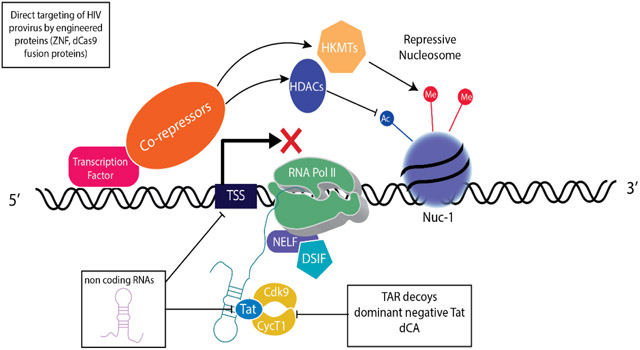
HIV-1 transcription is a combinatorial process that includes coordination of transcription factor recruitment, epigenetic regulation and RNA polymerase II (RNAPII) activity [10,11,13]. A number of cellular transcription factors are recruited to the HIV long terminal repeat (LTR) which functions as the promoter and enhancer for the HIV-1 provirus. More than 40 cellular factors have been reported to bind and influence HIV transcription [Los Alamos Data Base; URL: http://www.hiv.lanl.gov/] [27], suggesting that the LTR has a broad range of activities in different cells and is capable of responding to cellular signals and metabolic states. Therefore, HIV-1 expression versus latency will reflect the active transcriptional networks of the infected cell [28-30]. Latent HIV-1 is primarily harbored in quiescent CD4+ T cell subsets, including T memory stem cells, and central, transitional and effector memory cells, although macrophages and dendritic cells contribute to the latent reservoir in various tissues [24**,28,31,32]. These cell populations may lack transcription factors that mediate transcriptional activation, like NF-κB, and express a wide array of transcriptional repressors that are responsible for maintaining their quiescent state and facilitate transcriptional repression of HIV provirus. Examples of transcriptional repressors that are expressed in quiescent T cells and repress HIV-1 transcription include CBF-1 [33,34], Blimp-1 [35], AP-4 [36], YY-1 [37,38], Ets-2 [39] and Forkhead box proteins (FoxO) [39,40]. These transcriptional repressors bind the HIV-1 LTR and recruit complexes that modify chromatin, regulate RNAPII processiviness and restrict the availability of positive transcription factors such as P-TEFb. For example, BLIMP-1 represses HIV transcription by recruiting HDACs to the HIV LTR which mediated post-translational modification of the positioned nucleosome Nuc-1 as well as enhance negative elongation factor to pause the polymerase [35]. CBF-1 represses HIV-1 transcription through limiting P-TEFb accessibility and mediating nucleosome modifications through HDACs and methyl-transferases [33,34].
One function of cellular transcription factors is to recruit RNAPII and coactivator complexes to the HIV LTR. In addition, transcription factors recruit chromatin modifiers to the promoter, which through histone post-translational modifications including ubiquitination, methylation, acetylation, SUMOylation, ADP-ribosylation, phosphorylation and crotonylation [41,42], modify chromatin organization and interactions between transcriptional complexes; many of these post-translational modifications of histones have been reported to influence HIV-1 transcription and latency [11,43*-45]. The role of histone modifying complexes also underscore the importance of chromatin as a key check-point for HIV transcription and are consistent with observations in several latency models that repression of HIV transcription is associated with a positioned repressive nucleosome, Nuc-1, which presents a barrier to RNAPII processivity and transcription [11-13]. Best studied are the effects of histone acetylation/deacetylation and methylation acting as histone marks associated with transcriptional activation and repression. For example, histone acetylation, which is mediated by histone acetyltransferases (HATs), positively correlates with HIV transcription whereas histone deacetylation, which is directed by histone deacetyltransferase complexes (HDACs) is associated with HIV transcriptional repression. The activity of HATs and HDACs provided the initial rationale for exploring HDAC inhibitors as HIV latency reversing agents. Histone lysine methyltransferases (HKMTs) including SUV39H1, EZH2, G9a, SMYD2 are associated with HIV-1 transcriptional repression and if these HKMTs are inhibited reactivation of provirus transcription in cell lines and resting primary T cells isolated from HIV-1 infected patients on HAART has been observed suggesting these are potential druggable targets [45-50]. The ATP-dependent SWI/SNF chromatin remodeling complexes PBAF and BAF, influences nucleosome organization at the LTR and can be a transcriptional activators or repressors, respectively [51-55]. Despite the functionally distinct roles of SWI/SNF complexes in regulating HIV-1 transcription, BAF and PBAF complexes share many proteins including the key functional subunit, the BRG1 ATPase, making it challenging to specifically target their activities [56].
Specific histone marks can also be read by transcriptional coregulators to recruit specific complexes to promoters. One example relevant to HIV latency is the bromodomain and extraterminal domain (BET) family of protein, BRD4. BRD4 binds hyperacetylated H3 and recruits positive-transcription elongation factor b (P-TEFb) to promoters including the HIV-LTR in the absence of the HIV transactivator, Tat [57]. It has been proposed that BRD4 represses efficient HIV transcription by competing with the HIV transactivator, Tat, for P-TEFb binding which provided the rationale for using BET inhibitors such as JQ1 as latency reversing agents [58-60]. A short isoform of BRD4 binds BRG-1 to bridge BRD and SWI/SNF activities repressing HIV-1 transcription and possibly suggesting a pathway that can be usurped for a block and lock strategy [61*].
Robust HIV transcription requires the HIV encoded transactivator protein, Tat. Tat binds TAR, an RNA stem loop structure that is generated on the first 80 bases of the HIV RNA and promotes efficient transcription elongation by recruiting P-TEFb to the LTR [11,12,62]. P-TEFb enhances RNAPII activity by mediating phosphorylation of RNAPII carboxy terminal domain (CTD) and negative regulators of RNAPII activity, NELF and DSIF, as well as recruiting other coactivator complexes, in particular, the Super Elongation Complex to the active RNAPII complex [11-13,63]. Tat and TAR are required for HIV transcription and stochastic episodic changes in Tat expression has been proposed as a key determinant of HIV proviral transcription, repression and/or latency [64,65].
P-TEFb consists of Cyclin T1, which physically interacts with Tat, as well as Cdk9, which phosphorylates RNAPII CTD, DSIF, and NELF, releasing the paused RNAPII to enhance HIV-1 transcriptional elongation. In the absence of Tat, transcription is initiated but RNAPII will pause at +45 to +50, generating aborted transcripts [11,63,66]. The propensity of RNAPII to pause on the HIV LTR and the ability of Tat to interact with histone modifiers places Tat as a key regulator of active proviral transcription versus latency.
The necessity of Tat for efficient HIV transcriptional elongation also demonstrates how recruitment of P-TEFb to the HIV LTR is a limiting step for provirus transcription. P-TEFb is sequestered by an inhibitory complex, 7SK RNP, which includes HEXIM1, HEXIM2, 7SK RNA, MEPCE and LARP7 [63,66-68]. P-TEFb in complex with the 7SK RNP is sequestered in nuclear speckles [69,70] and its release is facilitated by phosphorylation of the T loop of CDK9 and exchange with Tat [71-73]. An alternative model is 7SK RNP is recruited to the LTR through interactions with BRD4 and KAP1, poising the HIV-1 LTR for the exchange of P-TEFb to Tat and transcriptional activation [74,75]. Exchange of P-TEFb from 7SK RNP to Tat is regulated by CDK7 and PP1 [63,71,76,77]. Strategies to target components of P-TEFb include engineering a dominant negative mutant CycT1 protein that inhibits HIV transcription by competing with wild-type CycT1 for binding to Tat as well as blocking Cdk9 activation and subsequent phosphorylation of RNAPII CTD and proviral transcriptional elongation [78]. Although, this approach works in latent cell lines, the efficacy and toxicity of this engineered CycT1 protein has not been examined in primary cells or animal models [78].
The importance of Tat and P-TEFb interactions for HIV transcription has led to efforts to develop specific inhibitors of this complex. One particularly promising compound, Didehydro-cortistatin A (dCA), an analog of a steroidal alkaloid found in the marine sponge Corticium simplex [79], has been shown by Valente et al. to be a potent Tat inhibitor [80**]. The primary mechanism of action for dCA is disrupting Tat-TAR interactions by directly binding the Tat basic domain and blocking P-TEFb recruitment and activity [81]. dCA inhibits HIV-1 transcription and reactivation in a variety of cell lines, primary cells and samples from patients treated with ARTs. Furthermore, dCA can be removed and the repression of HIV persists for nearly a month suggesting a long term mechanism of repression [81] which may reflect direct or indirect actions of dCA on histone post-translational modifications, recruitment of repressive BAF complexes and diminished recruitment of RNAPII [82]. dCA has also been reported to inhibit the expression of inflammatory cytokines suggesting additional benefits in treating HIV comorbidities [83], although how dCA is targeting inflammation has not been examined in detail. Importantly, recent studies with a humanized mouse model has shown efficacy in vivo with inhibition of HIV replication and reactivation upon antiretroviral therapy interruption [83].
Engineering Repressors of HIV-1 transcription
In addition to efforts to develop small molecules that target the Tat-TAR-P-TEFb axis engineering approaches utilizing chimeric proteins with dominant negative activities, inhibitory RNAs or gene editing have been explored.
Dominant Negative Proteins.
Tat has five key functional domains that facilitate its activity as a transcriptional activator: an acidic/proline rich domain; a zinc-finger/cystine rich domain that influences folding and structure; a core domain; a basic-arginine rich motif (ARM) that mediates RNA binding, nuclear localization and Cyclin T1 binding and; glutamic acid rich activation motif [12,13,62]. Efforts over the years to disrupt Tat activity by generating dominant negative Tat proteins and chimeric inhibitors have focused on these domains. Some recent approaches to disrupt Tat activity include the generation of the dominant negative Nullbasic, in which the ARM domain of Tat was mutated by introducing a stretch of glycines; this inhibits P-TEFb recruitment to the LTR by Tat and reduces HIV transcription and reactivation of latent HIV [84]. Similarly, a Tat variant that included two domains from HEXIM1, HT-1, was engineered to inhibit HIV transcription. HEXIM1 is part of a 7sK RNP which sequesters P-TEFb to this inhibitory complex. The chimeric Tat-HEXIM1 (HT-1) competed with functional Tat for TAR binding as well as reduced available P-TEFb. In cell lines HT-1 repress HIV transcription and reactivation of latent virus [85].
RNA strategies to Blocking Tat function.
In the context of HIV-1 infection, several miRNAs have been suggested to regulate HIV-1 transcription either indirectly by influencing T cell maturation and function or directly by generating antiviral RNAs. For example, miRNAs enriched in resting CD4 T cells potently inhibit HIV transcription by targeting 3’ ends of HIV-1 mRNA [86]. Similarly, several groups have reported that cellular miRNAs inhibit HIV-1 expression by interfering with the Tat-TAR-P-TEFb axis. In particular, the expression level of Cyclin T1 is regulated by several sncRNA, miRNA-198, miR-27b, and lncRNA, NEAT1, which when overexpressed reduces Cyclin T1 and repress HIV-1 transcription [87-89]. In addition to cellular RNAs, it has been reported that HIV generates miRNAs that target the HIV LTR in primary macrophages [90]; however, the role these RNAs play in controlling HIV-1 expression and latency is still not understood.
Given the importance of structured RNAs for the recruitment of Tat to the LTR and the regulation of P-TEFb availability coupled with the relative ease of engineering RNAs and their sequence specificity, several groups have engineered RNA inhibitors to target HIV transcription. Strategies have spanned siRNAs, anti-sense RNAs, aptamers, RNA TAR decoys and ribozymes to inhibit Tat and HIV replication in cell lines and primary cells [91-99]. HIV proviral transcriptional silencing and inhibition of latency reversal in cell lines has been demonstrated using shRNAs to key targets within the LTR and the HIV genome [100-102].
Engineering factors that directly target HIV.
Understanding the critical biochemical interactions and functional domains necessary for transcriptional control have allowed chimeric proteins with defined targets and specific activities to be engineered. Engineering proteins that target HIV provirus and its expression has been explored. For example, recent approaches have employed engineered proteins including Zinc finger (ZnF) nucleases, transcription activator-like effector nucleases (TALENs), homing nucleases and meganucleases to mediate site-directed genome editing to directly target HIV-1 proviruses or factors associated with HIV-1 infection [103-105]. Cells resistant to HIV-1 have been generated using these engineered proteins by targeting key entry receptors such as CCR5 [106] and there are clinical trials [107] to transplant HIV-1 resistant ΔCCR5 cells into HIV-1+ individuals to recapitulate cure protocols that utilize transplanted bone marrow cells from ΔCCR5 donors [108,109]. TALENs and ZnF nucleases have also been used to directly modify or target HIV-1 provirus and shown to reduce p24 expression [110] and HIV-1 DNA [111]. While use of ZnF nucleases and TALENs to specifically target HIV-1 have shown promise in vitro and are being explored, the designing and engineering of nucleases can be iterative and, in general, are less amenable for high-throughput screening.
The discovery and development of CRISPR/Cas9 technology has reshaped gene editing partly due to ease and flexibility of targeting the Cas9 nuclease to specific DNA sequences using complementary guide RNAs [112]. In the context of HIV-1, CRISPR/Cas9 tools have been designed to target LTRs and disrupt or eliminate HIV-1 in latently infected cells [113-115]. CRISPR/Cas9 has also been used to successfully excise HIV, in vivo with a humanized mouse model [116**]. For these experiments infected mice were treated with nanoparticles coated with antiretroviral drugs and a single injection of AAV-CRISPR-Cas9 with multiple gRNAs targeting conserved LTR and gag regions of the HIV genome [116**]. When mice were weaned from ART, two of nine mice showed no signs of viral rebound. Although this outcome seems modest, it is an important step towards showing the clinical potential of CRISPR/Cas9 as an HIV cure method. However, CRISPR/Cas9 induces double stranded breaks that are subsequently repaired through an error-prone non-homologous end joining DNA repair resulting in mutations or insertions and deletions at the targeted site [114,117]. While this can incapacitate the HIV-1 provirus, escape mutants can render CRISPR/Cas9 ineffective [118*-120*, 121, 122]. This HIV-1 escape can be mitigated by using multiple gRNAs that target the LTR making cells less susceptible to HIV infection [118*, 120*]
Cas9 has also been engineered to act as a sequence specific transcriptional activator or repressor rather than a nuclease. For example, a catalytically dead Cas9 (dCas9), which lacks endonuclease activity has been fused with transactivator or transrepressor domains and directed to specific promoters with gRNAs to modulate cellular gene regulation [123,124]. This approach has been utilized to induce HIV expression in latently infected cells by fusing dCas9 to a VP64 transcriptional activation domain and targeting the dCas9-VP64 transactivator to the HIV LTR [125,126]. dCas9 has also been repurposed to repress transcription by creating fusion proteins with repressor domains derived from Kruppel-associated box (KRAB) zinc-finger proteins [127,128**-131]. KRAB ZnF proteins have been proposed to maintain the integrity of the genome by silencing transcription of endogenous retroelements through the binding of their LTRs and facilitating post-translational modification of histones, recruitment of repressor complexes including KAP1 and promoting DNA methylation leading to heritable epigenetic repression [132-134]. It is interesting to speculate whether KRAB zinc finger proteins might be able to “block and lock” HIV provirus. KRAB zinc fingers have been reported to repress HIV transcription [135-140] and we are exploring using dCas9-KRAB to target the HIV-LTR. Preliminary studies have shown greater than 70% reduction in HIV-1 proviral expression in cell lines, correlating with epigenetic modifications such as post-translational modification of histones (unpublished observation). Utilizing dCas9 repressor proteins may provide a novel strategy to achieve a deep repression of the latent reservoirs.
Conclusion
HIV persistence and latency are the major challenges to the elimination of HIV infection. Our understanding of basic mechanisms of HIV-1 transcriptional regulation has translated into exciting new treatment strategies that may enhance “shock and kill” strategies or induce deep latency consistent with a block-and-lock strategy. Furthermore, advancements with gene editing approaches have made it realistic to consider directly targeting HIV-1 provirus and cofactors and coreceptors. Although these approaches have been demonstrated to work in vitro and, in a few cases, animal models, there are several practical issues that still exist in targeting HIV proviral transcription. Major challenges to implementing cure strategies include off-target effects since many of the factors are general transcriptional regulators, the ability of the virus to adapt and escape treatments and delivering expression vectors or reagents to the appropriate cells and tissues. Related to this latter issue is the paucity of latently infected cells and the lack of reliable biomarkers to monitor the reservoir. In addition, the breadth of cells that contribute to the reservoir in different tissues that harbor HIV-1 including the brain, gut and lymph nodes remains unknown. Ideally, any cure strategy would be durable or long-lasting as well as targeting a broad range of HIV subtypes and clades. Finally, any cure would need to balance risk with the current standard of care using ART which is a proven and effective way to control and limit HIV disease and transmissions. Despite the clear challenges for using engineered proteins and RNAs to either render cells resistant to HIV-1 infection or to target HIV-1 provirus, it is exciting to speculate, that once optimized, how these tools in combination with other cure strategies, including anti-retroviral therapies, latency reversal agents or cell mediated transplant approaches, could lead to eradication of HIV-1.
Acknowledgements
This work was in part funded by NIH R01 AI138960 and amfAR Innovation Grant 109603-62-RGRL. We thank Dr. Luis Agosto for critical input and suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTERST STATEMENT
None of the listed authors present a conflict of interest or any financial association that influences the objectivity, integrity or interpretation of context of this manuscript.
References
- 1.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R: HIV reservoirs: what, where and how to target them. Nat Rev Microbiol 2016, 14:55–60. [DOI] [PubMed] [Google Scholar]
- 2.Castro-Gonzalez S, Colomer-Lluch M, Serra-Moreno R: Barriers for HIV Cure: The Latent Reservoir. AIDS Res Hum Retroviruses 2018, 34:739–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitman MC, Lau JSY, McMahon JH, Lewin SR: Barriers and strategies to achieve a cure for HIV. Lancet HIV 2018, 5:e317–e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sengupta S, Siliciano RF: Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48:872–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Margolis DM, Archin NM: Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. J Infect Dis 2017, 215:S111–S118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margolis DM, Garcia JV, Hazuda DJ, Haynes BF: Latency reversal and viral clearance to cure HIV-1. Science 2016, 353:aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hakre S, Chavez L, Shirakawa K, Verdin E: Epigenetic regulation of HIV latency. Curr Opin HIV AIDS 2011, 6:19–24. [DOI] [PubMed] [Google Scholar]
- 8.Mbonye U, Karn J: Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455:328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiralli Lester GM, Henderson AJ: Mechanisms of HIV Transcriptional Regulation and Their Contribution to Latency. Mol Biol Int 2012, 2012:614120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, Van Lint C: The Molecular Biology of HIV Latency. Adv Exp Med Biol 2018, 1075:187–212. [DOI] [PubMed] [Google Scholar]
- 11.Mbonye U, Karn J: The Molecular Basis for Human Immunodeficiency Virus Latency. Annu Rev Virol 2017, 4:261–285. [DOI] [PubMed] [Google Scholar]
- 12.Mousseau G, Valente ST: Role of Host Factors on the Regulation of Tat-Mediated HIV-1 Transcription. Curr Pharm Des 2017, 23:4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cary DC, Fujinaga K, Peterlin BM: Molecular mechanisms of HIV latency. J Clin Invest 2016, 126:448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin AR, Siliciano RF: Progress Toward HIV Eradication: Case Reports, Current Efforts, and the Challenges Associated with Cure. Annu Rev Med 2016, 67:215–28. [DOI] [PubMed] [Google Scholar]
- 15.Jean MJ, Fiches G, Hayashi T, Zhu J: Current Strategies for Elimination of HIV-1 Latent Reservoirs Using Chemical Compounds Targeting Host and Viral Factors. AIDS Res Hum Retroviruses 2019, 35:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim Y, Anderson JL, Lewin SR: Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spivak AM, Planelles V: Novel Latency Reversal Agents for HIV-1 Cure. Annu Rev Med 2018, 69:421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darcis G, Van Driessche B, Van Lint C: HIV Latency: Should We Shock or Lock? Trends Immunol 2017, 38:217–228. [DOI] [PubMed] [Google Scholar]
- 19.Abner E, Jordan A: HIV “shock and kill” therapy: In need of revision. Antiviral Res 2019, 166:19–34. [DOI] [PubMed] [Google Scholar]
- 20.Pinzone MR, O’Doherty U: Measuring integrated HIV DNA ex vivo and in vitro provides insights about how reservoirs are formed and maintained. Retrovirology 2018, 15:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massanella M, Richman DD: Measuring the latent reservoir in vivo. J Clin Invest 2016, 126:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.*.Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, et al. : Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med 2016, 22:1043–1049.This study used viral outgrowth assays and limiting dilution PCR to determine the amount of intact HIV sequences capable of replication. They showed that the number of defective sequences accumulate within early stages of infection and that these are a substantial part of the reservoir.
- 23.Wong JK, Yukl SA: Tissue reservoirs of HIV. Curr Opin HIV AIDS 2016, 11:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.**.Telwatte S, Lee S, Somsouk M, Hatano H, Baker C, Kaiser P, Kim P, Chen T-H, Milush J, Hunt PW, et al. : Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog 2018, 14:e1007357.In this study FACS-sorted CD4+ T cells from matched peripheral blood and rectal biopsies obtained from ART-suppressed individuals were compared for expression of different HIV transcripts to get insights into mechanisms of HIV repression in different tissues. Their results showed differences in HIV transcripts between the blood and rectal CD4+ T cells and they suggest this reflects different mechanisms of latency in the different compartments.
- 25.Perreau M, Banga R, Pantaleo G: Targeted Immune Interventions for an HIV-1 Cure. Trends Mol Med 2017, 23:945–961. [DOI] [PubMed] [Google Scholar]
- 26.Kuhlmann A-S, Peterson CW, Kiem H-P: Chimeric antigen receptor T-cell approaches to HIV cure. Curr Opin HIV AIDS 2018, 13:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ: A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res 2000, 28:663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agosto LM, Henderson AJ: CD4+ T Cell Subsets and Pathways to HIV Latency. AIDS Res Hum Retroviruses 2018, 34:780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Churchill MJ, Cowley DJ, Wesselingh SL, Gorry PR, Gray LR: HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J Neurovirol 2015, 21:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marban C, Forouzanfar F, Ait-Ammar A, Fahmi F, El Mekdad H, Daouad F, Rohr O, Schwartz C: Targeting the Brain Reservoirs: Toward an HIV Cure. Front Immunol 2016, 7:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruel T, Schwartz O: Markers of the HIV-1 reservoir: facts and controversies. Curr Opin HIV AIDS 2018, 13:383–388. [DOI] [PubMed] [Google Scholar]
- 32.Lee GQ, Lichterfeld M: Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr Opin HIV AIDS 2016, 11:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyagi M, Karn J: CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J 2007, 26:4985–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyagi M, Pearson RJ, Karn J: Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 2010, 84:6425–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaczmarek Michaels K, Natarajan M, Euler Z, Alter G, Viglianti G, Henderson AJ: Blimp-1, an intrinsic factor that represses HIV-1 proviral transcription in memory CD4+ T cells. J Immunol Baltim Md 1950 2015, 194:3267–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai K, Okamoto T: Transcriptional Repression of Human Immunodeficiency Virus Type 1 by AP-4. J Biol Chem 2006, 281:12495–12505. [DOI] [PubMed] [Google Scholar]
- 37.Bernhard W, Barreto K, Raithatha S, Sadowski I: An upstream YY1 binding site on the HIV-1 LTR contributes to latent infection. PloS One 2013, 8:e77052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barton K, Margolis D: Selective targeting of the repressive transcription factors YY1 and cMyc to disrupt quiescent human immunodeficiency viruses. AIDS Res Hum Retroviruses 2013, 29:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oteiza A, Mechti N: FoxO4 negatively controls Tat-mediated HIV-1 transcription through the post-transcriptional suppression of Tat encoding mRNA. J Gen Virol 2017, 98:1864–1878. [DOI] [PubMed] [Google Scholar]
- 40.Roux A, Leroy H, Muylder BD, Bracq L, Oussous S, Dusanter-Fourt I, Chougui G, Tacine R, Randriamampita C, Desjardins D, et al. : FOXO1 transcription factor plays a key role in T cell—HIV-1 interaction. PLOS Pathog 2019, 15:e1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnes CE, English DM, Cowley SM: Acetylation & Co: an expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem 2019, 63:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Placek K, Schultze JL, Aschenbrenner AC: Epigenetic reprogramming of immune cells in injury, repair, and resolution. J Clin Invest 2019, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Jiang G, Nguyen D, Archin NM, Yukl SA, Méndez-Lagares G, Tang Y, Elsheikh MM, Thompson GR, Hartigan-O’Connor DJ, Margolis DM, et al. : HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest 2018, 128:1190–1198.This study demonstrated that crotonylation is a histone lysine post-translational modification that influences the epigenetic regulation of latent HIV
- 44.Kumar A, Darcis G, Van Lint C, Herbein G: Epigenetic control of HIV-1 post integration latency: implications for therapy. Clin Epigenetics 2015, 7:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeng MY, Ali I, Ott M: Manipulation of the host protein acetylation network by human immunodeficiency virus type 1. Crit Rev Biochem Mol Biol 2015, 50:314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehm D, Ott M: Host Methyltransferases and Demethylases: Potential New Epigenetic Targets for HIV Cure Strategies and Beyond. AIDS Res Hum Retroviruses 2017, 33:S-8–S-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boehm D, Jeng M, Camus G, Gramatica A, Schwarzer R, Johnson JR, Hull PA, Montano M, Sakane N, Pagans S, et al. : SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21:569–579.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen K, Das B, Dobrowolski C, Karn J: Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. mBio 2017, 8:e00133–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imai K, Togami H, Okamoto T: Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem 2010, 285:16538–16545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouchat S, Gatot J-S, Kabeya K, Cardona C, Colin L, Herbein G, De Wit S, Clumeck N, Lambotte O, Rouzioux C, et al. : Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS Lond Engl 2012, 26:1473–1482. [DOI] [PubMed] [Google Scholar]
- 51.Mahmoudi T: The BAF complex and HIV latency. Transcription 2012, 3:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T: Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. PLOS Biol 2011, 9:e1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Duyne R, Guendel I, Narayanan A, Gregg E, Shafagati N, Tyagi M, Easley R, Klase Z, Nekhai S, Kehn-Hall K, et al. : Varying modulation of HIV-1 LTR activity by Baf complexes. J Mol Biol 2011, 411:581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizutani T, Ishizaka A, Tomizawa M, Okazaki T, Yamamichi N, Kawana-Tachikawa A, Iwamoto A, Iba H: Loss of the Brm-type SWI/SNF chromatin remodeling complex is a strong barrier to the Tat-independent transcriptional elongation of human immunodeficiency virus type 1 transcripts. J Virol 2009, 83:11569–11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn-Hall K, et al. : Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology 2010, 405:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomar S, Ali I, Ott M: A BAF’ling Approach to Curing HIV. Cell Chem Biol 2018, 25:1441–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E: Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci 2007, 104:13690–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li P-C, Planelles V, Bradner JE, et al. : BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle Georget Tex 2013, 12:452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M: BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 2012, 92:1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Z, Guo J, Wu Y, Zhou Q: The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res 2013, 41:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.*.Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou M-M, Ott M: The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol Cell 2017, 67:1001–1012.e6.In this study, a short isoform of BRD4 is identified as a corepressor of HIV-1 transcription. Using genome wide ChIP-seq and ATAC-seq, the authors show that BRD4s binds to BRG1 subunit of BAF complex, a chromatin remodeler, and together, they mediate transcriptional repression of HIV-1 by maintaining a repressive nucleosome at HIV-1 promoter.
- 62.Rice AP: The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr Pharm Des 2017, 23:4098–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rice AP: Roles of CDKs in RNA polymerase II transcription of the HIV-1 genome. Transcription 2019, 10:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razooky BS, Cao Y, Hansen MMK, Perelson AS, Simpson ML, Weinberger LS: Nonlatching positive feedback enables robust bimodality by decoupling expression noise from the mean. PLoS Biol 2017, 15:e2000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV: Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122:169–182. [DOI] [PubMed] [Google Scholar]
- 66.Bacon CW, D’Orso I: CDK9: a signaling hub for transcriptional control. Transcription 2019, 10:57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muniz L, Egloff S, Ughy B, Jády BE, Kiss T: Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog 2010, 6:e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M: HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 2010, 38:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marcello A, Cinelli RA, Ferrari A, Signorelli A, Tyagi M, Pellegrini V, Beltram F, Giacca M: Visualization of in vivo direct interaction between HIV-1 TAT and human cyclin T1 in specific subcellular compartments by fluorescence resonance energy transfer. J Biol Chem 2001, 276:39220–39225. [DOI] [PubMed] [Google Scholar]
- 70.Herrmann CH, Mancini MA: The Cdk9 and cyclin T subunits of TAK/P-TEFb localize to splicing factor-rich nuclear speckle regions. J Cell Sci 2001, 114:1491–1503. [DOI] [PubMed] [Google Scholar]
- 71.Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J: Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J Biol Chem 2018, 293:10009–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Budhiraja S, Ramakrishnan R, Rice AP: Phosphatase PPM1A negatively regulates P-TEFb function in resting CD4(+) T cells and inhibits HIV-1 gene expression. Retrovirology 2012, 9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNamara RP, Bacon CW, D’Orso I: Transcription elongation control by the 7SK snRNP complex: Releasing the pause. Cell Cycle Georget Tex 2016, 15:2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.D’Orso I, Frankel AD: RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol 2010, 17:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McNamara RP, Reeder JE, McMillan EA, Bacon CW, McCann JL, D’Orso I: KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol Cell 2016, 61:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tyagi M, Iordanskiy S, Ammosova T, Kumari N, Smith K, Breuer D, Ilatovskiy AV, Kont YS, Ivanov A, Üren A, et al. : Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015, 12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ammosova T, Obukhov Y, Kotelkin A, Breuer D, Beullens M, Gordeuk VR, Bollen M, Nekhai S: Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PloS One 2011, 6:e18985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jadlowsky JK, Nojima M, Okamoto T, Fujinaga K: Dominant negative mutant cyclin T1 proteins that inhibit HIV transcription by forming a kinase inactive complex with Tat. J Gen Virol 2008, 89:2783–2787. [DOI] [PubMed] [Google Scholar]
- 79.Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M: Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J Am Chem Soc 2006, 128:3148–3149. [DOI] [PubMed] [Google Scholar]
- 80**.Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, Baran P, Fromentin R, Chomont N, Valente ST: An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12:97–108.This study described the small molecule dCA as an inhibitor of Tat function and HIV replication.
- 81.Mediouni S, Chinthalapudi K, Ekka MK, Usui I, Jablonski JA, Clementz MA, Mousseau G, Nowak J, Macherla VR, Beverage JN, et al. : Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li C, Mousseau G, Valente ST: Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mediouni S, Jablonski J, Paris JJ, Clementz MA, Thenin-Houssier S, McLaughlin JP, Valente ST: Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr HIV Res 2015, 13:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin H, Li D, Sivakumaran H, Lor M, Rustanti L, Cloonan N, Wani S, Harrich D: Shutdown of HIV-1 Transcription in T Cells by Nullbasic, a Mutant Tat Protein. mBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leoz M, Kukanja P, Luo Z, Huang F, Cary DC, Peterlin BM, Fujinaga K: HEXIM1-Tat chimera inhibits HIV-1 replication. PLoS Pathog 2018, 14:e1007402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H: Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med 2007, 13:1241–1247. [DOI] [PubMed] [Google Scholar]
- 87.Sung T-L, Rice AP: miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog 2009, 5:e1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chiang K, Sung T-L, Rice AP: Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J Virol 2012, 86:3244–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu H, Hu P-W, Couturier J, Lewis DE, Rice AP: HIV-1 replication in CD4+ T cells exploits the down-regulation of antiviral NEAT1 long non-coding RNAs following T cell activation. Virology 2018, 522:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li L, Feng H, Da Q, Jiang H, Chen L, Xie L, Huang Q, Xiong H, Luo F, Kang L, et al. : Expression of HIV-encoded microRNA-TAR and its inhibitory effect on viral replication in human primary macrophages. Arch Virol 2016, 161:1115–1123. [DOI] [PubMed] [Google Scholar]
- 91.Li M, Li H, Rossi JJ: RNAi in combination with a ribozyme and TAR decoy for treatment of HIV infection in hematopoietic cell gene therapy. Ann N Y Acad Sci 2006, 1082:172–179. [DOI] [PubMed] [Google Scholar]
- 92.Coburn GA, Cullen BR: Potent and Specific Inhibition of Human Immunodeficiency Virus Type 1 Replication by RNA Interference. J Virol 2002, 76:9225–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boden D, Pusch O, Silbermann R, Lee F, Tucker L, Ramratnam B: Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins. Nucleic Acids Res 2004, 32:1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anderson J, Li M-J, Palmer B, Remling L, Li S, Yam P, Yee J-K, Rossi J, Zaia J, Akkina R: Safety and Efficacy of a Lentiviral Vector Containing Three Anti-HIV Genes—CCR5 Ribozyme, Tat-rev siRNA, and TAR Decoy—in SCID-hu Mouse–Derived T Cells. Mol Ther 2007, 15:1182–1188. [DOI] [PubMed] [Google Scholar]
- 95.Michienzi A, Li S, Zaia JA, Rossi JJ: A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc Natl Acad Sci 2002, 99:14047–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gottfredsson M, Bohjanen PR: Human immunodeficiency virus type I as a target for gene therapy. Front Biosci J Virtual Libr 1997, 2:d619–634. [DOI] [PubMed] [Google Scholar]
- 97.Lo KM, Biasolo MA, Dehni G, Palú G, Haseltine WA: Inhibition of replication of HIV-1 by retroviral vectors expressing tat-antisense and anti-tat ribozyme RNA. Virology 1992, 190:176–183. [DOI] [PubMed] [Google Scholar]
- 98.Bala J, Chinnapaiyan S, Dutta RK, Unwalla H: Aptamers in HIV research diagnosis and therapy. RNA Biol 2018, 15:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Scarborough RJ, Gatignol A: RNA Interference Therapies for an HIV-1 Functional Cure. Viruses 2017, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Méndez C, Ledger s, Petoumenos K, Ahlenstiel C, Kelleher AD: RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Surabhi RM, Gaynor RB: RNA Interference Directed against Viral and Cellular Targets Inhibits Human Immunodeficiency Virus Type 1 Replication. J Virol 2002, 76:12963–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nielsen MH, Pedersen FS, Kjems J: Molecular strategies to inhibit HIV-1 replication. Retrovirology 2005, 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benjamin R, Berges BK, Solis-Leal A, Igbinedion O, Strong CL, Schiller MR: TALEN gene editing takes aim on HIV. Hum Genet 2016, 135:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gaj T, Gersbach CA, Barbas CF: ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kwarteng A, Ahuno ST, Kwakye-Nuako G: The therapeutic landscape of HIV-1 via genome editing. AIDS Res Ther 2017, 14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee Y-L, et al. : Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 2008, 26:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smith C: CCR5-modified CD4+ T cells for HIV infection (TRAILBLAZER). Identification no. NCT03666871. Retrieved from https://clinicaltrials.Gov/ct2/show/nct03666871. 2019.
- 108.Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, Martinez-Picado J, Nijhuis M, Wensing AMJ, Lee H, et al. : HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huetter G, Nowak D, Mossner M, Ganepola S, Muessig A, Allers K: Long-term control of HIV by CCR5Δ32/Δ32 stem-cell transplantaion. New Engl J Med 2009, 360. [DOI] [PubMed] [Google Scholar]
- 110.Ji H, Lu P, Liu B, Qu X, Wang Y, Jiang Z, Yang X, Zhong Y, Yang H, Pan H, et al. : Zinc-Finger Nucleases Induced by HIV-1 Tat Excise HIV-1 from the Host Genome in Infected and Latently Infected Cells. Mol Ther - Nucleic Acids 2018, 12:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ebina H, Kanemura Y, Misawa N, Sakuma T, Kobayashi T, Yamamoto T, Koyanagi Y: A High Excision Potential of TALENs for Integrated DNA of HIV-Based Lentiviral Vector. PLOS ONE 2015, 10:e0120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Doudna JA, Charpentier E: The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346:1258096–1258096. [DOI] [PubMed] [Google Scholar]
- 113.Tsui CK, Gupta A, Bassik MC: Finding host targets for HIV therapy. Nat Genet 2017, 49:175–176. [DOI] [PubMed] [Google Scholar]
- 114.Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-Beltran W, et al. : A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet 2017, 49:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X: In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther 2017, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.**.Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, Chen C, Mancuso P, Sariyer R, Ferrante P, et al. : Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat Commun 2019, 10:2753.In this study a combination treatment consisting of antiretroviral therapy (ART) and AAV delivery of CRISPR/Cas9 was employed to treat a humanized mouse model infected with HIV. A subset of mice had decreased HIV DNA and viral rebound was not detected with removal of ART providing a proof of concept that eradication of HIV in vivo is possible.
- 117.Li Z, Wu J, Chavez L, Hoh R, Deeks SG, Pillai SK, Zhou Q: Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog 2019, 15:e1007498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.*.Ebina H, Misawa N, Kanemura Y, Koyanagi Y: Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 2013, 3:2510.In this study, the authors excised an integrated HIV-1 provirus from infected cells by exploiting the CRISPR/Cas9 system with gRNAs targeting the HIV-1 LTRs. They also showed that cells harboring Cas9 and multiplex gRNAs targeting the HIV-1 LTRs are resistant to HIV-1 infection.
- 119.Wang G, Zhao N, Berkhout B, Das AT. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol Ther 2016, 24:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120*.Wang G, Zhao N, Berkhout B, Das AT. A combinatorial CRISPR-Cas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep 2016, 17:2819–2826.This study utilized multiple guide-RNAs to target HIV-1 and delay Cas9 mediated escape. Furthermore, the authors demonstrated that continuous Cas9 activity leads to detrimental hypermutations that inactivate the HIV-1 provirus.
- 121.Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, et al. : RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A 2014, 111:11461–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, Wainberg MA, Liang C: CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep 2016, 15:481–489. [DOI] [PubMed] [Google Scholar]
- 123.Agne M, Blank I, Emhardt AJ, Gäbelein CG, Gawlas F, Gillich N, Gonschorek P, Juretschke TJ, Krämer SD, Louis N, et al. : Modularized CRISPR/dCas9 effector toolkit for target-specific gene regulation. ACS Synth Biol 2014, 3:986–989. [DOI] [PubMed] [Google Scholar]
- 124.Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK: CRISPR RNA–guided activation of endogenous human genes. Nat Methods 2013, 10:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saayman SM, Lazar DC, Scott TA, Hart JR, Takahashi M, Burnett JC, Planelles V, Morris KV, Weinberg MS: Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol Ther J Am Soc Gene Ther 2016, 24:488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ji H, Jiang Z, Lu P, Ma L, Li C, Pan H, Fu Z, Qu X, Wang P, Deng J, et al. : Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol Ther J Am Soc Gene Ther 2016, 24:508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hou P, Chen S, Wang S, Yu X, Chen Y, Jiang M: Genome editing of CXCR4 by CRISPR/Cas9 confers cells resistant to HIV-1 infection. Sci Rep 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.**.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. : CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154:442–451.The authors show that CRISPR/Cas9 system provides a platform for highly specific RNA-guided transcriptional regulation by fusing a nuclease deficient Cas9 with effector domains that have distinct regulatory functions.
- 129.Gao X, Tsang JC, Gaba F, Wu D, Lu L, Liu P: Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res 2014, 42:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. : Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159:647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R: Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 2015, 12:401–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Friedli M, Trono D: The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol 2015, 31:429–51. [DOI] [PubMed] [Google Scholar]
- 133.Wolf G, Greenberg D, Macfarlan TS: Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Kruppel-associated box zinc finger protein family. Mob DNA 2015, 6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ecco G, Imbeault M, Trono D: KRAB zinc finger proteins. Dev Camb Engl 2017, 144:2719–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eberhardy SR, Goncalves J, Coelho S, Segal DJ, Berkhout B, Barbas CF 3rd: Inhibition of human immunodeficiency virus type 1 replication with artificial transcription factors targeting the highly conserved primer-binding site. J Virol 2006, 80:2873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Herchenroder O, Hahne JC, Meyer WK, Thiesen HJ, Schneider J: Repression of the human immunodeficiency virus type 1 promoter by the human KRAB domain results in inhibition of virus production. Biochim Biophys Acta 1999, 1445:216–23. [DOI] [PubMed] [Google Scholar]
- 137.Pengue G, Caputo A, Rossi C, Barbanti-Brodano G, Lania L: Transcriptional silencing of human immunodeficiency virus type 1 long terminal repeat-driven gene expression by the Kruppel-associated box repressor domain targeted to the transactivating response element. J Virol 1995, 69:6577–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reynolds L, Ullman C, Moore M, Isalan M, West MJ, Clapham P, Klug A, Choo Y: Repression of the HIV-1 5’ LTR promoter and inhibition of HIV-1 replication by using engineered zinc-finger transcription factors. Proc Natl Acad Sci U A 2003, 100:1615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rossi C, Gibellini D, Barbanti-Brodano G, Betti M, Boarini C, Pengue G, Lania L, Caputo A: Transiently transfected and stably integrated HIV-1 LTR responds differentially to the silencing activity of the Kruppel-associated box (KRAB) transcriptional repressor domain. J Med Virol 1999, 58:264–72. [DOI] [PubMed] [Google Scholar]
- 140.Segal DJ, Goncalves J, Eberhardy S, Swan CH, Torbett BE, Li X, Barbas CF 3rd: Attenuation of HIV-1 replication in primary human cells with a designed zinc finger transcription factor. J Biol Chem 2004, 279:14509–19. [DOI] [PubMed] [Google Scholar]